2.1. Measurement Principle and Theoretical Basis
The MHS consists of a tubular non-porous symmetric membrane which is permeable to gas. The inside of the tube can be considered as the measurement chamber, which can be closed on both ends by valves. Together with the membrane it comprises the measurement cell of the MHS.
If the composition of the gas of interest (outside the cell) differs from that gas inside the measurement chamber, the partial pressures of the various components of the gas there will change by permeation through the membrane. The operation principle of an MHS is based on these changes.
For a given partial pressure difference the permeation depends on the material-dependent permeability coefficient
of a gaseous compound
j (
(m
2/s)—diffusion coefficient,
(mol/mol)—solubility), which can be expressed in (m
2/s) assuming Henry’s law for gas solution to be valid (dimensionless solubility
equal to the inverse dimensionless Henry constant [
33]). The permeability of a gas component
j can be related to that of a second gas component
k defining its selectivity
.
Table 1 shows the permeability of gas components of air for a silicone rubber from Robb [
34] (converted) and gas selectivity coefficients
with respect to the gas component of interest water vapor (index
w).
In order to carry out a measurement, the measurement chamber is flushed with a gas of known composition long enough to establish steady-state molecular fluxes within the membrane (conditioning step). By closing the measurement chamber at time
t =
t0, the consecutive measurement step is started. The change of the number of gas molecules within the chamber can be expressed as a pressure change according to the ideal gas law and Dalton’s law of partial pressure. Early in the measurement step, the gas composition in the measurement chamber deviates little from the initial condition established in the conditioning step. Up to this time, called
tlin (s), the pressure changes linearly with time and can then be described by the approximate solution [
33]:
The superscripts “o” and “i” define parameters outside and within the measurement cell. are the mole fractions of gas component j within the measurement chamber and outside, and is the ratio of the inside (Pa) and outside (Pa) gas pressures.
The geometry factor
(m
−2) accounts for the cylindrical shape of the gas-selective cell (gas volume
(m
3), length
(m), outer
(m) and inner
(m) radius of the gas selective tube):
The composition of the outer gas is arbitrary. To highlight the contribution of the single gas component
k to
, we consider two cases that only differ in the molar composition
of that component. A difference
results in a dilution of the other gas components by the factor
, where
is the molar fraction of this gas in a reference state. We are now able to reformulate Equation (1) to indicate the contribution of
by:
where
is the Kronecker delta and
the gas-specific cell constant of the measurement chamber for the gas component
k.
In order to determine the mole fraction of water vapor
of ambient air (denoted
x), we expose one of two identical measurement chambers to this air, and the other to drier air with
, while both are purged by the same gas (e.g., air from a pump) during conditioning. Considering
and
in Equation (3) the difference of the pressure changes is:
The parameter
(Pa/s) in Equation (4) depends on the outer reference gas composition
and the specific cell constant of water vapor
. Equation (4) can be applied without having to known the exact composition of the outer reference gas by comparing two pairs of simultaneously operated identical chambers. Due to identical
of the pairs the following equality holds:
where
s indicates a reference states of water-saturated air. Assuming linearity between water vapor concentration and pressure change, the unknown mole fraction
follows for known quantities
and
with:
The use of the same dry air reference state in Equations (5) and (6) allows one to advantageously reduce the measuring system to three measuring chambers according to
Figure 1: Pressure differences
and
between the air of interest and the dryer air reference and between the moist and dryer air references, which are measured with the pressure sensors
and
, suffice to determine
and
.
From the structure of Equation (6) follows that the mole fractions used here can be replaced by any variable that is directly proportional to the mole fraction, e.g., by the water vapor pressure. For this case the vapor pressure of water vapor saturation
is given from the vapor-pressure curve developed already in 1871 by Lord Kelvin [
35] and improved later, e.g., in Appendix 4.B of [
36] for moist air over a pure water phase (−45 to 60 °C):
with
,
17.62 °C
−1 and
243.12 °C.
