
The Interplay between the Unfolded Protein Response, Inflammation and Infection in Cystic Fibrosis




Abstract
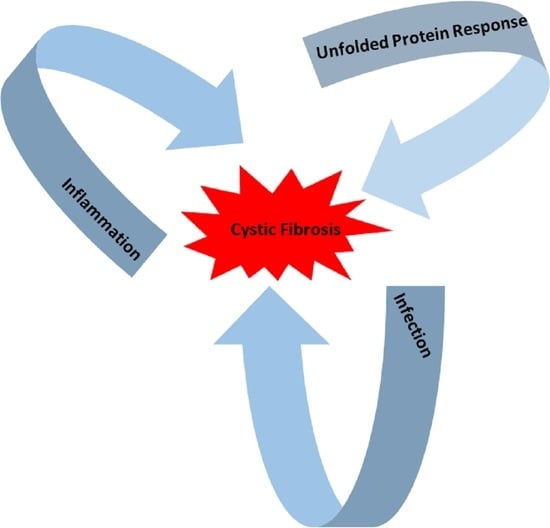
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
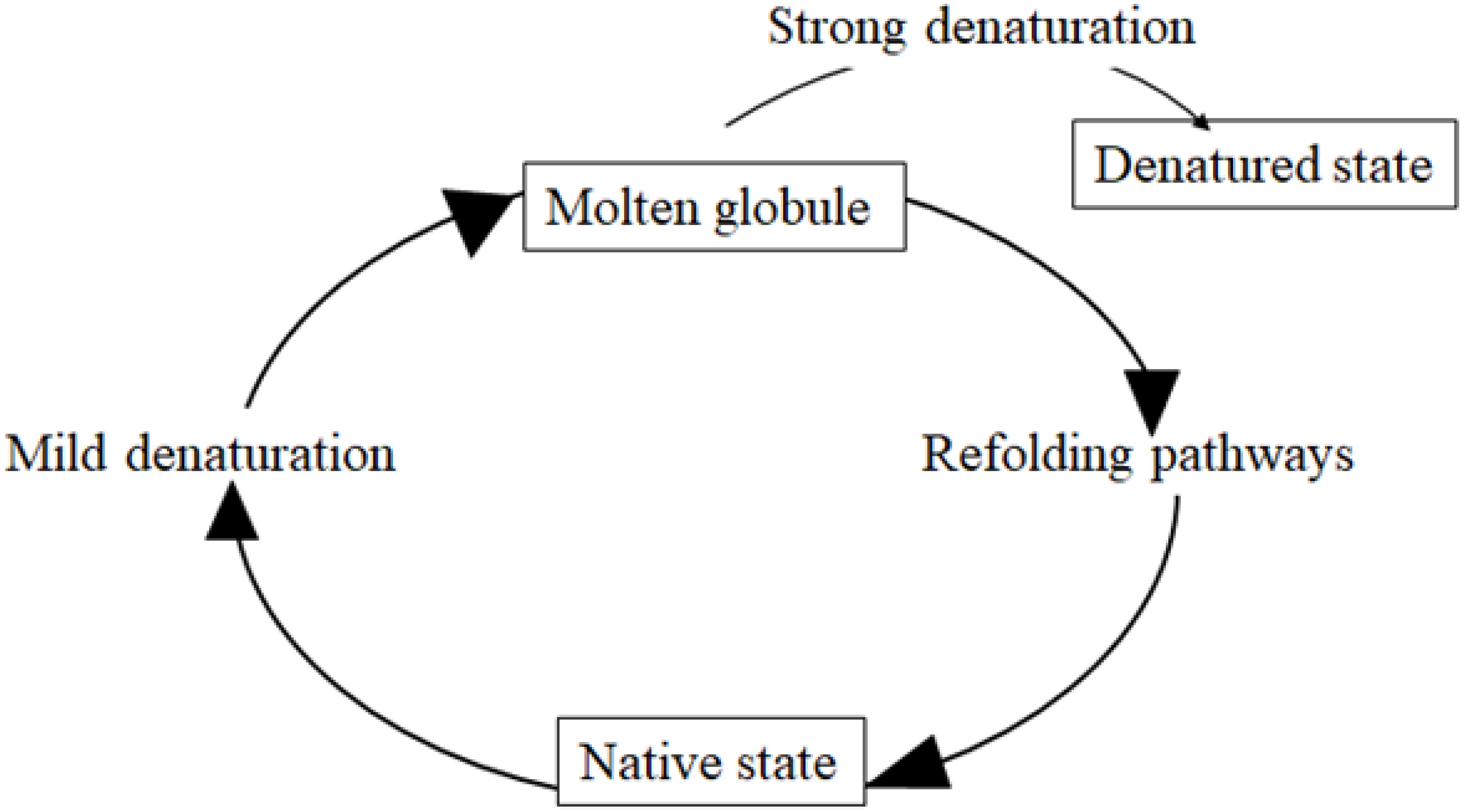
2. Misfolding and Unfolding of Proteins and Their Processing
3. The Unfolded Protein Response (UPR)
3.1. The Binding Immunoglobulin Protein (BiP)
3.2. ATF6 Signaling
3.3. IRE1 Signaling
3.4. PERK Signaling
3.5. Noncoding RNAs and PIWI Proteins
4. Inflammation and UPR in CF
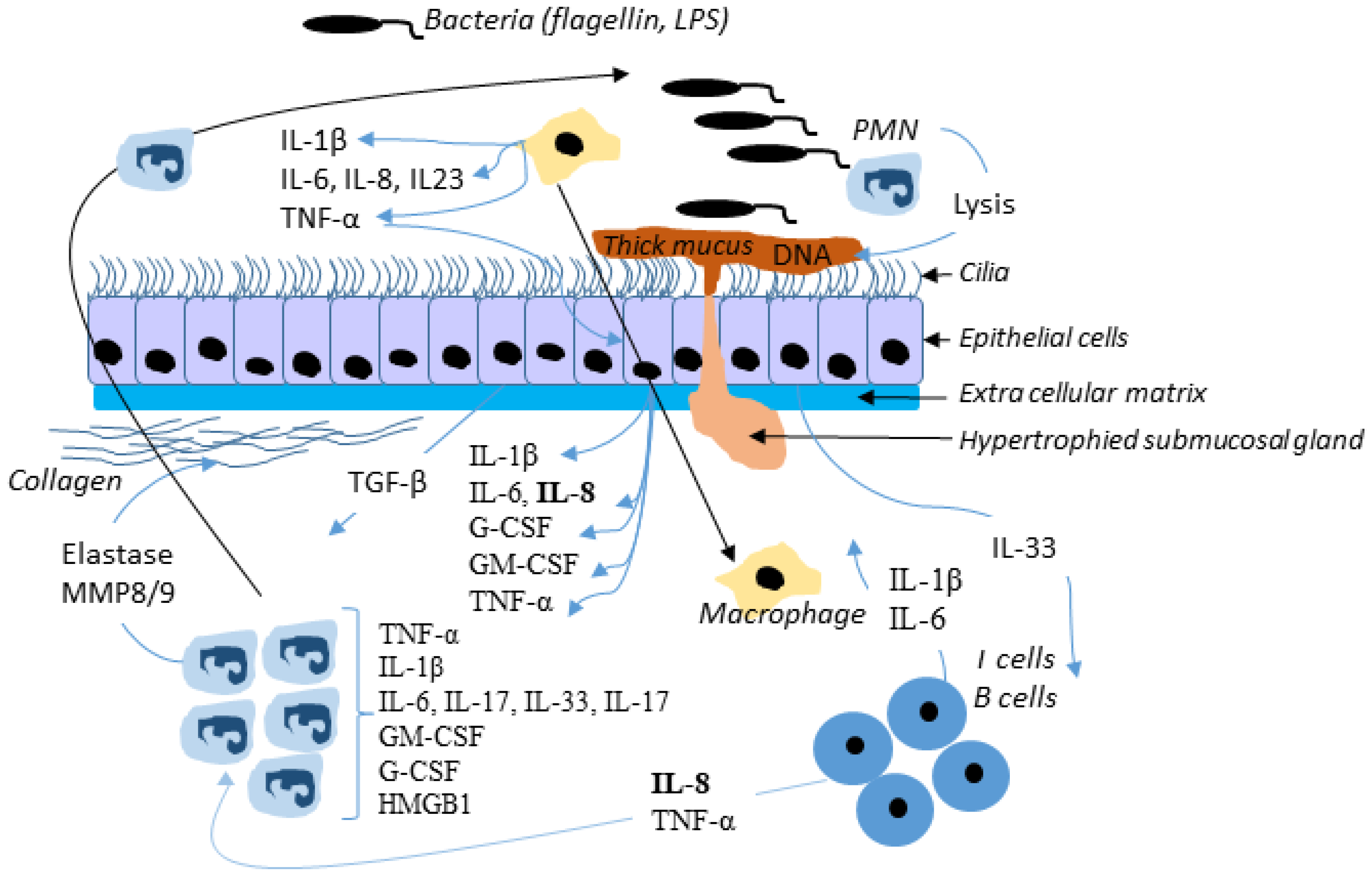
4.1. Inflammation
4.2. UPR and Inflammation Are Functionally Linked
5. Infection and UPR in CF
5.1. Infection in CF
5.2. UPR and Infection Are Functionally Linked
6. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| ATF6/4 | activating transcription factor 6/4 |
| BiP | Binding Immunoglobulin protein |
| CHOP | CAAT/enhancer-binding protein (C/EBP) homologous protein |
| COX-2 | cyclooxygenase-2 |
| cPLA2 | cytosolic phospholipase A2 |
| CREB | cAMP response element-binding protein |
| CREB3L3 | cAMP responsive element-binding protein 3 like 3 |
| eIF2α | eukaryotic translation initiation factor 2α |
| ERAD | Endoplasmic-reticulum-associated protein degradation |
| ERQC | ER Quality Control |
| ERSE | ER stress response element |
| GADD34 | growth arrest and DNA-damage-inducible 34 |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| GRP78 | glucose-regulated protein 78 |
| GSK-3β | Glycogen synthase kinase 3 beta |
| HMGB1 | High-mobility group box 1 |
| ICAM-1 | intercellular adhesion molecule-1 |
| IKK | IκB kinase |
| IRE1 | inositol-requiring enzyme 1 |
| IκB | inhibitor of nuclear factor kappa B |
| LUMAN | cAMP responsive element-binding protein 3 or CREB3 |
| MAMPs | microbial-associated molecular patterns |
| MMP-9 | matrix metalloproteinase-9 |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NLRs | NOD-like receptors |
| NOD1/2 | Nucleotide-binding oligomerization domain-containing protein 1/2 |
| PERK | protein kinase RNA (PKR)-like ER kinase |
| PPARγ | Peroxisome proliferator-activated receptor γ |
| PRRs | pattern recognition receptors |
| RIDD | IRE1-dependent decay |
| TLR3/TLR4 | Toll-like receptors 3/4 |
| TRADD | Tumor necrosis factor receptor type 1-associated DEATH domain protein |
| TRAF2 | TNFα receptor-associated factor 2 |
| TRAF2 | tumor necrosis factor receptor-associated factor 2 |
| VCAM-1 | vascular cell adhesion molecule-1 |
| XBP1 | X-box binding protein 1 |
References
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Rich, D.P.; Anderson, M.P.; Gregory, R.J.; Cheng, S.H.; Paul, S.; Jefferson, D.M.; McCann, J.D.; Klinger, K.W.; Smith, A.E.; Welsh, M.J. Expression of cystic fibrosis transmembrane conductance regulator corrects defective chloride channel regulation in cystic fibrosis airway epithelial cells. Nature 1990, 347, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.P.; Gregory, R.J.; Thompson, S.; Souza, D.W.; Paul, S.; Mulligan, R.C.; Smith, A.E.; Welsh, M.J. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science 1991, 253, 202–205. [Google Scholar] [CrossRef] [Green Version]
