Direct Electron Transfer of Dehydrogenases for Development of 3rd Generation Biosensors and Enzymatic Fuel Cells




Abstract
:1. Introduction
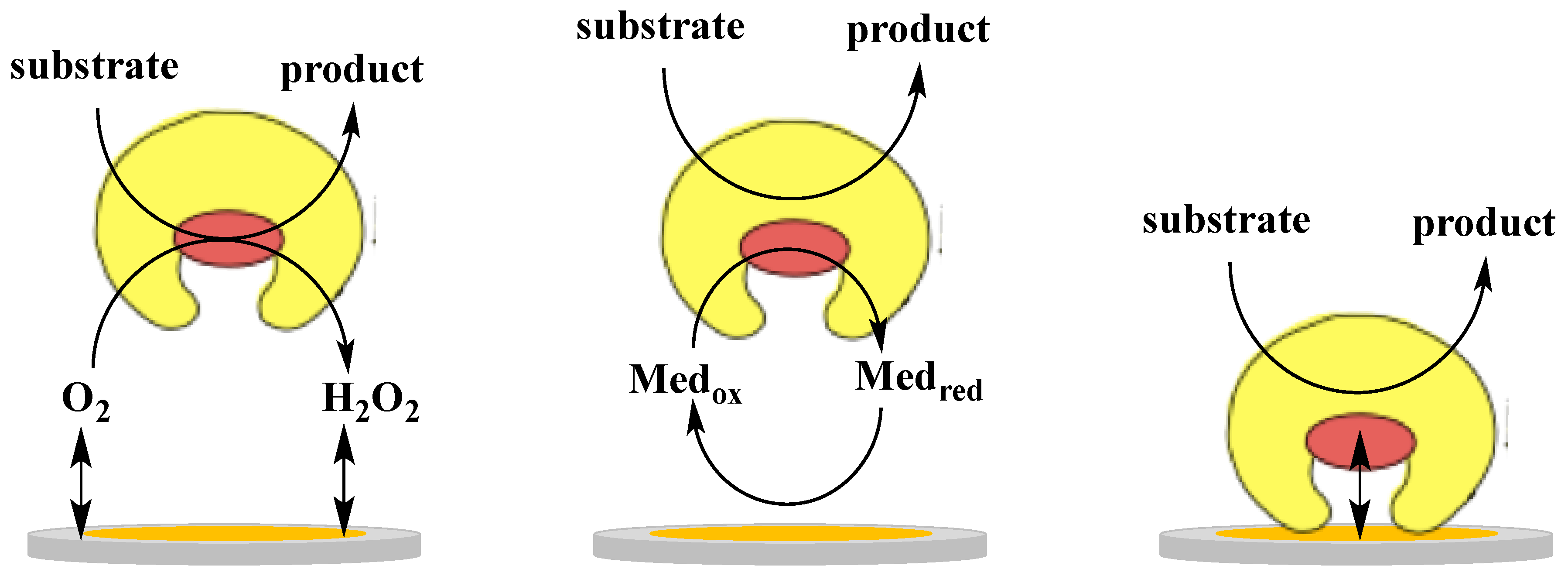
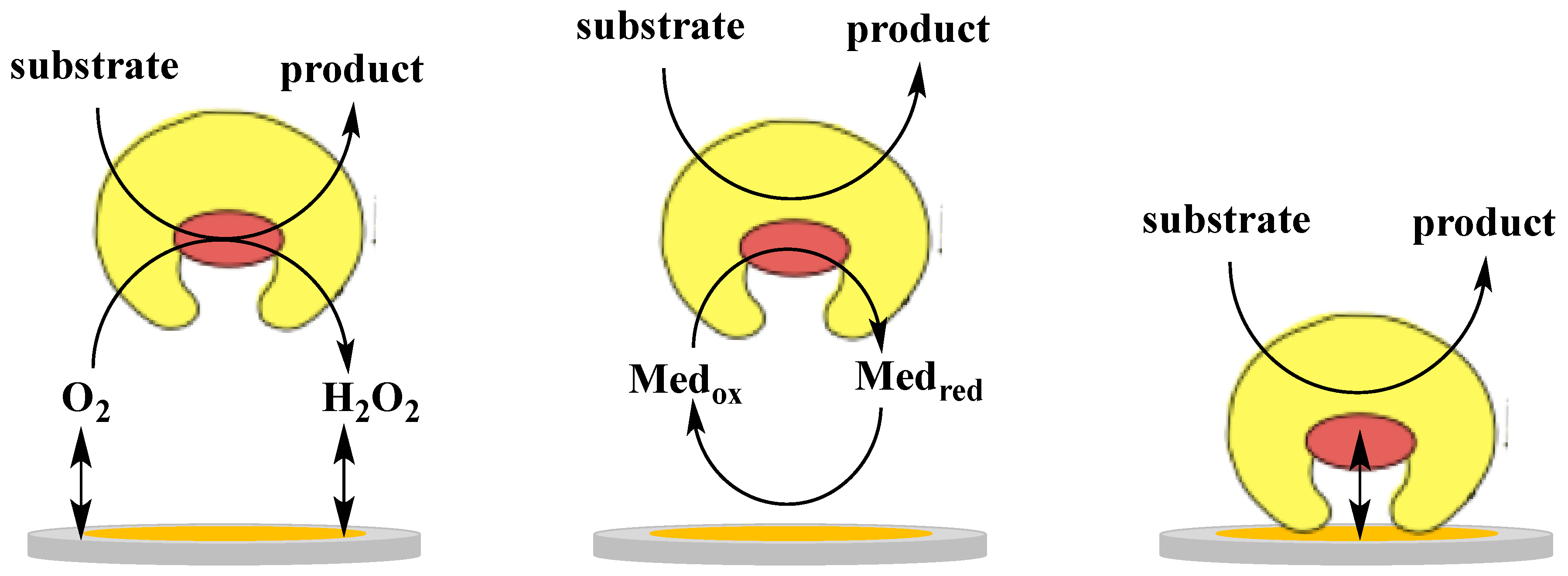
2. Direct Electron Transfer of Dehydrogenases
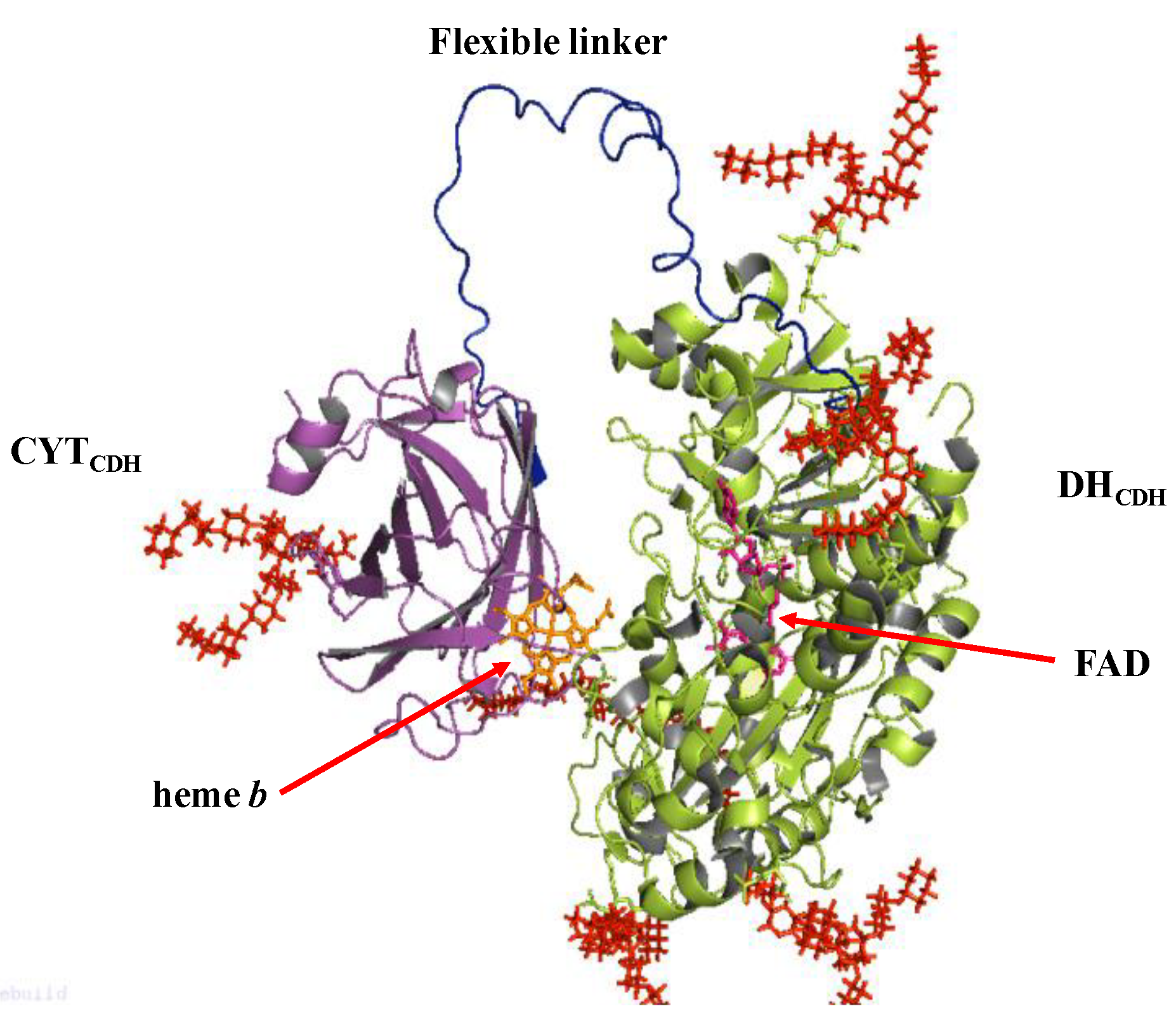
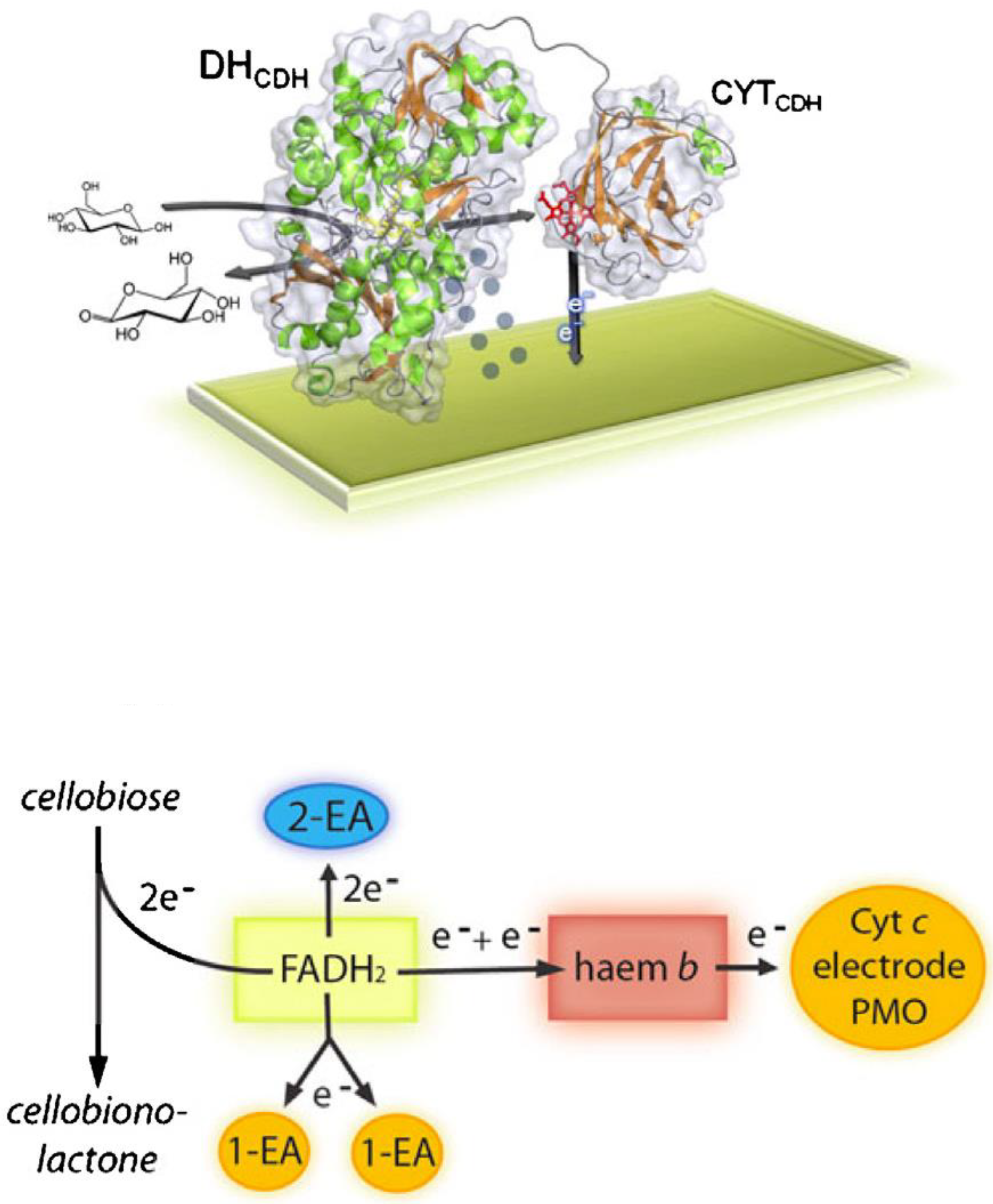
3. Cellobiose Dehydrogenase
3.1. Cellobiose Dehydrogenase Based Biosensors
3.1.1. CDH Based Lactose Biosensors
3.1.2. CDH Based Glucose Biosensors
3.2. CDH Based Biofuel Cells
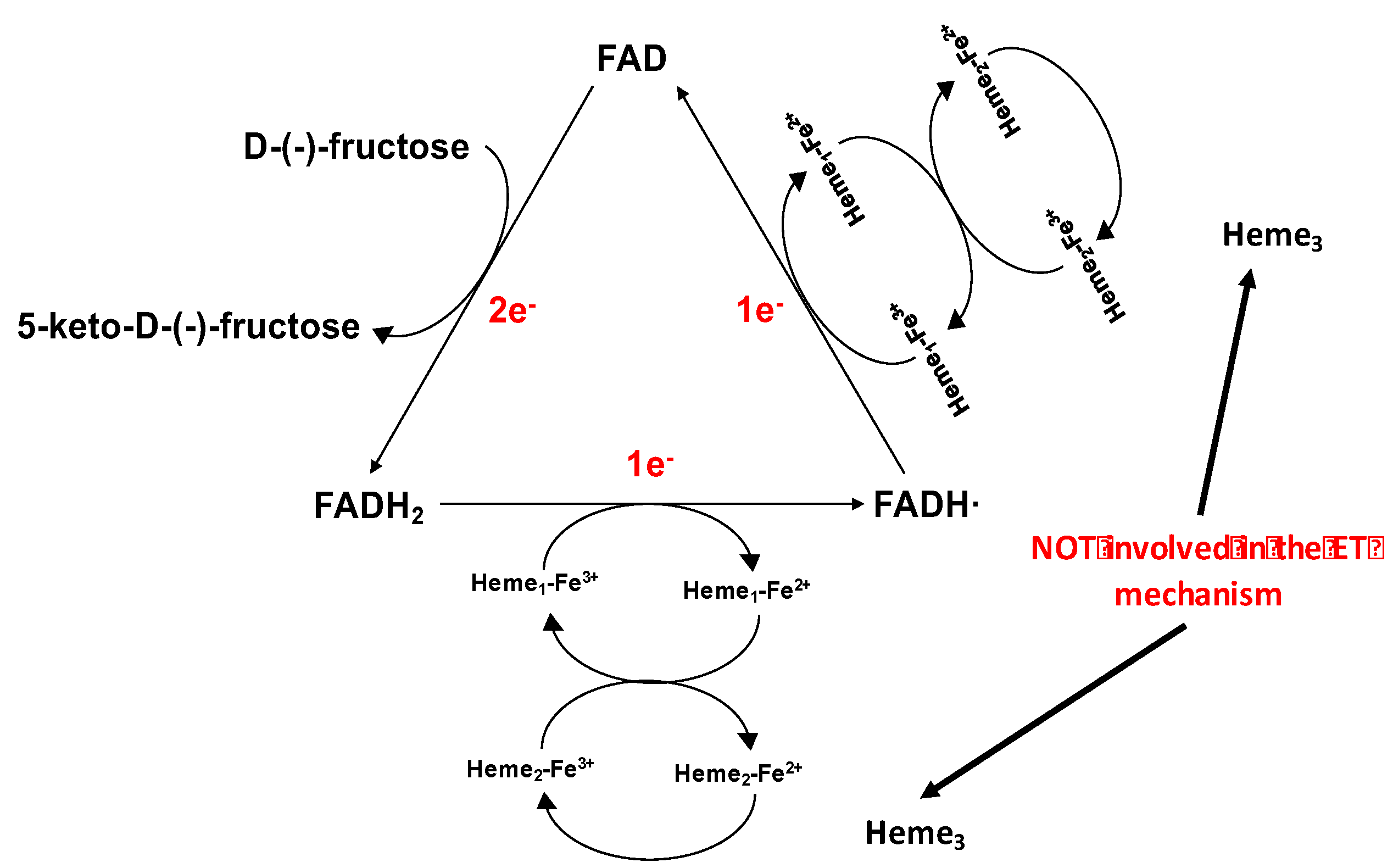
4. Fructose Dehydrogenase
- (1)
- D-(-)-fructose is oxidised to 5-keto-D-(-)-fructose involving 2e−/2H+ with the concomitant reduction of FAD to FADH2;
- (2)
- FADH2 is sequentially reoxidized in two separate 1 ET steps. In the first FADH2 is partially reoxidized to FADH· through the IET pathway between the DHFDH and CYTFDH domains, whereby one of the three heme c moieties (heme c1) is reduced. Next, the electron is transferred from heme c1 to a second heme c (heme c2) of the two hemes involved in the ET pathway and then to a final electron acceptor, which is the electrode when FDH is adsorbed onto the electrode surface;
- (3)
- FADH· is finally reoxidized to FAD by heme c1 and the electron is then transferred to heme c2 (which gives the second internal electron transfer (IET) step), which in turn is reoxidized by the electrode whereby FDH is returned to its fully oxidised state.
