Opportunities, Challenges, and Prospects in Electrochemical Biosensing of Circulating Tumor DNA and Its Specific Features

 , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Circulating Tumor DNA: Properties, Specific Features and Clinical Applications
3. Conventional Methodologies for the Determination of ctDNA and Characteristic Signatures
4. Electrochemical Biosensing of ctDNA and Surrogate Markers
4.1. Methods for ctDNA Quantification
4.2. Electrochemical Biosensing of Tumor-Specific Mutations
4.3. Electrochemical Biosensing of Methylation Changes in ctDNA
4.4. Electrochemical Biosensing of Cancer-Related Viral DNA Sequences
5. General Conclusions, Insights, and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, X.; Yea, M.; Zhang, W.; Tan, D.; Jaffrezic-Renault, N.; Yang, X.; Guo, Z. Liquid biopsy of circulating tumor DNA and biosensor applications. Biosens. Bioelectron. 2019, 126, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Gorgannezhad, L.; Umer, M.; Islam, N.; Nguyen, N.T.; Shiddiky, M.J.A. Circulating tumor DNA and liquid biopsy: Opportunities, challenges, and recent advances in detection technologies. Lab Chip 2018, 18, 1174–1196. [Google Scholar] [CrossRef] [PubMed]
- Warton, K.; Mahon, K.L.; Samimi, G. Methylated circulating tumor DNA in blood: power in cancer prognosis and response. Endocr. Relat. Cancer 2016, 23, R157–R171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Felden, J.; Craig, A.J.; Villanueva, A. Role of circulating tumor DNA to help decision-making in hepatocellular carcinoma. Oncoscience 2018, 5, 209–211. [Google Scholar] [PubMed]
- Rodda, A.E.; Parker, B.J.; Spencer, A.; Corrie, S.R. Extending circulating tumor DNA analysis to ultralow abundance mutations: Techniques and challenges. ACS Sens. 2018, 3, 540–560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liang, Y.; Li, S.; Zeng, F.; Meng, Y.; Chen, Z.; Liu, S.; Tao, Y.; Yu, F. The interplay of circulating tumor DNA and chromatin modification, therapeutic resistance, and metastasis. Mol. Cancer 2019, 18, 36. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, J.; Park, B.H. Circulating tumor DNA: Measurement and clinical utility. Annu. Rev. Med. 2018, 69, 223–234. [Google Scholar] [CrossRef]