- Berdiev, B.K.; Qadri, Y.J.; Benos, D.J. Assessment of the CFTR and ENaC association. Mol. Biosyst. 2009, 5, 123–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heeckeren, A.; Walenga, R.; Konstan, M.W.; Bonfield, T.; Davis, P.B.; Ferkol, T. Excessive inflammatory response of cystic fibrosis mice to bronchopulmonary infection with Pseudomonas Aeruginosa. J. Clin. Investig. 1997, 100, 2810–2815. [Google Scholar] [CrossRef] [PubMed]
- Pier, G.B.; Grout, M.; Zaidi, T.S.; Olsen, J.C.; Johnson, L.G.; Yankaskas, J.R.; Goldberg, J.B. Role of mutant CFTR in hypersusceptibility of cystic fibrosis patients to lung infections. Science 1996, 271, 64–67. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.H.; Gregory, R.J.; Marshall, J.; Paul, S.; Souza, D.W.; White, G.A.; O’Riordan, C.R.; Smith, A.E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990, 63, 827–834. [Google Scholar] [CrossRef]
- Liu, C.Y.; Kaufman, R.J. The unfolded protein response. J. Cell Sci. 2003, 116 Pt 10, 1861–1862. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, R.J. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes Dev. 1999, 13, 1211–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schröder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Ribeiro, C.M.P.; Boucher, R.C. Role of endoplasmic reticulum stress in cystic fibrosis-related airway inflammatory responses. Proc. Am. Thorac. Soc. 2010, 7, 387–394. [Google Scholar] [CrossRef]
- Van’t Wout, E.F.A.; van Schadewijk, A.; van Boxtel, R.; Dalton, L.E.; Clarke, H.J.; Tommassen, J.; Marciniak, S.J.; Hiemstra, P.S. Virulence factors of Pseudomonas Aeruginosa induce both the unfolded protein and integrated stress responses in airway epithelial cells. PLoS Pathog. 2015, 11, e1004946. [Google Scholar] [CrossRef] [Green Version]
- Nanua, S.; Sajjan, U.; Keshavjee, S.; Hershenson, M.B. Absence of typical unfolded protein response in primary cultured cystic fibrosis airway epithelial cells. Biochem. Biophys. Res. Commun. 2006, 343, 135–143. [Google Scholar] [CrossRef]
- Kerbiriou, M.; Le Drévo, M.-A.; Férec, C.; Trouvé, P. Coupling cystic fibrosis to endoplasmic reticulum stress: Differential role of Grp78 and ATF6. Biochim. Biophys. Acta 2007, 1772, 1236–1249. [Google Scholar] [CrossRef] [Green Version]
- Rab, A.; Bartoszewski, R.; Jurkuvenaite, A.; Wakefield, J.; Collawn, J.F.; Bebok, Z. Endoplasmic reticulum stress and the unfolded protein response regulate genomic cystic fibrosis transmembrane conductance regulator expression. Am. J. Physiol. Cell Physiol. 2007, 292, C756–C766. [Google Scholar] [CrossRef] [Green Version]
- Bartoszewski, R.; Rab, A.; Jurkuvenaite, A.; Mazur, M.; Wakefield, J.; Collawn, J.F.; Bebok, Z. Activation of the unfolded protein response by DeltaF508 CFTR. Am. J. Respir. Cell Mol. Biol. 2008, 39, 448–457. [Google Scholar] [CrossRef]
- Hull-Ryde, E.A.; Minges, J.T.; Martino, M.E.B.; Kato, T.; Norris-Drouin, J.L.; Ribeiro, C.M.P. IRE1α is a therapeutic target for cystic fibrosis airway inflammation. Int. J. Mol. Sci. 2021, 22, 3063. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, C.M.P.; Lubamba, B.A. Role of IRE1α/XBP-1 in cystic fibrosis airway inflammation. Int. J. Mol. Sci. 2017, 18, E118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakunts, A.; Orsi, A.; Vitale, M.; Cattaneo, A.; Lari, F.; Tadè, L.; Sitia, R.; Raimondi, A.; Bachi, A.; van Anken, E. Ratiometric sensing of BiP-client versus BiP levels by the unfolded protein response determines its signaling amplitude. eLife 2017, 6, e27518. [Google Scholar] [CrossRef] [PubMed]