4.1. FDH Based Biosensors
4.2. FDH Based Biofuel Cells
5. Enzyme Engineering to Enhance DET for Future Biosensors and BFC Developments
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Cardosi, M.; Liu, Z. Amperometric glucose sensors for whole blood measurement based on dehydrogenase enzymes. In Dehydrogenases; Canuto, R.A., Ed.; InTechOpen: London, UK, 2012; pp. 319–354. [Google Scholar]
- Anthony, C.; Ghosh, M. The structure and function of the PQQ-containing quinoprotein dehydrogenases. Prog. Biophys. Mol. Biol. 1998, 69, 1–21. [Google Scholar] [CrossRef]
- Bray, R.C. Molybdenum-containing enzymes. J. Less Common Met. 1977, 54, 527–536. [Google Scholar] [CrossRef]
- Hille, R.; Hall, J.; Basu, P. The mononuclear molybdenum enzymes. Chem. Rev. 2014, 114, 3963–4038. [Google Scholar] [CrossRef] [PubMed]
- Gorton, L.; Domínguez, E. Electrochemistry of NAD(P)+/NAD(P)H. Encyclopedia of Electrochemistry; Bard, A.J., Stratmann, M., Wilson, G.S., Eds.; Bioelectrochemistry, Wiley-VCH: Weinheim, Germany, 2002; Volume 9, pp. 67–143. [Google Scholar]
- Chenault, H.K.; Whitesides, G.M. Regeneration of nicotinamide cofactors for use in organic-synthesis. Appl. Biochem. Biotechnol. 1987, 14, 147–197. [Google Scholar] [CrossRef] [PubMed]
- Clark, W.M. Oxidation-Reduction Potentials of Organic Systems; Robert E. Krieger Publishing Company: Huntington, NY, USA, 1972. [Google Scholar]
- Gorton, L.; Domínguez, E. Electrocatalytic oxidation of NAD(P)H at mediator-modified electrodes. Rev. Mol. Biotechnol. 2002, 82, 371–392. [Google Scholar] [CrossRef]
- Gorton, L. Carbon-paste electrodes modified with enzymes, tissues and cells. Electroanalysis 1995, 7, 23–45. [Google Scholar] [CrossRef]
- Gorton, L.; Csöregi, E.; Domínguez, E.; Emnéus, J.; Jönsson-Pettersson, G.; Marko-Varga, G.; Persson, B. Selective detection in flow-analysis based on the combination of immobilized enzymes and chemically modified electrodes. Anal. Chim. Acta 1991, 250, 203–248. [Google Scholar] [CrossRef]
- Antiochia, R.; Lavagnini, I. Alcohol biosensor based on the immobilization of meldola blue and alcohol dehydrogenase into a carbon nanotube paste electrode. Anal. Lett. 2006, 39, 1643–1655. [Google Scholar] [CrossRef]
- Antiochia, R.; Gorton, L. Development of a carbon nanotube paste electrode osmium polymer-mediated biosensor for determination of glucose in alcoholic beverages. Biosens. Bioelectron. 2007, 22, 2611–2617. [Google Scholar] [CrossRef] [PubMed]
- Nikitina, O.; Shleev, S.; Gayda, G.; Demkiv, O.; Gonchar, M.; Gorton, L.; Csöregi, E.; Nistor, M. Bi-enzyme biosensor based on NAD+-and glutathione-dependent recombinant formaldehyde dehydrogenase and diaphorase for formaldehyde assay. Sens. Actuators B Chem. 2007, 125, 1–9. [Google Scholar] [CrossRef]
- Antiochia, R.; Palleschi, G. Atri-enzyme electrode probe for the sequential determination of fructose and glucose in the same sample. Anal. Lett. 1997, 30, 683–697. [Google Scholar] [CrossRef]
- Klinman, J.P.; Dove, J.E. Novel Cofactors; Gulf Professional Publishing: Houston, TX, USA, 2001; Volume 58. [Google Scholar]
- Ferri, S.; Kojima, K.; Sode, K. Review of glucose oxidases and glucose dehydrogenases: A bird’s eye view of glucose sensing enzymes. J. Diabetes Sci. Technol. 2011, 5, 1068–1076. [Google Scholar] [CrossRef] [PubMed]
- Heller, A. Electron-conducting redox hydrogels: Design, characteristics and synthesis. Curr. Opin. Chem. Biol. 2006, 10, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Heller, A.; Feldman, B. Electrochemical glucose sensors and their applications in diabetes management. Chem. Rev. 2008, 108, 2482–2505. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Hämmerle, M.; Olsthoorn, A.J.J.; Schuhmann, W.; Schmidt, H.-L.; Duine, J.A.; Heller, A. High current density “wired” quinoprotein glucose dehydrogenase electrode. Anal. Chem. 1993, 65, 238–241. [Google Scholar] [CrossRef]
- Zafar, M.N.; Beden, N.; Leech, D.; Sygmund, C.; Ludwig, R.; Gorton, L. Characterization of different FAD-dependent glucose dehydrogenases for possible use in glucose-based biosensors and biofuel cells. Anal. Bioanal. Chem. 2012, 402, 2069–2077. [Google Scholar] [CrossRef] [PubMed]
- Tsujimura, S.; Kojima, S.; Kano, K.; Ikeda, T.; Sato, M.; Sanada, H.; Omura, H. Novel FAD-dependent glucose dehydrogenase for a dioxygen-insensitive glucose biosensor. Biosci. Biotechnol. Biochem. 2006, 70, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Antiochia, R.; Vinci, G.; Gorton, L. Rapid and direct determination of fructose in food: A new osmium-polymer mediated biosensor. Food Chem. 2013, 140, 742–747. [Google Scholar] [CrossRef] [PubMed]
- Laurinavicius, V.; Kurtinaitiene, B.; Liauksminas, V.; Ramanavicius, A.; Meskys, R.; Rudomanskis, R.; Skotheim, T.; Boguslavsky, L. Oxygen insensitive glucose biosensor based on PQQ-dependent glucose dehydrogenase. Anal. Lett. 1999, 32, 299–316. [Google Scholar] [CrossRef]
- Tseng, T.-F.; Yang, Y.-L.; Lou, S.-L. Chromium Hexacyanoferrate Modified Biosensor Based on PQQ-Dependent Glucose Dehydrogenase. In Proceedings of the 29th Annual International Conference of the Engineering in Medicine and Biology Society, EMBS 2007, Lyon, France, 22–26 August 2007; pp. 2681–2684. [Google Scholar]
- Razumiene, J.; Cirbaite, E.; Razumas, V.; Laurinavicius, V. New mediators for biosensors based on PQQ-dependent alcohol dehydrogenases. Sens. Actuators B Chem. 2015, 207, 1019–1025. [Google Scholar] [CrossRef]
- Laurinavicius, V.; Razumiene, J.; Ramanavicius, A.; Ryabov, A.D. Wiring of PQQ-dehydrogenases. Biosens. Bioelectron. 2004, 20, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Wollenberger, U.; Scheller, F.W. PQQ as redox shuttle for quinoprotein glucose dehydrogenase. Biol. Chem. 1998, 379, 1207–1211. [Google Scholar] [PubMed]
- Pinyou, P.; Ruff, A.; Pöller, S.; Ma, S.; Ludwig, R.; Schuhmann, W. Design of an Os complex modified hydrogel with optimized redox potential for biosensors and biofuel cells. Chem.-A Eur. J. 2016, 22, 5319–5326. [Google Scholar] [CrossRef] [PubMed]
- Gorton, L.; Lindgren, A.; Larsson, T.; Munteanu, F.; Ruzgas, T.; Gazaryan, I. Direct electron transfer between heme-containing enzymes and electrodes as basis for third generation biosensors. Anal. Chim. Acta 1999, 400, 91–108. [Google Scholar] [CrossRef]
- Ikeda, T.; Kobayashi, D.; Matsushita, F.; Sagara, T.; Niki, K. Bioelectrocatalysis at electrodes coated with alcohol-dehydrogenase, a quinohemoprotein with heme c serving as a built-in mediator. J. Electroanal. Chem. 1993, 361, 221–228. [Google Scholar] [CrossRef]
- Milton, R.D.; Minteer, S.D. Direct enzymatic bioelectrocatalysis: Differentiating between myth and reality. J. R. Soc. Interface 2017, 14, 20170253. [Google Scholar] [CrossRef] [PubMed]
- Karyakin, A.A. Principles of direct (mediator free) bioelectrocatalysis. Bioelectrochemistry 2012, 88, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Matsumura, H.; Ishida, T.; Yoshida, M.; Igarashi, K.; Samejima, M.; Ohno, H.; Nakamura, N. pH-dependent electron transfer reaction and direct bioelectrocatalysis of the quinohemoprotein pyranose dehydrogenase. Biochem. Biophys. Res. Commun. 2016, 477, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Ferapontova, E.E.; Gorton, L. Direct electrochemistry of heme multicofactor-containing enzymes on alkanethiol-modified gold electrodes. Bioelectrochemistry 2005, 66, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Ferapontova, E.E.; Ruzgas, T.; Gorton, L. Direct electron transfer of heme- and molybdopterin cofactor-containing chicken liver sulfite oxidase on alkanethiol-modified gold electrodes. Anal. Chem. 2003, 75, 4841–4850. [Google Scholar] [CrossRef] [PubMed]
- Ferapontova, E.E.; Shipovskov, S.; Gorton, L. Bioelectrocatalytic detection of theophylline at theophylline oxidase electrodes. Biosens. Bioelectron. 2007, 22, 2508–2515. [Google Scholar] [CrossRef] [PubMed]