- Elazezy, M.; Joosse, S.A. Techniques of using circulating tumor DNA as a liquid biopsy component in cancer management. Comput. Struct. Biotech. J. 2018, 16, 370–378. [Google Scholar] [CrossRef]
- Lim, M.; Kim, C.J.; Sunkara, V.; Kim, M.H.; Cho, Y.K. Liquid biopsy in lung cancer: Clinical applications of circulating biomarkers (CTCs and ctDNA). Micromachines 2018, 9, 100. [Google Scholar] [CrossRef]
- Das, J.; Kelley, S.O. High-Performance Nucleic Acid Sensors for Liquid Biopsy Applications. Angew. Chem. Int. Ed. 2019, in press. [Google Scholar]
- Dai, Y.; Liu, C.C. Recent advances on electrochemical biosensing strategies toward universal point of care systems. Angew. Chem. Int. Ed. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Alix-Panabières, C. Liquid biopsy and minimal residual disease—Latest advances and implications for cure. Nat. Rev.Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Rowe, A.A.; White, R.J.; Bonham, A.J.; Plaxco, K.W. Fabrication of electrochemical-DNA biosensors for the reagentless detection of nucleic acids, proteins and small molecules. J Vis. Exp. 2011, 52, e2922. [Google Scholar] [CrossRef] [PubMed]
- Otandault, A.; Anker, P.; Al Amir Dache, Z.; Guillaumon, V.; Meddeb, R.; Pastor, B.; Pisareva, E.; Sanchez, C.; Tanos, R.; Tousch, G.; et al. Recent advances in circulating nucleic acids in oncology. Ann. Oncol. 2019, 30, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Su, Z.; Reynolds, N.P.; Arosio, P.; Hamley, I.W.; Gazit, E.; Mezzenga, R. Self-assembling peptide and protein amyloids: From structure to tailored function in nanotechnology. Chem. Soc. Rev. 2017, 46, 4661–4708. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Lin, D.; Wang, H.; Li, J.; Nienhaus, G.U.; Su, Z.; Wei, G.; Shang, L. Supramolecular self-assembly bioinspired synthesis of luminescent gold nanocluster-embedded peptide nanofibers for temperature sensing and cellular imaging. Bioconjug. Chem. 2017, 28, 2224–2229. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yu, X.; Lia, Y.; Su, Z.; Jandt, K.D.; Wei, G. Protein-mimetic peptide nanofibers: Motif design, self-assembly synthesis, and sequence-specific biomedical applications. Prog. Polym. Sci. 2018, 80, 94–124. [Google Scholar] [CrossRef]
- Song, H.; Zhang, X.; Liu, Y.; Su, Z. Developing Graphene-based nanohybrids for electrochemical sensing. Chem. Rec. 2019, 19, 524–549. [Google Scholar] [CrossRef]