- Vitale, M.; Bakunts, A.; Orsi, A.; Lari, F.; Tadè, L.; Danieli, A.; Rato, C.; Valetti, C.; Sitia, R.; Raimondi, A.; et al. Inadequate BiP availability defines endoplasmic reticulum stress. eLife 2019, 8, e41168. [Google Scholar] [CrossRef]
- Blohmke, C.J.; Mayer, M.L.; Tang, A.C.; Hirschfeld, A.F.; Fjell, C.D.; Sze, M.A.; Falsafi, R.; Wang, S.; Hsu, K.; Chilvers, M.A.; et al. Atypical activation of the unfolded protein response in cystic fibrosis airway cells contributes to P38 MAPK-mediated innate immune responses. J. Immunol. 2012, 189, 5467–5475. [Google Scholar] [CrossRef] [Green Version]
- Dobson, C.M. Principles of protein folding, misfolding and aggregation. Semin. Cell Dev. Biol. 2004, 15, 3–16. [Google Scholar] [CrossRef]
- Karplus, M.; Weaver, D.L. Protein-folding dynamics. Nature 1976, 260, 404–406. [Google Scholar] [CrossRef]
- Ohgushi, M.; Wada, A. “Molten-Globule State”: A compact form of globular proteins with mobile side-chains. FEBS Lett. 1983, 164, 21–24. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.J.; Ko, Y.H.; Pedersen, P.L. Altered protein folding may be the molecular basis of most cases of cystic fibrosis. FEBS Lett. 1992, 312, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Bychkova, V.E.; Ptitsyn, O.B. Folding intermediates are involved in genetic diseases? FEBS Lett. 1995, 359, 6–8. [Google Scholar] [CrossRef] [Green Version]
- Drumm, M.L.; Wilkinson, D.J.; Smit, L.S.; Worrell, R.T.; Strong, T.V.; Frizzell, R.A.; Dawson, D.C.; Collins, F.S. Chloride conductance expressed by Delta F508 and other mutant CFTRs in Xenopus Oocytes. Science 1991, 254, 1797–1799. [Google Scholar] [CrossRef] [PubMed]
- Dalemans, W.; Barbry, P.; Champigny, G.; Jallat, S.; Dott, K.; Dreyer, D.; Crystal, R.G.; Pavirani, A.; Lecocq, J.P.; Lazdunski, M. Altered chloride ion channel kinetics associated with the Delta F508 cystic fibrosis mutation. Nature 1991, 354, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.Y.; Partridge, A.W.; Daniels, C.; Du, K.; Lukacs, G.L.; Deber, C.M. Destabilization of the transmembrane domain induces misfolding in a phenotypic mutant of cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 2005, 280, 4968–4974. [Google Scholar] [CrossRef] [Green Version]
- Ward, C.L.; Kopito, R.R. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J. Biol. Chem. 1994, 269, 25710–25718. [Google Scholar] [CrossRef]
- Gottesman, S.; Wickner, S.; Maurizi, M.R. Protein quality control: Triage by chaperones and proteases. Genes Dev. 1997, 11, 815–823. [Google Scholar] [CrossRef] [Green Version]
- Pind, S.; Riordan, J.R.; Williams, D.B. Participation of the endoplasmic reticulum chaperone calnexin (P88, IP90) in the biogenesis of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 1994, 269, 12784–12788. [Google Scholar] [CrossRef]