- Sosna, M.; Bonamore, A.; Gorton, L.; Boffi, A.; Ferapontova, E.E. Direct electrochemistry and Os-polymer-mediated bioelectrocatalysis of NADH oxidation by Escherichia coli flavohemoglobin at graphite electrodes. Biosens. Bioelectron. 2013, 42, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Christenson, A.; Dock, E.; Gorton, L.; Ruzgas, T. Direct heterogeneous electron transfer of theophylline oxidase. Biosens. Bioelectron. 2004, 20, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.J.; McElhaney, A.E.; Feng, C.J.; Enemark, J.H.; Armstrong, F.A. Avoltammetric study of interdomain electron transfer within sulfite oxidase. J. Am. Chem. Soc. 2002, 124, 11612–11613. [Google Scholar] [CrossRef] [PubMed]
- Frasca, S.; Rojas, O.; Salewski, J.; Neumann, B.; Stiba, K.; Weidinger, I.M.; Tiersch, B.; Leimkühler, S.; Koetz, J.; Wollenberger, U. Human sulfite oxidase electrochemistry on gold nanoparticles modified electrode. Bioelectrochemistry 2012, 87, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Smutok, O.; Gayda, G.; Gonchar, M.; Schuhmann, W. A novel L-lactate-selective biosensor based on flavocytochrome b2 from methylotrophic yeast Hansenula polymorpha. Biosens. Bioelectron. 2005, 20, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Aguey-Zinsou, K.F.; Bernhardt, P.V.; Kappler, U.; McEwan, A.G. Direct electrochemistry of a bacterial sulfite dehydrogenase. J. Am. Chem. Soc. 2003, 125, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Miyaoka, S.; Matsushita, F.; Kobayashi, D.; Senda, M. Direct bioelectrocatalysis at metal and carbon electrodes modified with adsorbed D-gluconate dehydrogenase or adsorbed alcohol-dehydrogenase from bacterial-membranes. Chem. Lett. 1992, 21, 847–850. [Google Scholar] [CrossRef]
- Ludwig, R.; Harreither, W.; Tasca, F.; Gorton, L. Cellobiose dehydrogenase: A versatile catalyst for electrochemical applications. ChemPhysChem 2010, 11, 2674–2697. [Google Scholar] [CrossRef] [PubMed]
- Tasca, F.; Gorton, L.; Harreither, W.; Haltrich, D.; Ludwig, R.; Nöll, G. Comparison of direct and mediated electron transfer for cellobiose dehydrogenase from Phanerochaete sordida. Anal. Chem. 2009, 81, 2791–2798. [Google Scholar] [CrossRef] [PubMed]
- Kawai, S.; Yakushi, T.; Matsushita, K.; Kitazumi, Y.; Shirai, O.; Kano, K. The electron transfer pathway in direct electrochemical communication of fructose dehydrogenase with electrodes. Electrochem. Commun. 2014, 38, 28–31. [Google Scholar] [CrossRef]
- Kamitaka, Y.; Tsujimura, S.; Setoyama, N.; Kajino, T.; Kano, K. Fructose/dioxygen biofuel cell based on direct electron transfer-type bioelectrocatalysis. Phys. Chem. Chem. Phys. 2007, 9, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Kano, K. Bioelectrocatalysis-based application of quinoproteins and quinoprotein-containing bacterial cells in biosensors and biofuel cells. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2003, 1647, 121–126. [Google Scholar] [CrossRef]
- So, K.; Kawai, S.; Hamano, Y.; Kitazumi, Y.; Shirai, O.; Hibi, M.; Ogawa, J.; Kano, K. Improvement of a direct electron transfer-type fructose/dioxygen biofuel cell with a substrate-modified biocathode. Phys. Chem. Chem. Phys. 2014, 16, 4823–4829. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Andoralov, V.; Blum, Z.; Sotres, J.; Suyatin, D.B.; Ruzgas, T.; Arnebrant, T.; Shleev, S. Biofuel cell as a power source for electronic contact lenses. Biosens. Bioelectron. 2012, 37, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Krikstolaityte, V.; Lamberg, P.; Toscano, M.; Silow, M.; Eicher-Lorka, O.; Ramanavicius, A.; Niaura, G.; Abariute, L.; Ruzgas, T.; Shleev, S. Mediatorless carbohydrate/oxygen biofuel cells with improved cellobiose dehydrogenase based bioanode. Fuel Cells 2014, 14, 792–800. [Google Scholar] [CrossRef]
- Shleev, S. Quo vadis, implanted fuel cell? ChemPlusChem 2017, 82, 522–539. [Google Scholar] [CrossRef]
- Wang, X.; Falk, M.; Ortiz, R.; Matsumura, H.; Bobacka, J.; Ludwig, R.; Bergelin, M.; Gorton, L.; Shleev, S. Mediatorless sugar/oxygen enzymatic fuel cells based on gold nanoparticle-modified electrodes. Biosens. Bioelectron. 2012, 31, 219–225. [Google Scholar] [CrossRef] [PubMed]
- DirectSens Biosensors. Available online: http://www.directsens.com/ (accessed on 3 April 2018).
- Heller, A. Electrical wiring of redox enzymes. Acc. Chem. Res. 1990, 23, 128–134. [Google Scholar] [CrossRef]
- Ruff, A. Redox polymers in bioelectrochemistry: Common playgrounds and novel concepts. Curr. Opin. Electrochem. 2017, 5, 66–73. [Google Scholar] [CrossRef]
- Milton, R.D.; Wang, T.; Knoche, K.L.; Minteer, S.D. Tailoring biointerfaces for electrocatalysis. Langmuir 2016, 32, 2291–2301. [Google Scholar] [CrossRef] [PubMed]
- Prabhulkar, S.; Tian, H.; Wang, X.; Zhu, J.-J.; Li, C.-Z. Engineered proteins: Redox properties and their applications. Antioxid. Redox Signal. 2012, 17, 1796–1822. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M.; Mathias, J.P.; Seto, C.T. Molecular self-assembly and nanochemistry: A chemical strategy for the synthesis of nanostructures. Science 1991, 254, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Blum, Z.; Shleev, S. Direct electron transfer based enzymatic fuel cells. Electrochim. Acta 2012, 82, 191–202. [Google Scholar] [CrossRef]
- Flexer, V.; Mano, N. Wired pyrroloquinoline quinone soluble glucose dehydrogenase enzyme electrodes operating at unprecedented low redox potential. Anal. Chem. 2014, 86, 2465–2473. [Google Scholar] [CrossRef] [PubMed]
- Borgmann, S.; Hartwich, G.; Schulte, A.; Schuhmann, W. Amperometric Enzyme Sensors based on Direct and Mediated Electron Transfer. In Electrochemistry of Nucleic Acids and Proteins: Towards Electrochemical Sensors for Genomics and Proteomics; Palecek, E., Scheller, F., Wang, J., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; Volume 1, pp. 599–655. [Google Scholar]
- Ferapontova, E.E.; Shleev, S.; Ruzgas, T.; Stoica, L.; Christenson, A.; Tkac, J.; Yaropolov, A.I.; Gorton, L. Direct Electrochemistry of Proteins and Enzymes. In Electrochemistry of Nucleic Acids and Proteins: Towards Electrochemical Sensors for Genomics and Proteomics; Palecek, E., Scheller, F., Wang, J., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; Volume 1, pp. 517–598. [Google Scholar]
- Leger, C.; Bertrand, P. Direct electrochemistry of redox enzymes as a tool for mechanistic studies. Chem. Rev. 2008, 108, 2379–2438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghindilis, A.L.; Atanasov, P.; Wilkins, E. Enzyme-catalyzed direct electron transfer: Fundamentals and analytical applications. Electroanalysis 1997, 9, 661–674. [Google Scholar] [CrossRef]
- Wollenberger, U.; Spricigo, R.; Leimkühler, S.; Schröder, K. Protein electrodes with direct electrochemical communication. In Biosensing for the 21st Century; Renneberg, R., Lisdat, F., Eds.; Springer: Berlin, Germany, 2008; Volume 109, pp. 19–64. [Google Scholar]
- Wollenberger, U. Third Generation Biosensors—Integrating Recognition and Transduction in Electrochemical Sensors; Elsevier Science B.V.: Amsterdam, The Netherlands, 2005; pp. 65–130. [Google Scholar]
- Zhang, W.J.; Li, G.X. Third-generation biosensors based on the direct electron transfer of proteins. Anal. Sci. 2004, 20, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Das, M.; Chinnadayyala, S.R.; Singha, I.M.; Goswami, P. Recent advances on developing 3rd generation enzyme electrode for biosensor applications. Biosens. Bioelectron. 2016, 79, 386–397. [Google Scholar] [CrossRef] [PubMed]
- De Poulpiquet, A.; Ciaccafava, A.; Lojou, E. New trends in enzyme immobilization at nanostructured interfaces for efficient electrocatalysis in biofuel cells. Electrochim. Acta 2014, 126, 104–114. [Google Scholar] [CrossRef]
- de Poulpiquet, A.; Ranava, D.; Monsalve, K.; Giudici-Orticoni, M.-T.; Lojou, E. Biohydrogen for a new generation of H2/O2 biofuel cells: A sustainable energy perspective. ChemElectroChem 2014, 1, 1724–1750. [Google Scholar] [CrossRef]