- Wei, W.; Zhang, X.; Zhang, S.; Wei, G.; Su, Z. Biomedical and bioactive engineered nanomaterials for targeted tumor photothermal therapy: A review. Mat. Sci. Eng. C 2019, 104, 109891. [Google Scholar] [CrossRef]
- Wang, H.F.; Ma, R.N.; Sun, F.; Jia, L.P.; Zhang, W.; Shang, L.; Xue, Q.W.; Jia, W.L.; Wang, H.S. A versatile label-free electrochemical biosensor for circulating tumor DNA based on dual enzyme assisted multiple amplification strategy. Biosens. Bioelectron. 2018, 122, 224–230. [Google Scholar] [CrossRef]
- Wei, F.; Lin, C.C.; Joon, A.; Feng, Z.; Troche, G.; Lira, M.E.; Chia, D.; Mao, M.; Ho, C.L.; Su, W.C.; et al. Noninvasive saliva-based EGFR gene mutation detection in patients with lung cancer. Am. J. Respir. Crit. Care Med. 2014, 190, 1117–1126. [Google Scholar] [CrossRef]
- Esteban-Fernández de Ávila, B.; Araque, E.; Campuzano, S.; Pedrero, M.; Dalkiran, B.; Barderas, R.; Kilic, E.; Villalonga, R.; Pingarrón, J.M. Dual functional graphene derivative-based electrochemical platforms for detection of the TP53 gene with single nucleotide polymorphism selectivity in biological samples. Anal. Chem. 2015, 87, 2290–2998. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Fan, X.; Xu, S.; Davis, J.J.; Luo, X. Low fouling label-free DNA sensor based on polyethylene glycols decorated with gold nanoparticles for the detection of breast cancer biomarkers. Biosens. Bioelectron. 2015, 71, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Gao, Y.; Wang, S.; Qin, Y.; Xu, L.; Jin, D.; Yang, F.; Zhang, G.J. In situ hybridization chain reaction mediated ultrasensitive enzyme- free and conjugation-free electrochemical genosensor for BRCA-1 gene in complex matrices. Biosens. Bioelectron. 2016, 80, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Das, J.; Ivanov, I.; Sargent, E.H.; Kelley, S.O. DNA clutch probes for circulating tumor DNA analysis. J. Am. Chem. Soc. 2016, 138, 11009–11016. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wang, X.; Cao, X.; Zhao, Y. Accurate electrochemistry analysis of circulating methylated DNA from clinical plasma based on paired-end tagging and amplifications. Anal. Chem. 2017, 89, 10468–10473. [Google Scholar] [CrossRef] [PubMed]