- Harada, K.; Okiyoneda, T.; Hashimoto, Y.; Ueno, K.; Nakamura, K.; Yamahira, K.; Sugahara, T.; Shuto, T.; Wada, I.; Suico, M.A.; et al. Calreticulin negatively regulates the cell surface expression of cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 2006, 281, 12841–12848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knittler, M.R.; Dirks, S.; Haas, I.G. Molecular chaperones involved in protein degradation in the endoplasmic reticulum: Quantitative interaction of the heat shock cognate protein BiP with partially folded immunoglobulin light chains that are degraded in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 1995, 92, 1764–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCracken, A.A.; Brodsky, J.L. Recognition and delivery of ERAD substrates to the proteasome and alternative paths for cell survival. Curr. Top. Microbiol. Immunol. 2005, 300, 17–40. [Google Scholar] [CrossRef] [PubMed]
- El Khouri, E.; Le Pavec, G.; Toledano, M.B.; Delaunay-Moisan, A. RNF185 is a novel E3 ligase of endoplasmic reticulum-associated degradation (ERAD) that targets cystic fibrosis transmembrane conductance regulator (CFTR). J. Biol. Chem. 2013, 288, 31177–31191. [Google Scholar] [CrossRef] [Green Version]
- Rubenstein, R.C.; Zeitlin, P.L. Sodium 4-phenylbutyrate Downregulates Hsc70: Implications for Intracellular Trafficking of DeltaF508-CFTR. Available online: https://pubmed.ncbi.nlm.nih.gov/10666020/ (accessed on 21 August 2020).
- Meacham, G.C.; Lu, Z.; King, S.; Sorscher, E.; Tousson, A.; Cyr, D.M. The Hdj-2/Hsc70 Chaperone pair facilitates early steps in CFTR biogenesis. EMBO J. 1999, 18, 1492–1505. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Nijbroek, G.; Sullivan, M.L.; McCracken, A.A.; Watkins, S.C.; Michaelis, S.; Brodsky, J.L. Hsp70 molecular Chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol. Biol. Cell 2001, 12, 1303–1314. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Rab, A.; Tang, L.P.; Bebok, Z.; Rowe, S.M.; Bartoszewski, R.; Collawn, J.F. ΔF508 CFTR surface stability is regulated by DAB2 and CHIP-mediated ubiquitination in post-endocytic compartments. PLoS ONE 2015, 10, e0123131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Gething, M.J.; Sambrook, J. Protein folding in the cell. Nature 1992, 355, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Pobre, K.F.R.; Poet, G.J.; Hendershot, L.M. The endoplasmic reticulum (ER) Chaperone BiP is a master regulator of ER functions: Getting by with a little help from ERdj friends. J. Biol. Chem. 2019, 294, 2098–2108. [Google Scholar] [CrossRef] [Green Version]
- Ting, J.; Lee, A.S. Human Gene Encoding the 78,000-Dalton Glucose-Regulated Protein and Its Pseudogene: Structure, Conservation, and Regulation; DNA Mary Ann Liebert Inc.: Larchmont, NY, USA, 1988; Volume 7, pp. 275–286. [Google Scholar] [CrossRef]
- Melnyk, A.; Rieger, H.; Zimmermann, R. Co-Chaperones of the mammalian endoplasmic reticulum. Subcell. Biochem. 2015, 78, 179–200. [Google Scholar] [CrossRef] [PubMed]
- Preissler, S.; Rato, C.; Chen, R.; Antrobus, R.; Ding, S.; Fearnley, I.M.; Ron, D. AMPylation matches BiP activity to client protein load in the endoplasmic reticulum. eLife 2015, 4, e12621. [Google Scholar] [CrossRef] [PubMed]
- Gopal, U.; Pizzo, S.V. Cell surface GRP78 signaling: An emerging role as a transcriptional modulator in cancer. J. Cell. Physiol. 2021, 236, 2352–2363. [Google Scholar] [CrossRef]
- Okada, T.; Haze, K.; Nadanaka, S.; Yoshida, H.; Seidah, N.G.; Hirano, Y.; Sato, R.; Negishi, M.; Mori, K. A Serine Protease Inhibitor Prevents Endoplasmic Reticulum Stress-Induced Cleavage but Not Transport of the Membrane-Bound Transcription Factor ATF6. Available online: https://pubmed.ncbi.nlm.nih.gov/12782636/ (accessed on 15 October 2021).