If all measurement cells and their surroundings have the same temperature
(°C), Equation (6) can be rearranged with respect to the relative humidity
of the dryer reference state to give the relative humidity
:
For a measurement with respect to a dry reference cell with , Equation (8) simplifies to , i.e., the parameter ranges between 0 and 1 ().
For compensating the influence of temperature difference
of the measurement cells we consider the temperature-dependent relative difference of saturated vapor pressures
. To this, we can apply Equation (7) on
and
, which results in
. For
the exponent can be approximated by
. Because
, small differences between the cell-temperatures enable a series expansion of the exponential function (Equation 4.2.19, p. 105 in [
37]). Keeping only the first two terms results in the coefficient
:
and analogously for
when considering a temperature difference
to the dryer reference cell. Combining Equations (8) and (9) results in the equation of a MHS:
If all three measurement cells have the same environmental temperature, Equation (10) simplifies to Equation (8), i.e., the measurement will be temperature-independent.
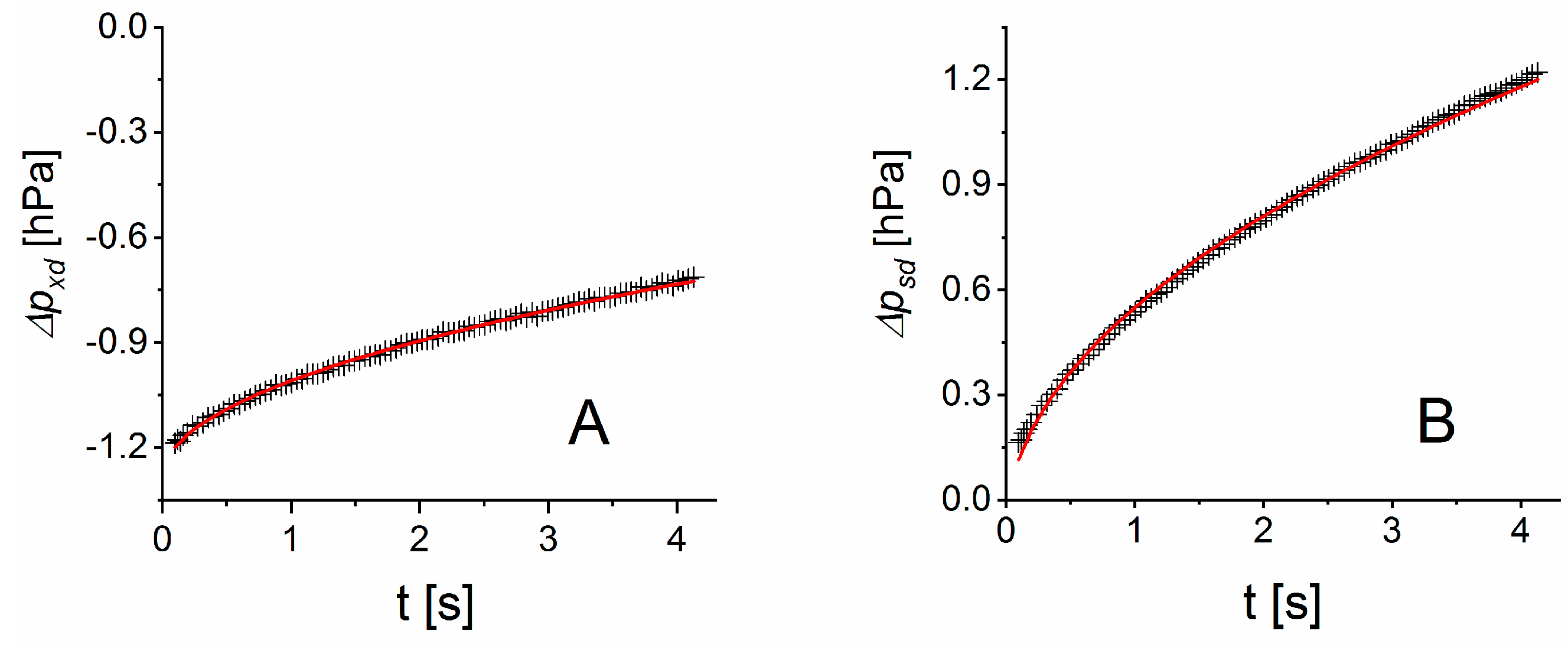
The differential pressure changes of
and
in Equation (5) ideally need to be determined within the time
after the measurement step has been initiated. Presuming a pressure build-up with
in first approximation, [
27], the time
can be estimated to
, e.g., with
c = 0.044 for an approximation error smaller than 10
−3. Depending on the geometry factor
of a measurement cell, the high permeability of water vapor can cause