- Daneshpour, M.; Syedmorad, L.; Izadi, P.; Omidfar, K. Femtomolar level detection of RASSF1A tumor suppressor gene methylation by electrochemical nano-genosensor based on Fe3O4/TMC/Au nanocomposite and PT-modified electrode. Biosens. Bioelectron. 2016, 77, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, F.; Zhang, D.; Zhao, Y.; Wei, J.; Wang, L.; Song, S.; Fan, C.; Zhao, Y. Single copy-sensitive electrochemical assay for circulating methylated DNA in clinical samples with ultrahigh specificity based on a sequential discrimination–amplification strategy. Chem. Sci. 2017, 8, 4764–4770. [Google Scholar] [CrossRef]
- Povedano, E.; Vargas, E.; Ruiz-Valdepeñas Montiel, V.; Torrente-Rodríguez, R.M.; Pedrero, M.; Barderas, R.; San Segundo-Acosta, P.; Peláez-García, A.; Mendiola, M.; Hardisson, D.; et al. Electrochemical affinity biosensors for fast detection of gene-specific methylations with no need for bisulfite and amplification treatments. Sci. Rep. 2018, 8, 6418. [Google Scholar] [CrossRef]
- Povedano, E.; Ruiz-Valdepeñas Montiel, V.; Valverde, A.; Navarro-Villoslada, F.; Yáñez-Sedeño, P.; Pedrero, M.; Montero-Calle, A.; Barderas, R.; Peláez-García, A.; Mendiola, M.; et al. Versatile electroanalytical bioplatforms for simultaneous determination of cancer-related DNA 5-methyl- and 5-hydroxymethyl-cytosines at global and gene-specific levels in human serum and tissues. ACS Sens. 2019, 4, 227–234. [Google Scholar] [CrossRef]
- Cai, C.; Guo, Z.; Cao, Y.; Zhang, W.; Chen, Y. A dual biomarker detection platform for quantitating circulating tumor DNA (ctDNA). Nanotheranostics 2018, 2, 12–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahangar, L.E.; Mehrgardi, M.A. Amplified detection of hepatitis B virus using an electrochemical DNA biosensor on a nanoporous gold platform. Bioelectrochemistry 2017, 117, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.M.T.; Beck, V.A.; Pierce, N.A. Next-generation in situ hybridization chain reaction: Higher gain, lower cost, greater durability. ACS Nano 2014, 8, 4284–4294. [Google Scholar] [CrossRef] [PubMed]
- Das, J.; Ivanov, I.; Montermini, L.; Janusz, R.; Sargent, E.H.; Kelley, S.O. An electrochemical clamp assay for direct, rapid analysis of circulating nucleic acids in serum. Nat. Chem. 2015, 7, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, S.; Pedrero, M.; Yáñez-Sedeño, P.; Pingarrón, J.M. Advances in electrochemical (bio)sensing targeting epigenetic modifications of nucleic acids. Electroanalysis 2019, in press. [Google Scholar] [CrossRef]