- Shen, J.; Chen, X.; Hendershot, L.; Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of golgi localization signals. Dev. Cell 2002, 3, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Hai, T.W.; Liu, F.; Coukos, W.J.; Green, M.R. Transcription factor ATF CDNA clones: An extensive family of leucine zipper proteins able to selectively form DNA-binding heterodimers. Genes Dev. 1989, 3, 2083–2090. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Niemeijer, M.; van de Water, B.; Beltman, J.B. ATF6 is a critical determinant of CHOP dynamics during the unfolded protein response. iScience 2020, 23, 100860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, B.; Lee, A.S. The mammalian endoplasmic reticulum stress response element consists of an evolutionarily conserved tripartite structure and interacts with a novel stress-inducible complex. Nucleic Acids Res. 1999, 27, 1437–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, H.; Haze, K.; Yanagi, H.; Yura, T.; Mori, K. Identification of the Cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 1998, 273, 33741–33749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haze, K.; Okada, T.; Yoshida, H.; Yanagi, H.; Yura, T.; Negishi, M.; Mori, K. Identification of the G13 (CAMP-response-element-binding protein-related protein) gene product related to activating transcription factor 6 as a transcriptional activator of the mammalian unfolded protein response. Biochem. J. 2001, 355 Pt 1, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Thuerauf, D.J.; Marcinko, M.; Belmont, P.J.; Glembotski, C.C. Effects of the isoform-specific characteristics of ATF6 alpha and ATF6 beta on endoplasmic reticulum stress response gene expression and cell viability. J. Biol. Chem. 2007, 282, 22865–22878. [Google Scholar] [CrossRef] [Green Version]
- Asada, R.; Kanemoto, S.; Kondo, S.; Saito, A.; Imaizumi, K. The signalling from endoplasmic reticulum-resident BZIP transcription factors involved in diverse cellular physiology. J. Biochem. 2011, 149, 507–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwawaki, T.; Hosoda, A.; Okuda, T.; Kamigori, Y.; Nomura-Furuwatari, C.; Kimata, Y.; Tsuru, A.; Kohno, K. Translational control by the ER transmembrane kinase/ribonuclease IRE1 under ER stress. Nat. Cell Biol. 2001, 3, 158–164. [Google Scholar] [CrossRef]
- Martino, M.B.; Jones, L.; Brighton, B.; Ehre, C.; Abdulah, L.; Davis, C.W.; Ron, D.; O’Neal, W.K.; Ribeiro, C.M.P. The ER stress transducer IRE1β is required for airway epithelial mucin production. Mucosal Immunol. 2013, 6, 639–654. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.M.U.; Bagratuni, T.; Davenport, E.L.; Nowak, P.R.; Silva-Santisteban, M.C.; Hardcastle, A.; McAndrews, C.; Rowlands, M.G.; Morgan, G.J.; Aherne, W.; et al. Structure of the Ire1 autophosphorylation complex and implications for the unfolded protein response. EMBO J. 2011, 30, 894–905. [Google Scholar] [CrossRef] [Green Version]
- Sanches, M.; Duffy, N.M.; Talukdar, M.; Thevakumaran, N.; Chiovitti, D.; Canny, M.D.; Lee, K.; Kurinov, I.; Uehling, D.; Al-awar, R.; et al. Structure and mechanism of action of the hydroxy-aryl-aldehyde class of IRE1 endoribonuclease inhibitors. Nat. Commun. 2014, 5, 4202. [Google Scholar] [CrossRef] [Green Version]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [Green Version]
- Sidrauski, C.; Walter, P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates MRNA splicing in the unfolded protein response. Cell 1997, 90, 1031–1039. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 MRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Travers, K.J.; Patil, C.K.; Wodicka, L.; Lockhart, D.J.; Weissman, J.S.; Walter, P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000, 101, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Iwakoshi, N.N.; Lee, A.-H.; Glimcher, L.H. The X-box binding protein-1 transcription factor is required for plasma cell differentiation and the unfolded protein response. Immunol. Rev. 2003, 194, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Sriburi, R.; Jackowski, S.; Mori, K.; Brewer, J.W. XBP1: A link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 2004, 167, 35–41. [Google Scholar] [CrossRef] [Green Version]
- So, J.-S.; Hur, K.Y.; Tarrio, M.; Ruda, V.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Lichtman, A.H.; Iwawaki, T.; Glimcher, L.H.; et al. Silencing of lipid metabolism genes through IRE1α-mediated MRNA decay lowers plasma lipids in mice. Cell Metab. 2012, 16, 487–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Lee, J.; Reno, C.M.; Sun, C.; Park, S.W.; Chung, J.; Lee, J.; Fisher, S.J.; White, M.F.; Biddinger, S.B.; et al. Regulation of Glucose Homeostasis through a XBP-1-FoxO1 Interaction. Nat. Med. 2011, 17, 356–365. [Google Scholar] [CrossRef] [Green Version]
- Tao, R.; Chen, H.; Gao, C.; Xue, P.; Yang, F.; Han, J.-D.J.; Zhou, B.; Chen, Y.-G. Xbp1-mediated histone H4 deacetylation contributes to DNA double-strand break repair in yeast. Cell Res. 2011, 21, 1619–1633. [Google Scholar] [CrossRef] [Green Version]
- Tirasophon, W.; Lee, K.; Callaghan, B.; Welihinda, A.; Kaufman, R.J. The endoribonuclease activity of mammalian IRE1 autoregulates its MRNA and is required for the unfolded protein response. Genes Dev. 2000, 14, 2725–2736. [Google Scholar] [CrossRef] [Green Version]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef]
- Imagawa, Y.; Hosoda, A.; Sasaka, S.-I.; Tsuru, A.; Kohno, K. RNase domains determine the functional difference between IRE1alpha and IRE1beta. FEBS Lett. 2008, 582, 656–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Vattem, K.M.; Sood, R.; An, J.; Liang, J.; Stramm, L.; Wek, R.C. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol. Cell. Biol. 1998, 18, 7499–7509. [Google Scholar] [CrossRef] [Green Version]
- McQuiston, A.; Diehl, J.A. Recent insights into PERK-dependent signaling from the stressed endoplasmic reticulum. F1000Research 2017, 6, 1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd, M.A.; Osborne, J.C.; Safer, B.; Powell, G.M.; Merrick, W.C. Characteristics of eukaryotic initiation factor 2 and its subunits. J. Biol. Chem. 1980, 255, 1189–1193. [Google Scholar] [CrossRef]
- Ernst, H.; Duncan, R.F.; Hershey, J.W. Cloning and sequencing of complementary DNAs encoding the alpha-subunit of translational initiation factor EIF-2. characterization of the protein and its messenger RNA. J. Biol. Chem. 1987, 262, 1206–1212. [Google Scholar] [CrossRef]
- Adams, S.L.; Safer, B.; Anderson, W.F.; Merrick, W.C. Eukaryotic initiation complex formation. Evidence for two distinct pathways. J. Biol. Chem. 1975, 250, 9083–9089. [Google Scholar] [CrossRef]