- Shleev, S.; Jarosz-Wilkolazka, A.; Khalunina, A.; Morozova, O.; Yaropolov, A.; Ruzgas, T.; Gorton, L. Direct electron transfer reactions of laccases from different origins on carbon electrodes. Bioelectrochemistry 2005, 67, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Shleev, S.; Tkac, J.; Christenson, A.; Ruzgas, T.; Yaropolov, A.I.; Whittaker, J.W.; Gorton, L. Direct electron transfer between copper-containing proteins and electrodes. Biosens. Bioelectron. 2005, 20, 2517–2554. [Google Scholar] [CrossRef] [PubMed]
- Cracknell, J.A.; Vincent, K.A.; Armstrong, F.A. Enzymes as working or inspirational electrocatalysts for fuel cells and electrolysis. Chem. Rev. 2008, 108, 2439–2461. [Google Scholar] [CrossRef] [PubMed]
- Vincent, K.A.; Parkin, A.; Armstrong, F.A. Investigating and exploiting the electrocatalytic properties of hydrogenases. Chem. Rev. 2007, 107, 4366–4413. [Google Scholar] [CrossRef] [PubMed]
- Ruzgas, T.; Csöregi, E.; Emnéus, J.; Gorton, L.; Marko-Varga, G. Peroxidase-modified electrodes: Fundamentals and application. Anal. Chim. Acta 1996, 330, 123–138. [Google Scholar] [CrossRef]
- Christenson, A.; Dimcheva, N.; Ferapontova, E.E.; Gorton, L.; Ruzgas, T.; Stoica, L.; Shleev, S.; Yaropolov, A.L.; Haltrich, D.; Thorneley, R.N.F.; Aust, S.D. Direct electron transfer between ligninolytic redox enzymes and electrodes. Electroanalysis 2004, 16, 1074–1092. [Google Scholar] [CrossRef]
- Guo, L.-H.; Allen, H.; Hill, O. Direct electrochemistry of proteins and enzymes. In Advances in Inorganic Chemistry; Academic Press: Cambridge, MA, USA, 1991; Volume 36, pp. 341–375. [Google Scholar]
- Ikeda, T.; Matsushita, F.; Senda, M. Amperometric fructose sensor based on direct bioelectrocatalysis. Biosens. Bioelectron. 1991, 6, 299–304. [Google Scholar] [CrossRef]
- Christenson, A.; Gustavsson, T.; Gorton, L.; Hägerhäll, C. Direct and mediated electron transfer between intact succinate:quinone oxidoreductase from Bacillus subtilis and a surface modified gold electrode reveals redox state-dependent conformational changes. Biochim. Biophys. Acta-Bioenerg. 2008, 1777, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J.; Sucheta, A.; Ackrell, B.A.; Armstrong, F.A. Electrocatalytic voltammetry of succinate dehydrogenase: Direct quantification of the catalytic properties of a complex electron-transport enzyme. J. Am. Chem. Soc. 1996, 118, 5031–5038. [Google Scholar] [CrossRef]
- Bollella, P.; Ludwig, R.; Gorton, L. Cellobiose dehydrogenase: Insights on the nanostructuration of electrodes for improved development of biosensors and biofuel cells. Appl. Mater. Today 2017, 9, 319–332. [Google Scholar] [CrossRef]
- Ludwig, R.; Ortiz, R.; Schulz, C.; Harreither, W.; Sygmund, C.; Gorton, L. Cellobiose dehydrogenase modified electrodes: Advances by materials science and biochemical engineering. Anal. Bioanal. Chem. 2013, 405, 3637–3658. [Google Scholar] [CrossRef] [PubMed]
- Tsujimura, S.; Nishina, A.; Hamano, Y.; Kano, K.; Shiraishi, S. Electrochemical reaction of fructose dehydrogenase on carbon cryogel electrodes with controlled pore sizes. Electrochem. Commun. 2010, 12, 446–449. [Google Scholar] [CrossRef]
- Xia, H.-q.; Hibino, Y.; Kitazumi, Y.; Shirai, O.; Kano, K. Interaction between D-fructose dehydrogenase and methoxy-substituent-functionalized carbon surface to increase productive orientations. Electrochim. Acta 2016, 218, 41–46. [Google Scholar] [CrossRef]
- Treu, B.L.; Minteer, S.D. Isolation and purification of PQQ-dependent lactate dehydrogenase from Gluconobacter and use for direct electron transfer at carbon and gold electrodes. Bioelectrochemistry 2008, 74, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Ramanavicius, A.; Habermüller, K.; Csöregi, E.; Laurinavicius, V.; Schuhmann, W. Polypyrrole-entrapped quinohemoprotein alcohol dehydrogenase. Evidence for direct electron transfer via conducting-polymer chains. Anal. Chem. 1999, 71, 3581–3586. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Miyaoka, S.; Miki, K. Enzyme-catalysed electrochemical oxidation of D-gluconate at electrodes coated with D-gluconate dehydrogenase, a membrane-bound flavohemoprotein. J. Electroanal. Chem. 1993, 352, 267–278. [Google Scholar] [CrossRef]
- Shiota, M.; Yamazaki, T.; Yoshimatsu, K.; Kojima, K.; Tsugawa, W.; Ferri, S.; Sode, K. An Fe-S cluster in the conserved Cys-rich region in the catalytic subunit of FAD-dependent dehydrogenase complexes. Bioelectrochemistry 2016, 112, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Tuurala, S.; Lau, C.; Atanassov, P.; Smolander, M.; Minteer, S.D. Characterization and stability study of immobilized PQQ-dependent aldose dehydrogenase bioanodes. Electroanalysis 2012, 24, 229–238. [Google Scholar] [CrossRef]
- Treu, B.L.; Sokic-Lazic, D.; Minteer, S. Bioelectrocatalysis of pyruvate with PQQ-dependent pyruvate dehydrogenase. ECS Trans. 2010, 25, 1–11. [Google Scholar]
- Xu, S.; Minteer, S.D. Investigating the impact of multi-heme pyrroloquinoline quinone-aldehyde dehydrogenase orientation on direct bioelectrocatalysis via site specific enzyme immobilization. ACS Catal. 2013, 3, 1756–1763. [Google Scholar] [CrossRef]
- Zamocky, M.; Ludwig, R.; Peterbauer, C.; Hallberg, B.; Divne, C.; Nicholls, P.; Haltrich, D. Cellobiose dehydrogenase-a flavocytochrome from wood-degrading, phytopathogenic and saprotropic fungi. Curr. Protein Pept. Sci. 2006, 7, 255–280. [Google Scholar] [CrossRef] [PubMed]
- Sutzl, L.; Laurent, C.V.F.P.; Abrera, A.T.; Schutz, G.; Ludwig, R.; Haltrich, D. Multiplicity of enzymatic functions in the CAZy AA3 family. Appl. Microbiol. Biotechnol. 2018, 102, 2477–2492. [Google Scholar] [CrossRef] [PubMed]
- Henriksson, G.; Johansson, G.; Pettersson, G. Acritical review of cellobiose dehydrogenases. J. Biotechnol. 2000, 78, 93–113. [Google Scholar] [CrossRef]
- Stoica, L.; Ludwig, R.; Haltrich, D.; Gorton, L. Third-generation biosensor for lactose based on newly discovered cellobiose dehydrogenase. Anal. Chem. 2006, 78, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Cameron, M.D.; Aust, S.D. Cellobiose dehydrogenase—An extracellular fungal flavocytochrome. Enzym. Microb. Technol. 2001, 28, 129–138. [Google Scholar] [CrossRef]
- Schulz, C.; Ludwig, R.; Micheelsen, P.O.; Silow, M.; Toscano, M.D.; Gorton, L. Enhancement of enzymatic activity and catalytic current of cellobiose dehydrogenase by calcium ions. Electrochem. Commun. 2012, 17, 71–74. [Google Scholar] [CrossRef]
- Kielb, P.; Sezer, M.; Katz, S.; Lopez, F.; Schulz, C.; Gorton, L.; Ludwig, R.; Wollenberger, U.; Zebger, I.; Weidinger, I.M. Spectroscopic observation of calcium-induced reorientation of cellobiose dehydrogenase immobilized on electrodes and its effect on electrocatalytic activity. ChemPhysChem 2015, 16, 1960–1968. [Google Scholar] [CrossRef] [PubMed]
- Kracher, D.; Zahma, K.; Schulz, C.; Sygmund, C.; Gorton, L.; Ludwig, R. Inter-domain electron transfer in cellobiose dehydrogenase: Modulation by pH and divalent cations. FEBS J. 2015, 282, 3136–3148. [Google Scholar] [CrossRef] [PubMed]
- Tavahodi, M.; Ortiz, R.; Schulz, C.; Ekhtiari, A.; Ludwig, R.; Haghighi, B.; Gorton, L. Direct electron transfer of cellobiose dehydrogenase on positively charged polyethyleneimine gold nanoparticles. ChemPlusChem 2017, 82, 546–552. [Google Scholar] [CrossRef]
- Igarashi, K.; Yoshida, M.; Matsumura, H.; Nakamura, N.; Ohno, H.; Samejima, M.; Nishino, T. Electron transfer chain reaction of the extracellular flavocytochrome cellobiose dehydrogenase from the basidiomycete Phanerochaete chrysosporium. FEBS J. 2005, 272, 2869–2877. [Google Scholar] [CrossRef] [PubMed]