- Chen, Y.; Hong, T.; Wang, S.; Mo, J.; Tian, T.; Zhou, X. Epigenetic modification of nucleic acids: From basic studies to medical applications. Chem. Soc. Rev. 2017, 46, 2844–2872. [Google Scholar] [CrossRef] [PubMed]
- Bartosik, M.; Jirakova, L.; Anton, M.; Vojtesek, B.; Hrstka, R. Genomagnetic LAMP-based electrochemical test for determination of high-risk HPV16 and HPV18 in clinical samples. Anal. Chim. Acta 2018, 1042, 37–43. [Google Scholar] [CrossRef]
- Bartosik, M.; Durikova, H.; Vojtesek, B.; Anton, M.; Jandakova, E.; Hrstka, R. Electrochemical chip-based genomagnetic assay for detection of high-risk human papillomavirus DNA. Biosens. Bioelectron. 2016, 83, 300–305. [Google Scholar] [CrossRef]
- Campuzano, S.; Pedrero, M.; Pingarron, J.M. Viral protein-based bioanalytical tools for small RNA biosensing. Trends Anal. Chem. 2016, 79, 335–343. [Google Scholar] [CrossRef]
- Campuzano, S.; Yáñez-Sedeño, P.; Pingarrón, J.M. Reagentless and reusable electrochemical affinity biosensors for near real-time and/or continuous operation. Advances and prospects. Curr. Opin. Electrochem. 2019, 16, 35–41. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode | Fundamentals | Target | Technique | Linear range | LOD | Assay Time | Sample | Ref. |
|---|---|---|---|---|---|---|---|---|
| Determination of ctDNA | ||||||||
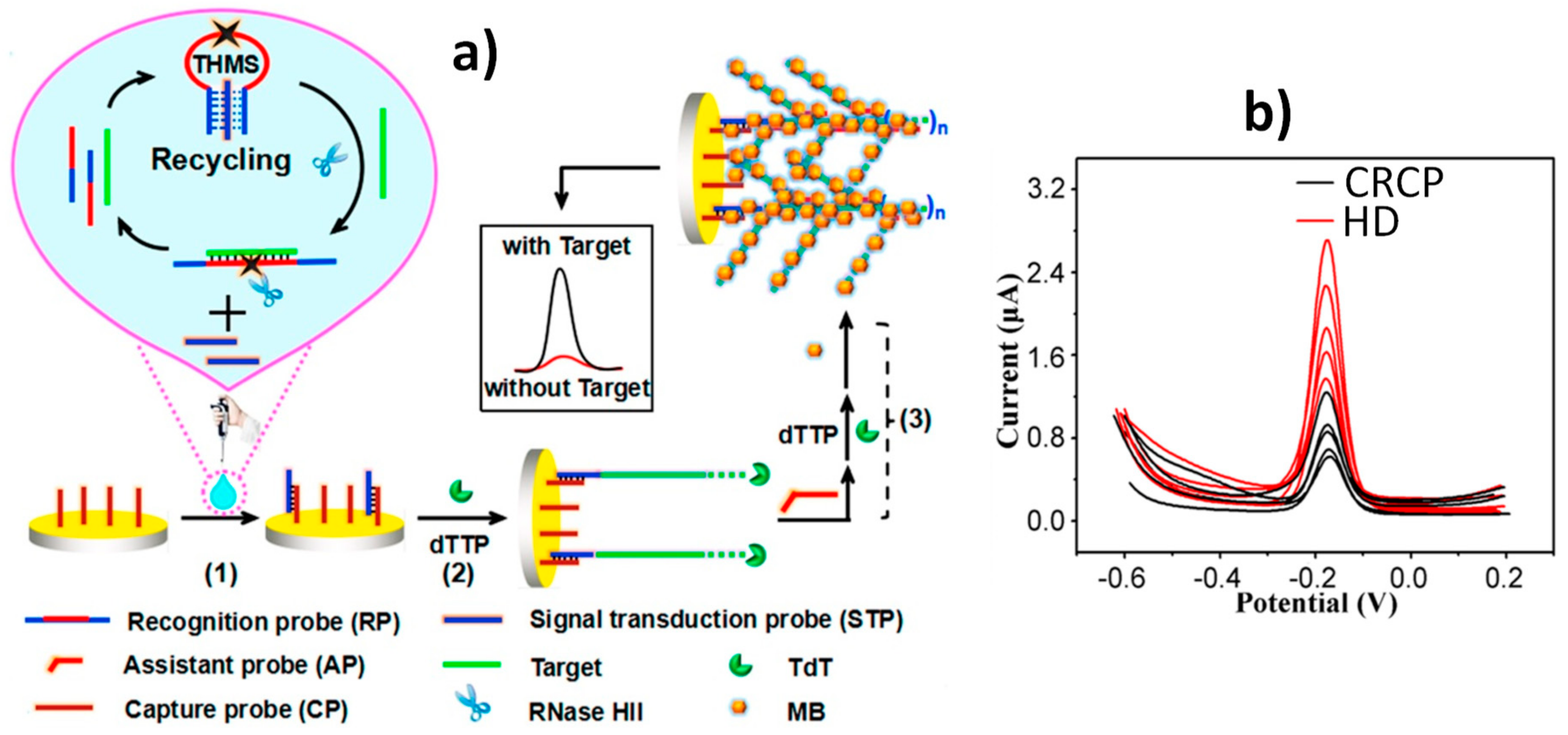
| AuE | THMS probe and TdT and RNase HII dual amplification | KRAS G12DM ss-ctDNA | DPV (MB) | 0.01 fM-1 pM | 2.4 aM | ~6.5 h + Cp-AuE (4 h) | DNA extracted from plasma of CRCP and HD | [20] |
| Tumor-specific mutations in ctDNA | ||||||||
| Array of 16 bare AuE chips | Sandwich hybridization format using paired capture and FITC-Dp further conjugated with HRP-anti-FITC Fab fragments | EGFR mutations | Chronoamperometry (TMB/H2O2) | — | — | ≤ 10 min | Saliva and plasma samples of NSCLC patients | [21] |
| rGO-CMC-modified SPCE | Direct hybridization using an amino and biotin dually labeled hairpin specific DNA Cp | Single base mutation in TP53 | Amperometry (TMB/H2O2) | 0.01–0.1 μM | 2.9 nM (29 fmol in 10 μL) | 45 min + Cp-SPCE (2 h 15 min) | Spiked untreated human serum and saliva samples and cDNA from MCF-10A, MCF-7 and SK-BR-7 cells | [22] |