- Rowlands, A.G.; Panniers, R.; Henshaw, E.C. The catalytic mechanism of guanine nucleotide exchange factor action and competitive inhibition by phosphorylated eukaryotic initiation factor 2. J. Biol. Chem. 1988, 263, 5526–5533. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Vallejo, M.; Ron, D.; Miller, C.P.; Habener, J.F. C/ATF, a member of the activating transcription factor family of DNA-binding proteins, dimerizes with CAAT/enhancer-binding proteins and directs their binding to CAMP response elements. Proc. Natl. Acad. Sci. USA 1993, 90, 4679–4683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palam, L.R.; Baird, T.D.; Wek, R.C. Phosphorylation of EIF2 facilitates ribosomal bypass of an inhibitory upstream ORF to enhance CHOP translation. J. Biol. Chem. 2011, 286, 10939–10949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, M.; Samali, A.; Chevet, E. Regulation of the unfolded protein response by noncoding RNA. Am. J. Physiol. Cell Physiol. 2017, 313, C243–C254. [Google Scholar] [CrossRef] [Green Version]
- Bartoszewska, S.; Kochan, K.; Madanecki, P.; Piotrowski, A.; Ochocka, R.; Collawn, J.F.; Bartoszewski, R. Regulation of the unfolded protein response by MicroRNAs. Cell. Mol. Biol. Lett. 2013, 18, 555–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisognin, A.; Sales, G.; Coppe, A.; Bortoluzzi, S.; Romualdi, C. MAGIA2: From MiRNA and genes expression data integrative analysis to MicroRNA-transcription factor mixed regulatory circuits (2012 Update). Nucleic Acids Res. 2012, 40, W13–W21. [Google Scholar] [CrossRef] [Green Version]
- Bartoszewski, R.; Brewer, J.W.; Rab, A.; Crossman, D.K.; Bartoszewska, S.; Kapoor, N.; Fuller, C.; Collawn, J.F.; Bebok, Z. The unfolded protein response (UPR)-activated transcription factor X-box-binding protein 1 (XBP1) induces MicroRNA-346 expression that targets the human antigen peptide transporter 1 (TAP1) MRNA and governs immune regulatory genes. J. Biol. Chem. 2011, 286, 41862–41870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebert, M.; Bartoszewska, S.; Janaszak-Jasiecka, A.; Moszyńska, A.; Cabaj, A.; Króliczewski, J.; Madanecki, P.; Ochocka, R.J.; Crossman, D.K.; Collawn, J.F.; et al. PIWI proteins contribute to apoptosis during the UPR in human airway epithelial cells. Sci. Rep. 2018, 8, 16431. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, Y.W.; Siomi, M.C.; Siomi, H. PIWI-Interacting RNA: Its biogenesis and functions. Annu. Rev. Biochem. 2015, 84, 405–433. [Google Scholar] [CrossRef]
- Feghali, C.A.; Wright, T.M. Cytokines in acute and chronic inflammation. Front. Biosci. J. Virtual Libr. 1997, 2, d12–d26. [Google Scholar] [CrossRef] [Green Version]
- Pillarisetti, N.; Williamson, E.; Linnane, B.; Skoric, B.; Robertson, C.F.; Robinson, P.; Massie, J.; Hall, G.L.; Sly, P.; Stick, S.; et al. Infection, inflammation, and lung function decline in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 75–81. [Google Scholar] [CrossRef]
- Ranganathan, S.C.; Parsons, F.; Gangell, C.; Brennan, S.; Stick, S.M.; Sly, P.D.; Australian Respiratory Early Surveillance Team for Cystic Fibrosis. Evolution of pulmonary inflammation and nutritional status in infants and young children with cystic fibrosis. Thorax 2011, 66, 408–413. [Google Scholar] [CrossRef] [Green Version]
- Grommes, J.; Soehnlein, O. Contribution of neutrophils to acute lung injury. Mol. Med. Camb. Mass 2011, 17, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.-T.; Yang, C.-M. Inflammatory signalings involved in airway and pulmonary diseases. Mediat. Inflamm. 2013, 2013, 791231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aghasafari, P.; George, U.; Pidaparti, R. A Review of inflammatory mechanism in airway diseases. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. AI 2019, 68, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Bonfield, T.L.; Panuska, J.R.; Konstan, M.W.; Hilliard, K.A.; Hilliard, J.B.; Ghnaim, H.; Berger, M. Inflammatory cytokines in cystic fibrosis lungs. Am. J. Respir. Crit. Care Med. 1995, 152 Pt 1, 2111–2118. [Google Scholar] [CrossRef]
- Bonfield, T.L.; Konstan, M.W.; Berger, M. Altered respiratory epithelial cell cytokine production in cystic fibrosis. J. Allergy Clin. Immunol. 1999, 104, 72–78. [Google Scholar] [CrossRef]
- Nichols, D.P.; Chmiel, J.F. Inflammation and its genesis in cystic fibrosis. Pediatr. Pulmonol. 2015, 50 (Suppl. 40), S39–S56. [Google Scholar] [CrossRef]
- Bardin, P.; Sonneville, F.; Corvol, H.; Tabary, O. Emerging MicroRNA therapeutic approaches for cystic fibrosis. Front. Pharmacol. 2018, 9, 1113. [Google Scholar] [CrossRef] [Green Version]
- Bardin, P.; Marchal-Duval, E.; Sonneville, F.; Blouquit-Laye, S.; Rousselet, N.; Le Rouzic, P.; Corvol, H.; Tabary, O. Small RNA and transcriptome sequencing reveal the role of MiR-199a-3p in inflammatory processes in cystic fibrosis airways. J. Pathol. 2018, 245, 410–420. [Google Scholar] [CrossRef]
- White, N.M.; Jiang, D.; Burgess, J.D.; Bederman, I.R.; Previs, S.F.; Kelley, T.J. Altered cholesterol homeostasis in cultured and in vivo models of cystic fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L476–L486. [Google Scholar] [CrossRef]
- Brennan, S.; Sly, P.D.; Gangell, C.L.; Sturges, N.; Winfield, K.; Wikstrom, M.; Gard, S.; Upham, J.W.; AREST, C.F. Alveolar macrophages and CC chemokines are increased in children with cystic fibrosis. Eur. Respir. J. 2009, 34, 655–661. [Google Scholar] [CrossRef] [Green Version]
- Bruscia, E.M.; Zhang, P.-X.; Ferreira, E.; Caputo, C.; Emerson, J.W.; Tuck, D.; Krause, D.S.; Egan, M.E. Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator−/− mice. Am. J. Respir. Cell Mol. Biol. 2009, 40, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Del Porto, P.; Cifani, N.; Guarnieri, S.; Di Domenico, E.G.; Mariggiò, M.A.; Spadaro, F.; Guglietta, S.; Anile, M.; Venuta, F.; Quattrucci, S.; et al. Dysfunctional CFTR alters the bactericidal activity of human macrophages against Pseudomonas Aeruginosa. PLoS ONE 2011, 6, e19970. [Google Scholar] [CrossRef] [Green Version]
- Painter, R.G.; Valentine, V.G.; Lanson, N.A.; Leidal, K.; Zhang, Q.; Lombard, G.; Thompson, C.; Viswanathan, A.; Nauseef, W.M.; Wang, G.; et al. CFTR expression in human neutrophils and the phagolysosomal chlorination defect in cystic fibrosis. Biochemistry 2006, 45, 10260–10269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moss, R.B.; Bocian, R.C.; Hsu, Y.P.; Dong, Y.J.; Kemna, M.; Wei, T.; Gardner, P. Reduced IL-10 secretion by CD4+ T lymphocytes expressing mutant cystic fibrosis transmembrane conductance regulator (CFTR). Clin. Exp. Immunol. 1996, 106, 374–388. [Google Scholar] [CrossRef]