- Harreither, W.; Sygmund, C.; Augustin, M.; Narciso, M.; Rabinovich, M.L.; Gorton, L.; Haltrich, D.; Ludwig, R. Catalytic properties and classification of cellobiose dehydrogenases from ascomycetes. Appl. Environ. Microbiol. 2011, 77, 1804–1815. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.M.; Henriksson, G.; Pettersson, G.; Divne, C. Crystal structure of the flavoprotein domain of the extracellular flavocytochrome cellobiose dehydrogenase. J. Mol. Biol. 2002, 315, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Hallberg, B.M.; Bergfors, T.; Bäckbro, K.; Pettersson, G.; Henriksson, G.; Divne, C. A new scaffold for binding haem in the cytochrome domain of the extracellular flavocytochrome cellobiose dehydrogenase. Structure 2000, 8, 79–88. [Google Scholar] [CrossRef]
- Tan, T.-C.; Kracher, D.; Gandini, R.; Sygmund, C.; Kittl, R.; Haltrich, D.; Hallberg, B.M.; Ludwig, R.; Divne, C. Structural basis for cellobiose dehydrogenase action during oxidative cellulose degradation. Nat. Commun. 2015, 6, 7542. [Google Scholar] [CrossRef] [PubMed]
- Isaksen, T.; Westereng, B.; Aachmann, F.L.; Agger, J.W.; Kracher, D.; Kittl, R.; Ludwig, R.; Haltrich, D.; Eijsink, V.G.H.; Horn, S.J. AC4-oxidizing lytic polysaccharide monooxygenase cleaving both cellulose and cello-oligosaccharides. J. Biol. Chem. 2014, 289, 2632–2642. [Google Scholar] [CrossRef] [PubMed]
- Kracher, D.; Scheiblbrandner, S.; Felice, A.K.G.; Breslmayr, E.; Preims, M.; Ludwicka, K.; Haltrich, D.; Eijsink, V.G.H.; Ludwig, R. Extracellular electron transfer systems fuel cellulose oxidative degradation. Science 2016, 352, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Kracher, D.; Ludwig, R. Cellobiose dehydrogenase: An essential enzyme for lignocellulose degradation in nature—A review/Cellobiosedehydrogenase: Ein essentielles Enzym für den Lignozelluloseabbau in der Natur-Eine Übersicht. Bodenkultur 2016, 67, 145–163. [Google Scholar] [CrossRef]
- Safina, G.; Ludwig, R.; Gorton, L. Asimple and sensitive method for lactose detection based on direct electron transfer between immobilised cellobiose dehydrogenase and screen-printed carbon electrodes. Electrochim. Acta 2010, 55, 7690–7695. [Google Scholar] [CrossRef]
- Tasca, F.; Harreither, W.; Ludwig, R.; Gooding, J.J.; Gorton, L. Cellobiose dehydrogenase aryl diazonium modified single walled carbon nanotubes: Enhanced direct electron transfer through a positively charged surface. Anal. Chem. 2011, 83, 3042–3049. [Google Scholar] [CrossRef] [PubMed]
- Bozorgzadeh, S.; Hamidi, H.; Ortiz, R.; Ludwig, R.; Gorton, L. Direct electron transfer of Phanerochaete chrysosporium cellobiose dehydrogenase at platinum and palladium nanoparticles decorated carbon nanotubes modified electrodes. Phys. Chem. Chem. Phys. 2015, 17, 24157–24165. [Google Scholar] [CrossRef] [PubMed]
- Bollella, P.; Mazzei, F.; Favero, G.; Fusco, G.; Ludwig, R.; Gorton, L.; Antiochia, R. Improved DET communication between cellobiose dehydrogenase and a gold electrode modified with a rigid self-assembled monolayer and green metal nanoparticles: The role of an ordered nanostructuration. Biosens. Bioelectron. 2017, 88, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Tasca, F.; Zafar, M.N.; Harreither, W.; Nöll, G.; Ludwig, R.; Gorton, L. Athird generation glucose biosensor based on cellobiose dehydrogenase from Corynascus thermophilus and single-walled carbon nanotubes. Analyst 2011, 136, 2033–2036. [Google Scholar] [CrossRef] [PubMed]
- Zafar, M.N.; Safina, G.; Ludwig, R.; Gorton, L. Characteristics of third-generation glucose biosensors based on Corynascus thermophilus cellobiose dehydrogenase immobilized on commercially available screen-printed electrodes working under physiological conditions. Anal. Biochem. 2012, 425, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Ammam, M.; Fransaer, J. Two-enzyme lactose biosensor based on β-galactosidase and glucose oxidase deposited by AC-electrophoresis: Characteristics and performance for lactose determination in milk. Sens. Actuators B 2010, 148, 583–589. [Google Scholar] [CrossRef]
- Loğoğlu, E.; Sungur, S.; Yildiz, Y. Development of lactose biosensor based on β-galactosidase and glucose oxidase immobilized into gelatin. J. Macromol. Sci. Part A Pure Appl. Chem. 2006, 43, 525–533. [Google Scholar] [CrossRef]
- Sharma, S.K.; Kumar, A.; Chaudhary, R.; Pundir, S.; Pundir, C.S.; Sehgal, N. Lactose biosensor based on lactase and galactose oxidase immobilized in polyvinyl formal. Artif. Cells Blood Substit. Biotechnol. 2007, 35, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, D.; Ralis, E.V.; Makower, A.; Scheller, F.W. Amperometric Bi-enzyme based biosensor for the detection of lactose—Characterization and application. J. Chem. Technol. Biotechnol. 1990, 49, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Glithero, N.; Clark, C.; Gorton, L.; Schuhmann, W.; Pasco, N. At-line measurement of lactose in dairy-processing plants. Anal. Bioanal. Chem. 2013, 405, 3791–3799. [Google Scholar] [CrossRef] [PubMed]
- Lopez, F.; Ma, S.; Ludwig, R.; Schuhmann, W.; Ruff, A. A polymer multilayer based amperometric biosensor for the detection of lactose in the presence of high concentrations of glucose. Electroanalysis 2017, 29, 154–161. [Google Scholar] [CrossRef]
- Tasca, F.; Ludwig, R.; Gorton, L.; Antiochia, R. Determination of lactose by a novel third generation biosensor based on a cellobiose dehydrogenase and aryl diazonium modified single wall carbon nanotubes electrode. Sens. Actuators B-Chem. 2013, 177, 64–69. [Google Scholar] [CrossRef]
- Ortiz, R.; Ludwig, R.; Gorton, L. Highly efficient membraneless glucose bioanode based on Corynascus thermophilus cellobiose dehydrogenase on aryl diazonium-activated single-walled carbon nanotubes. ChemElectroChem 2014, 1, 1948–1956. [Google Scholar] [CrossRef]
- Laviron, E. General expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems. J. Electroanal. Chem. 1979, 101, 19–28. [Google Scholar] [CrossRef]
- Bollella, P.; Schulz, C.; Favero, G.; Mazzei, F.; Ludwig, R.; Gorton, L.; Antiochia, R. Green synthesis and characterization of gold and silver nanoparticles and their application for development of a third generation lactose biosensor. Electroanalysis 2017, 29, 77–86. [Google Scholar] [CrossRef]
- Bartlett, P.N.; Al-Lolage, F.A. There is no evidence to support literature claims of direct electron transfer (DET) for native glucose oxidase (GOx) at carbon nanotubes or graphene. J. Electroanal. Chem. 2017. [Google Scholar] [CrossRef]
- Wilson, G.S. Native glucose oxidase does not undergo direct electron transfer. Biosens. Bioelectron. 2016, 82, VII–VIII. [Google Scholar] [CrossRef] [PubMed]
- Harreither, W.; Coman, V.; Ludwig, R.; Haltrich, D.; Gorton, L. Investigation of graphite electrodes modified with cellobiose dehydrogenase from the ascomycete Myriococcum thermophilum. Electroanalysis 2007, 19, 172–180. [Google Scholar] [CrossRef]
- Tasca, F.; Gorton, L.; Harreither, W.; Haltrich, D.; Ludwig, R.; Noll, G. Highly efficient and versatile anodes for biofuel cells based on cellobiose dehydrogenase from Myriococcum thermophilum. J. Phys. Chem. C 2008, 112, 13668–13673. [Google Scholar] [CrossRef]
- Sygmund, C.; Harreither, W.; Haltrich, D.; Gorton, L.; Ludwig, R. A new generation of glucose biosensors-engineering cellobiose dehydrogenase for increased direct electron transfer. New Biotechnol. 2009, 25, S115. [Google Scholar] [CrossRef]
- Cosnier, S.; Le Goff, A.; Holzinger, M. Towards glucose biofuel cells implanted in human body for powering artificial organs. Electrochem. Commun. 2014, 38, 19–23. [Google Scholar] [CrossRef]
- Leech, D.; Kavanagh, P.; Schuhmann, W. Enzymatic fuel cells: Recent progress. Electrochim. Acta 2012, 84, 223–234. [Google Scholar] [CrossRef]
- Coman, V.; Vaz-Domínguez, C.; Ludwig, R.; Harreither, W.; Haltrich, D.; De Lacey, A.L.; Ruzgas, T.; Gorton, L.; Shleev, S. A membrane-, mediator-, cofactor-less glucose/oxygen biofuel cell. Phys. Chem. Chem. Phys. 2008, 10, 6093–6096. [Google Scholar] [CrossRef] [PubMed]