| GCE | Direct hybridization at Cp/PEG/AuNPs/GCE | Single base mutation in BRCA1 | EIS ([Fe(CN)6]3-/4-) | 50.0 fM–1.0 nM | 1.72 fM | 2 h + Cp/PEG/AuNPs/GCEs (33 h) | Spiked human serum samples | [23] |
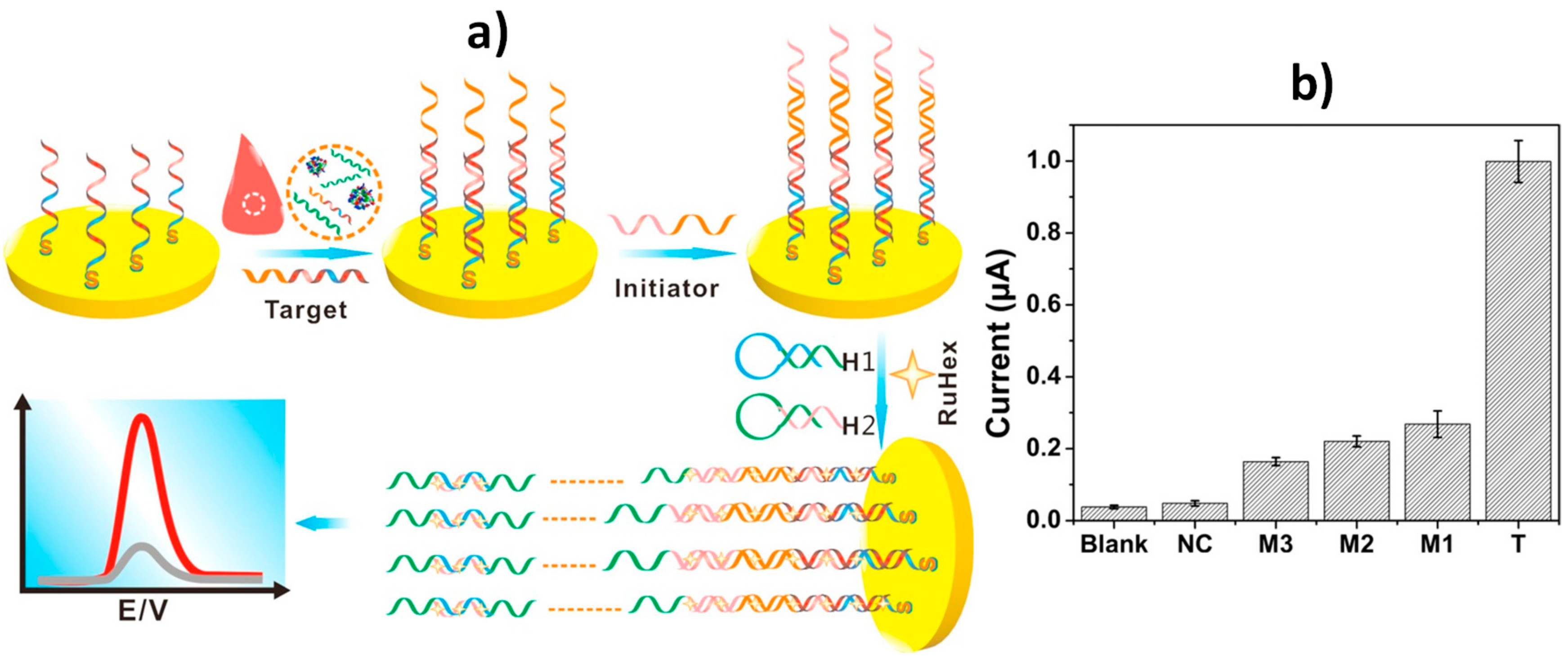
| AuE | HCR at Cp-AuE | Single base mutation in BRCA1 | DPV (RuHex) | 1 aM–10 pM | 1 aM | 4 h 45 min + Cp-AuE (3 h) | Spiked human serum samples | [24] |
| NMEs modified with PNA probes | Direct hybridization in connection with a clutch probe strategy | Single base mutation in KRAS and BRAF in ctDNAs | DPV (Ru(NH3)63+/Fe(CN)63−) | — | 0.01% mutation in wild-type DNA | 50 min + PNA probes-NMEs (12 h) | ctDNA from serum collected from lung cancer and melanoma patients | [25] |
| Epigenetic changes in ctDNA | ||||||||
| AuE | Paired-end tagging amplification | 5-mC (—) | Chronoamperometry (H2O2/TMB) | — | 40 pg (genomic DNA) | ~1.5 h (once the modified electrode was prepared) | gDNA extracted from plasma of NSCLC patients | [26] |
| SPCE | Immunosensor and DNA Dp-modified Fe3O4/TMC/Au nanocomposite as tracing tags | 5-mC/RASSF1A | DPV (AuNPs) | 1 × 10−14-5 × 10−9 M | 2 × 10−15 M | 2 h 40 min (once the PT/anti-5mC -SPCE was prepared) | Spiked plasma | [27] |
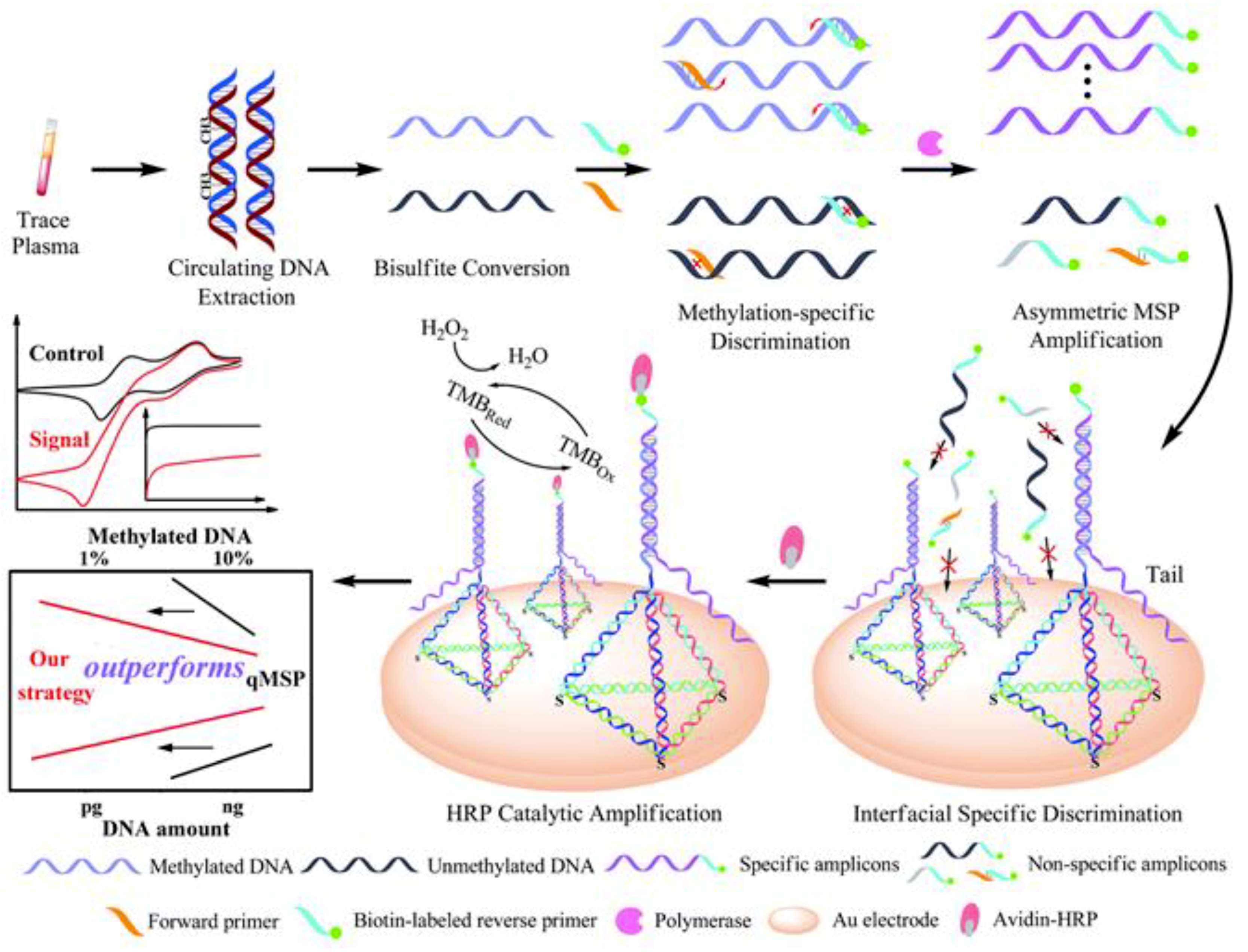
| AuE | Bisulfite + self-assembled tetrahedral DNA probes to capture amplicons generated by aMSP | 5-mC/p16INK4a | Chronoamperometry (H2O2/TMB) | 3–150 pg synthetic target methylated DNA | One methylated DNA molecule in the presence of a 1000-fold excess of unmethylated alleles | aMSP (~42 min) + 45 min (once the AuE was modified with the tetrahedral probes) | cDNA extracted from plasma samples of lung cancer patients | [28] |
| SPCE | Immunopurification (anti-5-mC-MBs) Immunodetection(b-DNA-Cp-MBs) | 5-mC/global (anti-5-mC) and gene-specific (b-DNA-Cp-MBs, MGMT and RASSF1A) | Amperometry (H2O2/HQ) | Global (anti-5-mC-MBs): 23−24,000 pM Gene-specific (b-DNA-Cp-MBs): 139−5000 pM (RASSF1A) 87–2500 pM (MGMT) | Global (anti-5-mC-MBs): 6.8 pM Gene-specific (b-DNA-Cp-MBs): 42 pM (RASSF1A) 26 pM (MGMT) | Global: 45 min (once the anti-5-mC-MBs were prepared) Gene-specific: 1 h (once b-DNA-Cp-MBs were prepared) | Spiked urine, plasma and saliva | [29] |