- Gao, Z.; Su, X. CFTR regulates acute inflammatory responses in macrophages. QJM Mon. J. Assoc. Phys. 2015, 108, 951–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Cebotaru, L.; Lee, H.W.; Yang, Q.; Pollard, B.S.; Pollard, H.B.; Guggino, W.B. CFTR controls the activity of NF-ΚB by enhancing the degradation of TRADD. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 40, 1063–1078. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, S.; Hiramatsu, N.; Hayakawa, K.; Saito, Y.; Kato, H.; Huang, T.; Yao, J.; Paton, A.W.; Paton, J.C.; Kitamura, M. Selective abrogation of BiP/GRP78 blunts activation of NF-ΚB through the ATF6 branch of the UPR: Involvement of C/EBPβ and MTOR-dependent dephosphorylation of Akt. Mol. Cell. Biol. 2011, 31, 1710–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Zhang, L.; Liu, Q.; Tang, L.; Sun, H.; Guo, H. Endoplasmic reticulum stress preconditioning antagonizes low-density lipoprotein-induced inflammation in human mesangial cells through upregulation of XBP1 and suppression of the IRE1α/IKK/NF-ΚB pathway. Mol. Med. Rep. 2015, 11, 2048–2054. [Google Scholar] [CrossRef] [Green Version]
- Chiang, S.-H.; Bazuine, M.; Lumeng, C.N.; Geletka, L.M.; Mowers, J.; White, N.M.; Ma, J.-T.; Zhou, J.; Qi, N.; Westcott, D.; et al. The protein kinase IKKepsilon regulates energy balance in obese mice. Cell 2009, 138, 961–975. [Google Scholar] [CrossRef] [Green Version]
- Hu, P.; Han, Z.; Couvillon, A.D.; Kaufman, R.J.; Exton, J.H. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-KappaB activation and down-regulation of TRAF2 expression. Mol. Cell. Biol. 2006, 26, 3071–3084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tufanli, O.; Telkoparan Akillilar, P.; Acosta-Alvear, D.; Kocaturk, B.; Onat, U.I.; Hamid, S.M.; Çimen, I.; Walter, P.; Weber, C.; Erbay, E. Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl. Acad. Sci. USA 2017, 114, E1395–E1404. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.-Y.; Wek, S.A.; McGrath, B.C.; Scheuner, D.; Kaufman, R.J.; Cavener, D.R.; Wek, R.C. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-KappaB in response to diverse cellular stresses. Mol. Cell. Biol. 2003, 23, 5651–5663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keestra-Gounder, A.M.; Byndloss, M.X.; Seyffert, N.; Young, B.M.; Chávez-Arroyo, A.; Tsai, A.Y.; Cevallos, S.A.; Winter, M.G.; Pham, O.H.; Tiffany, C.R.; et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature 2016, 532, 394–397. [Google Scholar] [CrossRef] [Green Version]
- Janssens, S.; Pulendran, B.; Lambrecht, B.N. Emerging functions of the unfolded protein response in immunity. Nat. Immunol. 2014, 15, 910–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, B.M.; Pincus, D.; Gotthardt, K.; Gallagher, C.M.; Walter, P. Endoplasmic Reticulum Stress Sensing in the Unfolded Protein Response. Available online: https://pubmed.ncbi.nlm.nih.gov/23388626/ (accessed on 24 August 2020).
- Cui, W.; Li, J.; Ron, D.; Sha, B. The structure of the PERK kinase domain suggests the mechanism for its activation. Acta Crystallogr. D Biol. Crystallogr. 2011, 67 Pt 5, 423–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrara, M.; Prischi, F.; Nowak, P.R.; Ali, M.M. Crystal structures reveal transient PERK luminal domain tetramerization in endoplasmic reticulum stress signaling. EMBO J. 2015, 34, 1589–1600. [Google Scholar] [CrossRef] [PubMed]
- Holcik, M.; Sonenberg, N. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 318–327. [Google Scholar] [CrossRef]
- Proud, C.G. EIF2 and the control of cell physiology. Semin. Cell Dev. Biol. 2005, 16, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.C.; Saferali, A.; He, G.; Sandford, A.J.; Strug, L.J.; Turvey, S.E. Endoplasmic reticulum stress and chemokine production in cystic fibrosis airway cells: Regulation by STAT3 modulation. J. Infect. Dis. 2017, 215, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.B.; Mercado, E.L.; Hoffmann, A.; Niwa, M. ER stress activates NF-ΚB by integrating functions of basal IKK activity, IRE1 and PERK. PLoS ONE 2012, 7, e45078. [Google Scholar] [CrossRef] [Green Version]
- Paramasivan, S.; Bassiouni, A.; Shiffer, A.; Dillon, M.R.; Cope, E.K.; Cooksley, C.; Ramezanpour, M.; Moraitis, S.; Ali, M.J.; Bleier, B.; et al. The international sinonasal microbiome study: A multicentre, multinational characterization of sinonasal bacterial ecology. Allergy 2020, 75, 2037–2049. [Google Scholar] [CrossRef]
- Coburn, B.; Wang, P.W.; Diaz Caballero, J.; Clark, S.T.; Brahma, V.; Donaldson, S.; Zhang, Y.; Surendra, A.; Gong, Y.; Elizabeth Tullis, D.; et al. Lung microbiota across age and disease stage in cystic fibrosis. Sci. Rep. 2015, 5, 10241. [Google Scholar] [CrossRef]
- Vongthilath, R.; Richaud Thiriez, B.; Dehillotte, C.; Lemonnier, L.; Guillien, A.; Degano, B.; Dalphin, M.-L.; Dalphin, J.-C.; Plésiat, P. Clinical and microbiological characteristics of cystic fibrosis adults never colonized by Pseudomonas Aeruginosa: Analysis of the French CF registry. PLoS ONE 2019, 14, e0210201. [Google Scholar] [CrossRef]
- Lipuma, J.J. The changing microbial epidemiology in cystic fibrosis. Clin. Microbiol. Rev. 2010, 23, 299–323. [Google Scholar] [CrossRef] [Green Version]
- Stecenko, A.A.; King, G.; Torii, K.; Breyer, R.M.; Dworski, R.; Blackwell, T.S.; Christman, J.W.; Brigham, K.L. Dysregulated cytokine production in human cystic fibrosis bronchial epithelial cells. Inflammation 2001, 25, 145–155. [Google Scholar] [CrossRef]
- Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature 2007, 449, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Moreira, L.O.; Zamboni, D.S. NOD1 and NOD2 signaling in infection and inflammation. Front. Immunol. 2012, 3, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chmiel, J.F.; Berger, M.; Konstan, M.W. The role of inflammation in the pathophysiology of CF lung disease. Clin. Rev. Allergy Immunol. 2002, 23, 5–27. [Google Scholar] [CrossRef]
- Bedi, B.; Maurice, N.M.; Ciavatta, V.T.; Lynn, K.S.; Yuan, Z.; Molina, S.A.; Joo, M.; Tyor, W.R.; Goldberg, J.B.; Koval, M.; et al. Peroxisome proliferator-activated receptor-γ agonists attenuate biofilm formation by Pseudomonas Aeruginosa. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 3608–3621. [Google Scholar] [CrossRef] [Green Version]
- Ahmadian, M.; Myoung Suh, J.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ Signaling and Metabolism: The Good, the Bad and the Future. Available online: https://pubmed.ncbi.nlm.nih.gov/23652116/ (accessed on 24 August 2020).