to be small, which can restrict the time for pressure analysis. Short measurement intervals will lead to larger measurement uncertainties. Hence, the optimum time range for pressure measurement may be larger than
. Our experiments show that the pressure build-up between the measurement cells scales with
for times
. Thus, a differential pressure evolution of the form
can be expected with fitting parameters
and
. Taking the time derivative gives for
the approximation
. Hence, parameter
according to Equation (5) is given by the ratio of
and
fitted to the differential pressure curves
and
.
We can develop correction procedures for unavoidable minor differences between the construction/properties of the three cells of MHS and the response characteristic of the pressure sensors. When exposing the measurement cell (the top cell in
Figure 1) and the dry reference cells (the middle cell in
Figure 1) to the same air, the readings for the top pressure sensor in
Figure 1 (denoted
) should result in a value of zero for the pressure change between the two cells. If it does not, an offset
can simply be determined and used to correct
. Exposing both reference cells (the middle and bottom cells in
Figure 1) to the same air gives analogously the offset determined from the record of the second pressure sensor (labeled
in
Figure 1), which can then be used to correct
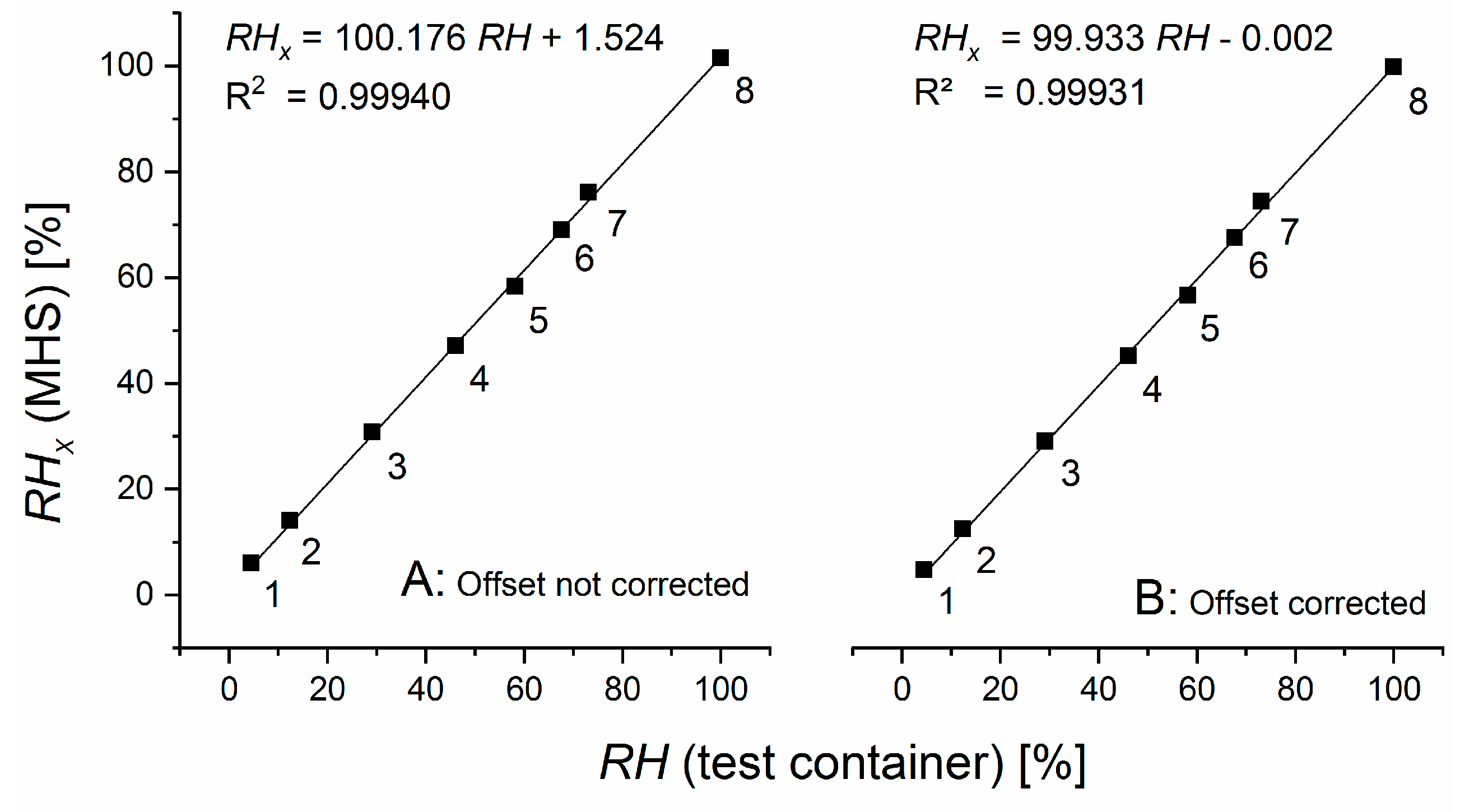
. If the measurement cell is exposed to the same, vapor-saturated air as the wet reference cell, the ratio of
over
should be equal to one when the offset-corrected values are used. Any deviations can be used to correct the ratio