- Coman, V.; Ludwig, R.; Harreither, W.; Haltrich, D.; Gorton, L.; Ruzgas, T.; Shleev, S. Adirect electron transfer-based glucose/oxygen biofuel cell operating in human serum. Fuel Cells 2010, 10, 9–16. [Google Scholar]
- Falk, M.; Pankratov, D.; Lindh, L.; Arnebrant, T.; Shleev, S. Miniature direct electron transfer based enzymatic fuel cell operating in human sweat and saliva. Fuel Cells 2014, 14, 1050–1056. [Google Scholar] [CrossRef]
- Kawai, S.; Goda-Tsutsumi, M.; Yakushi, T.; Kano, K.; Matsushita, K. Heterologous overexpression and characterization of a flavoprotein-cytochrome c complex fructose dehydrogenase of Gluconobacter japonicus NBRC3260. Appl. Environ. Microbiol. 2013, 79, 1654–1660. [Google Scholar] [CrossRef] [PubMed]
- Kamitaka, Y.; Tsujimura, S.; Kano, K. High current density bioelectrolysis of D-fructose at fructose dehydrogenase-adsorbed and Ketjen black-modified electrodes without a mediator. Chem. Lett. 2006, 36, 218–219. [Google Scholar] [CrossRef]
- Hamano, Y.; Tsujimura, S.; Shirai, O.; Kano, K. Micro-cubic monolithic carbon cryogel electrode for direct electron transfer reaction of fructose dehydrogenase. Bioelectrochemistry 2012, 88, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Bollella, P.; Hibino, Y.; Kano, K.; Gorton, L.; Antiochia, R. The influence of pH and divalent/monovalent cations on the internal electron transfer (IET), enzymatic activity and structure of fructose dehydrogenase. Anal. Bioanal. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kizling, M.; Bilewicz, R. Fructose dehydrogenase electron transfer pathway in bioelectrocatalytic reactions. ChemElectroChem 2018, 5, 166–174. [Google Scholar] [CrossRef]
- Bilewicz, R.; Kizling, M.; Dzwonek, M.; Wieckowska, A. Size does matter—Mediation of electron transfer by gold clusters in bioelectrocatalysis. ChemCatChem 2018. [Google Scholar] [CrossRef]
- Miyake, T.; Haneda, K.; Nagai, N.; Yatagawa, Y.; Onami, H.; Yoshino, S.; Abe, T.; Nishizawa, M. Enzymatic biofuel cells designed for direct power generation from biofluids in living organisms. Energy Environ. Sci. 2011, 4, 5008–5012. [Google Scholar] [CrossRef]
- Khan, G.F.; Shinohara, H.; Ikariyama, Y.; Aizawa, M. Electrochemical behaviour of monolayer quinoprotein adsorbed on the electrode surface. J. Electroanal. Chem. 1991, 315, 263–273. [Google Scholar] [CrossRef]
- Khan, G.F.; Kobatake, E.; Shinohara, H.; Ikariyama, Y.; Aizawa, M. Molecular interface for an activity controlled enzyme electrode and its application for the determination of fructose. Anal. Chem. 1992, 64, 1254–1258. [Google Scholar] [CrossRef]
- Khan, G.; Shinohara, H.; Ikariyama, Y.; Aizawa, M. An amperometric biosensor for fructose using a PQQ enzyme. Sens. Actuators B Chem. 1993, 14, 673–674. [Google Scholar] [CrossRef]
- Ameyama, M.; Shinagawa, E.; Matsushita, K.; Adachi, O. D-fructose dehydrogenase of Gluconobacter industrius: Purification, characterization and application to enzymatic microdetermination of D-fructose. J. Bacteriol. 1981, 145, 814–823. [Google Scholar] [PubMed]
- Parellada, J.; Domínguez, E.; Fernández, V.M. Amperometric flow injection determination of fructose in honey with a carbon paste sensor based on fructose dehydrogenase. Anal. Chim. Acta 1996, 330, 71–77. [Google Scholar] [CrossRef]
- Yabuki, S.; Mizutani, F. D-Fructose sensing electrode based on electron transfer of D-fructose dehydrogenase at colloidal gold-enzyme modified electrode. Electroanalysis 1997, 9, 23–25. [Google Scholar] [CrossRef]
- Begum, A.; Kobatake, E.; Suzawa, T.; Ikariyama, Y.; Aizawa, M. New electrocatalytic biomolecular interface for fabricating a fructose dehydrogenase-based sensing system. Anal. Chim. Acta 1993, 280, 31–36. [Google Scholar] [CrossRef]
- Mizutani, F.; Yabuki, S. Highly-sensitive measurement of dihydroxyphenols using carbon felt electrode impregnated with fructose dehydrogenase-containing solution. Chem. Lett. 1994, 23, 1569–1572. [Google Scholar] [CrossRef]
- Kinnear, K.T.; Monbouquette, H.G. An amperometric fructose biosensor based on fructose dehydrogenase immobilized in a membrane mimetic layer on gold. Anal. Chem. 1997, 69, 1771–1775. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, M.; Nomura, S.; Taniguchi, I. d-Fructose detection based on the direct heterogeneous electron transfer reaction of fructose dehydrogenase adsorbed onto multi-walled carbon nanotubes synthesized on platinum electrode. Biosens. Bioelectron. 2009, 24, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Nazaruk, E.; Landau, E.M.; Bilewicz, R. Membrane bound enzyme hosted in liquid crystalline cubic phase for sensing and fuel cells. Electrochim. Acta 2014, 140, 96–100. [Google Scholar] [CrossRef]
- Šakinytė, I.; Barkauskas, J.; Gaidukevič, J.; Razumienė, J. Thermally reduced graphene oxide: The study and use for reagentless amperometric D-fructose biosensors. Talanta 2015, 144, 1096–1103. [Google Scholar] [CrossRef] [PubMed]
- Siepenkoetter, T.; Salaj-Kosla, U.; Magner, E. The immobilization of fructose dehydrogenase on nanoporous gold electrodes for the detection of fructose. ChemElectroChem 2017, 4, 905–912. [Google Scholar] [CrossRef]
- Murata, K.; Kajiya, K.; Nakamura, N.; Ohno, H. Direct electrochemistry of bilirubin oxidase on three-dimensional gold nanoparticle electrodes and its application in a biofuel cell. Energy Environ. Sci. 2009, 2, 1280–1285. [Google Scholar] [CrossRef]
- Haneda, K.; Yoshino, S.; Ofuji, T.; Miyake, T.; Nishizawa, M. Sheet-shaped biofuel cell constructed from enzyme-modified nanoengineered carbon fabric. Electrochim. Acta 2012, 82, 175–178. [Google Scholar] [CrossRef]
- Miyake, T.; Haneda, K.; Yoshino, S.; Nishizawa, M. Flexible, layered biofuel cells. Biosens. Bioelectron. 2013, 40, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Kizling, M.; Stolarczyk, K.; Kiat, J.S.S.; Tammela, P.; Wang, Z.; Nyholm, L.; Bilewicz, R. Pseudocapacitive polypyrrole–nanocellulose composite for sugar-air enzymatic fuel cells. Electrochem. Commun. 2015, 50, 55–59. [Google Scholar] [CrossRef]
- Holland, J.T.; Lau, C.; Brozik, S.; Atanassov, P.; Banta, S. Engineering of glucose oxidase for direct electron transfer via site-specific gold nanoparticle conjugation. J. Am. Chem. Soc. 2011, 133, 19262–19265. [Google Scholar] [CrossRef] [PubMed]
- Kamitaka, Y.; Tsujimura, S.; Kataoka, K.; Sakurai, T.; Ikeda, T.; Kano, K. Effects of axial ligand mutation of the type I copper site in bilirubin oxidase on direct electron transfer-type bioelectrocatalytic reduction of dioxygen. J. Electroanal. Chem. 2007, 601, 119–124. [Google Scholar] [CrossRef]
- Campàs, M.; Prieto-Simón, B.; Marty, J.-L. A Review of the Use of Genetically Engineered Enzymes in Electrochemical Biosensors. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2009; pp. 3–9. [Google Scholar]
- Yamazaki, T.; Kojima, K.; Sode, K. Extended-range glucose sensor employing engineered glucose dehydrogenases. Anal. Chem. 2000, 72, 4689–4693. [Google Scholar] [CrossRef] [PubMed]
- Okuda, J.; Sode, K. PQQ glucose dehydrogenase with novel electron transfer ability. Biochem. Biophys. Res. Commun. 2004, 314, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Algov, I.; Grushka, J.; Zarivach, R.; Alfonta, L. Highly efficient flavin-adenine dinucleotide glucose dehydrogenase fused to a minimal cytochrome c domain. J. Am. Chem. Soc. 2017, 139, 17217–17220. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, R.; Rahman, M.; Zangrilli, B.; Sygmund, C.; Micheelsen, P.O.; Silow, M.; Toscano, M.D.; Ludwig, R.; Gorton, L. Engineering of cellobiose dehydrogenases for improved glucose sensitivity and reduced maltose affinity. ChemElectroChem 2017, 4, 846–855. [Google Scholar] [CrossRef]
- Bollella, P.; Gorton, L.; Ludwig, R.; Antiochia, R. Athird generation glucose biosensor based on cellobiose dehydrogenase immobilized on a glassy carbon electrode decorated with electrodeposited gold nanoparticles: Characterization and application in human saliva. Sensors 2017, 17, 1912. [Google Scholar] [CrossRef] [PubMed]