- Smith, J.A. Regulation of cytokine production by the unfolded protein response; implications for infection and autoimmunity. Front. Immunol. 2018, 9, 422. [Google Scholar] [CrossRef]
- Bedi, B.; Lin, K.-C.; Maurice, N.M.; Yuan, Z.; Bijli, K.; Koval, M.; Hart, C.M.; Goldberg, J.B.; Stecenko, A.; Sadikot, R.T. UPR modulation of host immunity by Pseudomonas Aeruginosa in cystic fibrosis. Clin. Sci. Lond. Engl. 2020, 134, 1911–1934. [Google Scholar] [CrossRef]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef] [Green Version]
- Paton, A.W.; Srimanote, P.; Talbot, U.M.; Wang, H.; Paton, J.C. A new family of potent AB(5) cytotoxins produced by shiga toxigenic Escherichia Coli. J. Exp. Med. 2004, 200, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Shenderov, K.; Riteau, N.; Yip, R.; Mayer-Barber, K.D.; Oland, S.; Hieny, S.; Fitzgerald, P.; Oberst, A.; Dillon, C.P.; Green, D.R.; et al. Cutting edge: Endoplasmic reticulum stress licenses macrophages to produce mature IL-1β in response to TLR4 stimulation through a caspase-8- and TRIF-dependent pathway. J. Immunol. 2014, 192, 2029–2033. [Google Scholar] [CrossRef] [PubMed]
- Duvigneau, J.C.; Luís, A.; Gorman, A.M.; Samali, A.; Kaltenecker, D.; Moriggl, R.; Kozlov, A.V. Crosstalk between inflammatory mediators and endoplasmic reticulum stress in liver diseases. Cytokine 2019, 124, 154577. [Google Scholar] [CrossRef]
- Dalet, A.; Argüello, R.J.; Combes, A.; Spinelli, L.; Jaeger, S.; Fallet, M.; Vu Manh, T.-P.; Mendes, A.; Perego, J.; Reverendo, M.; et al. Protein synthesis inhibition and GADD34 control IFN-β heterogeneous expression in response to DsRNA. EMBO J. 2017, 36, 761–782. [Google Scholar] [CrossRef] [Green Version]
- Cláudio, N.; Dalet, A.; Gatti, E.; Pierre, P. Mapping the crossroads of immune activation and cellular stress response pathways. EMBO J. 2013, 32, 1214–1224. [Google Scholar] [CrossRef] [Green Version]
- Tilney, L.G.; Harb, O.S.; Connelly, P.S.; Robinson, C.G.; Roy, C.R. How the parasitic bacterium Legionella Pneumophila modifies its phagosome and transforms it into rough ER: Implications for conversion of plasma membrane to the ER membrane. J. Cell Sci. 2001, 114 Pt 24, 4637–4650. [Google Scholar] [CrossRef] [PubMed]
- Hempstead, A.D.; Isberg, R.R. Inhibition of host cell translation elongation by Legionella Pneumophila blocks the host cell unfolded protein response. Proc. Natl. Acad. Sci. USA 2015, 112, E6790–E6797. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, G.K.; Harms, J.S.; Splitter, G.A. Modulation of microtubule dynamics by a TIR domain protein from the intracellular pathogen Brucella Melitensis. Biochem. J. 2011, 439, 79–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shima, K.; Klinger, M.; Schütze, S.; Kaufhold, I.; Solbach, W.; Reiling, N.; Rupp, J. The role of endoplasmic reticulum-related BiP/GRP78 in interferon gamma-induced persistent Chlamydia Pneumoniae infection. Cell. Microbiol. 2015, 17, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Webster, S.J.; Ellis, L.; O’Brien, L.M.; Tyrrell, B.; Fitzmaurice, T.J.; Elder, M.J.; Clare, S.; Chee, R.; Gaston, J.S.H.; Goodall, J.C. IRE1α mediates PKR activation in response to Chlamydia Trachomatis infection. Microbes Infect. 2016, 18, 472–483. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trouvé, P.; Férec, C.; Génin, E. The Interplay between the Unfolded Protein Response, Inflammation and Infection in Cystic Fibrosis. Cells 2021, 10, 2980. https://doi.org/10.3390/cells10112980
Trouvé P, Férec C, Génin E. The Interplay between the Unfolded Protein Response, Inflammation and Infection in Cystic Fibrosis. Cells. 2021; 10(11):2980. https://doi.org/10.3390/cells10112980
Chicago/Turabian StyleTrouvé, Pascal, Claude Férec, and Emmanuelle Génin. 2021. "The Interplay between the Unfolded Protein Response, Inflammation and Infection in Cystic Fibrosis" Cells 10, no. 11: 2980. https://doi.org/10.3390/cells10112980