in Equation (5).
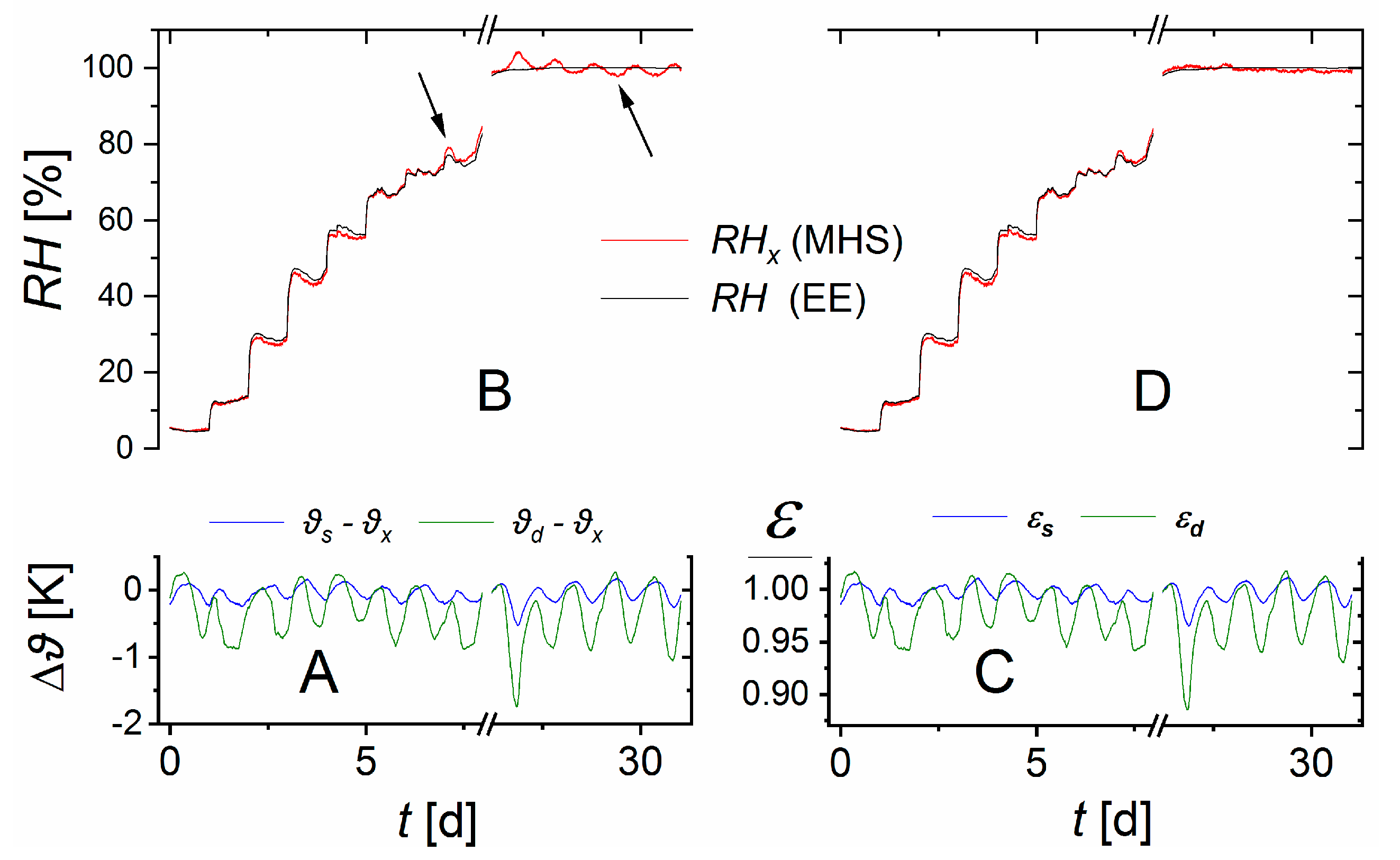
The sensor theory developed above is based on the assumption of linearity between the measured pressure changes and the external water vapor pressure. To test the validity of this assumption, a MHS must be run without calibration.
2.2. Experimental Setup
We prepared a humidity sensor consisting of three tubular measurement cells equal in size using silicone tubing (Silicone Peroxide, Fisher Bioblock Scientific, Illkirch, France). The tube (
= 1.6 mm,
= 2.4 mm) was cut into pieces of 1 m length. For individual adjustment of test conditions these tubes were placed separately on grids in three sealed plastic containers 295 × 230 × 84 mm
3 shown in
Figure 2 (tag 3). For pressure equilibration (
) with the outer air pressure
, each container was equipped with a mini syringe filter (pore size 0.2 µm, filter diameter 4 mm). One of the three containers was designated the test container (represented by the top cell in
Figure 1) and was modified to allow the adjustment of the relative humidity level within it. One of the remaining containers was designed to have relatively dry air in it (dry container), the other one to hold air saturated with water vapor (wet container). These reference containers are represented by the middle and the bottom cell in
Figure 1, respectively. Gas-tight tubes (about 20 cm long,
= 1 mm) were used to connect the gas-selective silicone tubes in the containers to external pinch valves (108P8NO12-01B, Bio-Chem Fluidics, Inc., Boonton, NJ, USA). In order to control the cyclic measurement of the MHS (see