- Bollella, P.; Fusco, G.; Stevar, D.; Gorton, L.; Ludwig, R.; Ma, S.; Boer, H.; Koivula, A.; Tortolini, C.; Favero, G. Aglucose/oxygen enzymatic fuel cell based on gold nanoparticles modified graphene screen-printed electrode. Proof-of-concept in human saliva. Sens. Actuators B Chem. 2018, 256, 921–930. [Google Scholar] [CrossRef]
- Hibino, Y.; Kawai, S.; Kitazumi, Y.; Shirai, O.; Kano, K. Construction of a protein-engineered variant of D-fructose dehydrogenase for direct electron transfer-type bioelectrocatalysis. Electrochem. Commun. 2017, 77, 112–115. [Google Scholar] [CrossRef]
- Ortiz, R.; Matsumura, H.; Tasca, F.; Zahma, K.; Samejima, M.; Igarashi, K.; Ludwig, R.; Gorton, L. Effect of deglycosylation of cellobiose dehydrogenases on the enhancement of direct electron transfer with electrodes. Anal. Chem. 2012, 84, 10315–10323. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dehydrogenases | Cofactor | Substrate | Ref. |
|---|---|---|---|
| Cellobiose dehydrogenase | FAD-heme | D-glucose, cellobiose, lactose | [44,45,82,83] |
| D-Fructose dehydrogenase | FAD-heme | D-fructose | [46,49,84,85] |
| Pyranose dehydrogenase | FAD-heme | aldoses | [33] |
| Lactate dehydrogenase | PQQ-heme | lactate | [86] |
| Lactate dehydrogenase/cyt b2 | FMN-heme | lactate | [41] |
| Alcohol dehydrogenase | PQQ-heme | ethanol | [30,87] |
| Succinate dehydrogenase | FAD-Fe-S cluster-heme | succinate | [80,81] |
| D-gluconate dehydrogenase | FAD-heme-Fe-S cluster | D-gluconate | [43,88] |
| D-glucose dehydrogenase | FAD-heme-Fe-S cluster | D-glucose | [89] |
| Aldose dehydrogenase | PQQ-heme | D-glucose | [90] |
| Pyruvate dehydrogenase | PQQ-heme | pyruvate | [91] |
| Aldehyde dehydrogenase | PQQ-heme | aldehyde | [92] |
| Sulphite oxidase | Moco-heme | sulphite | [35,39] |
| Sulphite dehydrogenase | Moco-heme | sulphite | [42] |
| Theophylline oxidase | ?-heme | theophylline | [34,36,38] |
| Lactose Biosensors | |||||||
|---|---|---|---|---|---|---|---|
| Electrode Platforms | Linear Range/(µM) | LOD/(µM) | Sensitivity/(µA mM−1 cm−2) | Class | Applied Potential/V vs. Ag|AgClsat | Stability | Ref. |
| TvCDH/PEDGE/MWCNTs/SPE | 0.5–200 | 0.25 | - | I | +0.198 | 100% of initial response after 8 h | [110] |
| PsCDH/PEDGE/MWCNTs/SPE | 0.5–100 | I | |||||
| PsCDH/NH2-PD/SWCNTs-GC | 1–150 | 0.5 | 476.8 | I | +0.2 | 85% of the initial response after 50 h | [111] |
| PsCDH/ PEI@AuNPs/AuE | 1–100 | 0.3 | 196.5 | I | +0.25 | 95% of the initial response after 24 h | [101] |
| PcCDH/PtNPs–MWCNTs/SPGE | - | - | 43.5 | I | +0.29 | 75% of their initial response after 10 h | [112] |
| PcCDH/PdNPs–MWCNTs/SPGE | - | - | 46.4 | I | +0.29 | ||
| CtCDH/AuNPs/BPDT/AuE | 5–400 | 3 | 27.5 | II | +0.25 | 85% of initial response after 20 days | [113] |
| Glucose Biosensors | |||||||
| CtCDH/PEDGE/MWCNTs/GC | 0.1–30 | 0.05 | 222 | II | +0.190 | - | [114] |
| CtCDH/PEDGE/MWCNTs-SPE | 0.025–30 | 0.01 | - | II | +0.198 | 90% of initial response after 7 h | [115] |
| CtCDH/PEDGE/SWCNTs-SPE | 0.025–30 | 0.01 | II | ||||
| BFC | Conditions | OCV/(V) | Power Output/Limiting Element (l.e.) | Operational Stability | Ref. |
|---|---|---|---|---|---|
| DcCDH/ThLac SPGE-based | 100 mM citrate–phosphate air-saturated buffer, pH 4.5 containing 5 mM glucose | 0.73 | 5 µW cm−2 at 0.5 V (l.e.: anode) | Half-life > 38 h | [133] |
| CtCDH/MvBOx SPGE-based | 50 mM PBS buffer pH 7.4 containing 5 mM glucose and 150 mM NaCl | 0.62 | ~3 µW cm−2 at 0.37 V (l.e.: anode) | Half-life > 6 h | [134] |
| Human serum | 0.58 | ~4 µW cm−2 at 0.19 V (l.e. cathode) | Half-life < 2 h | ||
| CtCDH/MvBOx AuNPs/AuE-based | 50 mM PBS buffer air-saturated pH 7.4 containing 5 mM glucose and 150 mM NaCl | 0.68 | 3.3 µW cm−2 at 0.52 V (l.e. anode) | ~20% drop in 12 h of continuous operation | [53] |
| 50 mM PBS buffer air saturated pH 7.4 containing 5 mM lactose | 0.68 | 14.9 µW cm−2 at 0.52 V (l.e. anode) | Half-life > 12 h | ||
| Human blood | 0.66 | 2.8 µW cm−2 at 0.45 V (l.e. cathode) | Half-life < 3 h | ||
| Human plasma | 0.63 | 3 µW cm−2 at 0.47 V (l.e. cathode) | Half-life < 8 h | ||
| CtCDH/MvBOx AuNPs/AuE-based (contact lenses) | Human tears | 0.57 | 1 µW cm−2 at 0.5 V (l.e. cathode) | Half-life > 20 h | [50] |
| CtCDH/MvBOx AuNPs/AuMWs-based | Sweat | 0.58 | 0.26 µW cm−2 at 0.5 V (l.e. cathode) | Half-life > 10 h | [135] |
| Sweat + 500 µM glucose | 0.61 | 0.47 µW cm−2 at 0.5 V (l.e. cathode) | - | ||
| Saliva before lunch | 0.56 | 0.1 µW cm−2 at 0.5 V (l.e. cathode) | - | ||
| Saliva after lunch | 0.56 | 0.2 µW cm−2 at 0.5 V (l.e. cathode) | - | ||
| Saliva after lunch + 500 µM glucose | 0.60 | 0.46 µW cm−2 at 0.5 V (l.e. cathode) | - | ||
| HiCDH/MHP/AuNPs/AuE-based MvBOx AuNPs/AuE-based | 50 mM PBS air-saturated buffer pH 7.4 containing 5 mM glucose | 0.65 | 4.77 µW cm−2 at 0.50 V (l.e. anode) | Half-life > 13 h | [51] |
| 50 mM PBS air-saturated buffer pH 7.4 containing 10 mM lactose | 0.67 | 8.64 µW cm−2 at 0.50 V (l.e. anode) | Half-life > 44 h |
| Fructose Biosensors | |||||
|---|---|---|---|---|---|
| Electrode Platforms | Linear Range/(mM) | LOD/(mM) | Sensitivity/(µA mM−1 cm−2) | Applied Potential/V vs. Ag|AgClsat | Ref. |
| FDH/CP | 0.2–30 | - | - | +0.2 | [79] |
| FDH/PEI/CP | Up to 10 | 75 | 385 | +0.4 | [147] |
| FDH/AuNPs/GC | Up to 0.5 | - | - | +0.5 | [148] |
| FDH/MWCNTs/GC | Up to 40 | 5 | - | - | [152] |
| FDH/LCP/SWCNTs/GC | Up to 10 | - | 4 | + 0.2 | [153] |
| FDH/TRGO1/GC | 0.7–8.8 | 0.7 | 14.5 | + 0.4 | [154] |
| FDH/MPA-NPD/Aunanoporous | 0.05–0.3 | 0.0012 | 3.7 | + 0.15 | [155] |
| BFC | Conditions | OCV (V) | Power Output/Limiting Element | References |
|---|---|---|---|---|
| FDH/KB/CP TsLac/CG/CP | 0.1 M McIlvaine O2-satured buffer (pH 5.0) containing 200 mM fructose | 0.79 | 850 mW cm−2 at 0.41 V under stirring (l.e.: cathode) | [47] |
| FDH/ME-AuNPs/CP MvBOx/AuNPs/CP | 0.1 M acetate O2-satured buffer (pH 6.0) containing 200 mM fructose | 0.73 | 0.66 mW cm−2 at 0.36 V without stirring (l.e.: cathode) | [156] |
| 0.87 mW cm−2 at 0.3 V under stirring (l.e.: anode) | ||||
| FDH/MvBOx KB-sheet shaped electrodes | 0.15 M McIlvaine O2-satured buffer solution (pH 5.0) containing 200 mM fructose. | 0.70 | 0.55 mW cm−2 at 0.4 V (l.e.: cathode) | [157] |
| FDH/MvBOx KB-carbon strips electrodes | 0.25 M McIlvaine O2-satured buffer solution (pH 5.0) containing 500 mM fructose | 2.09 | 0.64 mW at 1.2 V (l.e.: cathode) | [158] |
| FDH/ThLac LCP-SWCNTs based GC | 0.15 M McIlvaine O2-satured buffer (pH 5.0) containing 40 mM fructose | 0.70 | 0.85 mW cm−2 at 0.25 V under stirring (l.e.: anode) | [153] |
| FDH/MvBOx CCG based electrodes | 1 M citrate O2-satured buffer (pH 5.0) containing 500 mM fructose | 0.79 | 2.6 mW cm−2 at 0.46 V (l.e: cathode) | [49] |
| FDH/cell./PPy/MWCNTs/GC ThLac/NQ/MWCNTs/GC | 0.1 M McIlvaine O2-satured buffer solution (pH 5.3) containing 100 mM fructose | 0.76 | 1.6 mW cm−2 at 0.33 V (l.e.: cathode) | [159] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bollella, P.; Gorton, L.; Antiochia, R. Direct Electron Transfer of Dehydrogenases for Development of 3rd Generation Biosensors and Enzymatic Fuel Cells. Sensors 2018, 18, 1319. https://doi.org/10.3390/s18051319
Bollella P, Gorton L, Antiochia R. Direct Electron Transfer of Dehydrogenases for Development of 3rd Generation Biosensors and Enzymatic Fuel Cells. Sensors. 2018; 18(5):1319. https://doi.org/10.3390/s18051319
Chicago/Turabian StyleBollella, Paolo, Lo Gorton, and Riccarda Antiochia. 2018. "Direct Electron Transfer of Dehydrogenases for Development of 3rd Generation Biosensors and Enzymatic Fuel Cells" Sensors 18, no. 5: 1319. https://doi.org/10.3390/s18051319
APA StyleBollella, P., Gorton, L., & Antiochia, R. (2018). Direct Electron Transfer of Dehydrogenases for Development of 3rd Generation Biosensors and Enzymatic Fuel Cells. Sensors, 18(5), 1319. https://doi.org/10.3390/s18051319