| SPCE | Immunopurification (anti-5-mC or anti-5-hmC-MBs) | 5-mC and 5-hmC/global and gene-specific (MGMT and RASSF1A) | Amperometry (H2O2/HQ) | Global: 5-mC: 14–2500 pg 5-hmC: 0.04–0.55% Gene-specific: 5-mC: 4.0−250 pM (MGMT) 5-hmC: 1.44−100 pM (MGMT) | Global: 5-mC: 4.0 pg 5-hmC: 0.004% Gene-specific: 5-mC: 1.2 pM (MGMT) 5-hmC: 0.43 pM (MGMT) | Global: 45 min (once the anti-5-mC or anti-5-hmC-MBs were prepared) Gene-specific: 90 min (once the anti-5-mC or anti-5-hmC-MBs were prepared) | gDNA extracted from cell lines paraffin-embedded tissues from CRCP and direct determination 1/5 diluted serum from breast and lung cancer patients | [30] |
| SPCE | Sandwich structure based on PNA probe and anti-5-mC antibody AuNPs and LPA for double signal amplification | Tumor-specific mutations and 5-mC methylation of PIK3CA gene | SWV (lead ions) | 50 fM–10000 fM | 10 fM | 1 h 5 min + PNA-AuNPs conjugates (68.5 h) + LPA-anti-5-mC bioconjugates (6 h) | Spiked human plasma samples | [31] |
| Cancer-related viral DNA sequences | ||||||||
| NPGE | Sandwich hybridization approach involving a thiolated Cp and amino-labeled Dp further conjugated with Fc | HBV DNA | DPV (Fc) | 3 × 10−5–1 × 10−3 M | 0.8 μM | 9 h 20 min + Cp-NPGE (1 h 15 min) | Blood samples from infected people | [32] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campuzano, S.; Serafín, V.; Gamella, M.; Pedrero, M.; Yáñez-Sedeño, P.; Pingarrón, J.M. Opportunities, Challenges, and Prospects in Electrochemical Biosensing of Circulating Tumor DNA and Its Specific Features. Sensors 2019, 19, 3762. https://doi.org/10.3390/s19173762
Campuzano S, Serafín V, Gamella M, Pedrero M, Yáñez-Sedeño P, Pingarrón JM. Opportunities, Challenges, and Prospects in Electrochemical Biosensing of Circulating Tumor DNA and Its Specific Features. Sensors. 2019; 19(17):3762. https://doi.org/10.3390/s19173762
Chicago/Turabian StyleCampuzano, Susana, Verónica Serafín, Maria Gamella, María Pedrero, Paloma Yáñez-Sedeño, and José M. Pingarrón. 2019. "Opportunities, Challenges, and Prospects in Electrochemical Biosensing of Circulating Tumor DNA and Its Specific Features" Sensors 19, no. 17: 3762. https://doi.org/10.3390/s19173762
APA StyleCampuzano, S., Serafín, V., Gamella, M., Pedrero, M., Yáñez-Sedeño, P., & Pingarrón, J. M. (2019). Opportunities, Challenges, and Prospects in Electrochemical Biosensing of Circulating Tumor DNA and Its Specific Features. Sensors, 19(17), 3762. https://doi.org/10.3390/s19173762