Section 2.1), an actuator unit was designed containing the microprocessor controlled pinch valves (label 4,
Figure 2). In addition, the microprocessor (ATMEGA328P-PU, Atmel Corp., San Jose, CA, USA) was used for registration of digital pressure data, conversion into pressure units, offset-correction, and averaging, generation of the time stamps for each measured/averaged pressure, and transmitting the data via a TTL/USB-converter to a computer. Self-written C codes were used to run the microprocessor and for data storage on the PC. For pressure measurement pressure sensors of type AMS 5812-0000-D-B were used (pressure range ± 5.17 hPa, precision 2% of full scale within a temperature range of −25 to 85 °C, Amsys GmbH & Co, Mainz, Germany). The time span for the conditioning step was set to 110 s, and that for the measurement step to 4 s. The conditioning step caused a formation of a pressure gradient within the tubular measurement chambers, which equilibrated after closing the measurement chambers at the start of the measurement. To minimize the influence of this relaxation process on the measurement signal, an offset time of 100 ms was allowed between the closing of measurement chambers and starting the pressure registration. Dry air from a compressor was used as purge gas. Its flow through the measurement chambers was pre-adjusted by mass flow controllers (MFC 8710, range 0–5 L/min for air, Bürkert Fluid Control Systems, Ingelfingen, Germany) (label 1,
Figure 2) and controlled by a pressure buildup of 20 hPa upstream of the pinch valves. For pressure adjustment a glass tube dipped into a water-filled bottle (label 5,
Figure 2) was used.
Water vapor saturation was established within the wet container by a layer of distilled water on the container bottom well below the grid that supported the MHS tube. To adjust the humidity levels in the other two containers we fitted diffusive gas exchangers below the grids within these containers that were made from silicone tubes (length: 4.5 m, inner diameter: 9 mm, outer diameter: 11 mm) with walls that were highly permeable to water vapor (see
Table 1). The tube within the dry container was upstream connected to a mass flow controller (MFC 8710, range 0–5 L/min, Bürkert Fluid Control Systems) (label 1,
Figure 2) fed with dry compressor air. The tube in the test container was connected to a simple humidity generator (label 2,
Figure 2). The first component of the humidity generator consisted of water-filled vessels through which air bubbled up to saturate it with water vapor for ambient room temperature. The maximum relative humidity that could be achieved in the test container was approx. 80%. In the second component, the humidity of the air entering the test container was controlled by mixing this water-saturated air with dry air from the compressor. Each of the two air flows was controlled by a mass flow controller (MFC 8710, range 0–5 L/min, Bürkert Fluid Control Systems) (label 1,
Figure 2) that allowed the mixing ratio to be regulated to achieve any desired relative humidity of the air entering the test container. The desired air mixtures were fed into the diffuser tubes that were laid out in the dry and the test container. Water vapor diffused through the tube walls until the relative humidity in the containers reached that inside the diffuser tubes. This process avoided air flow and pressure oscillation inside the containers that could disturb the measurements and the entry of contaminants, e.g., oil, within the compressor air. Moreover, this diffusive equilibration of humidity within the containers guaranties the comparatively fast adjustment of the MHS to a slowly changing
RH value.
The three containers were placed on top of each other and wrapped in several layers of cloth to approximate temperature equilibration. Nevertheless, small temperature differences existed. These were used to test the performance of Equation (10).
In each container we installed a sensor (EE60, E + E Elektronik, Engerwitzdorf, Austria) that measured both the humidity and the temperature to serve as an independent reference (denoted EE below). For data acquisition and control of reference sensors and MFCs an ADLink ND-6000 series network (ADLINK Technology GmbH, Mannheim, Germany) (label 6,
Figure 2) was run via PC. The EE’s measurement range for humidity ranged from zero to full saturation with a precision of 2.5% of the measured value. The temperature range was between −40 and 60 °C, with a precision of 0.3 °C. Instead of using the factory calibration curve, we calibrated the temperature and
readings of the EE sensors against a certified resistive-electrolytic portable meter (XP201, Lufft Mess- und Regeltechnik GmbH, Fellbach, Germany). The XP201’s humidity range was also between zero and saturation, with a precision of 0.8%
(equivalent to two standard deviations for 15–30 °C temperature range). This precision is superimposed to the accuracy of the sensor’s reference standard of 0.3 to 0.7%
(calibration mark: 1867, D-K-15202-01-00, 2019-01). The temperature range was between 0 and 70 °C with a precision of 0.15 °C.
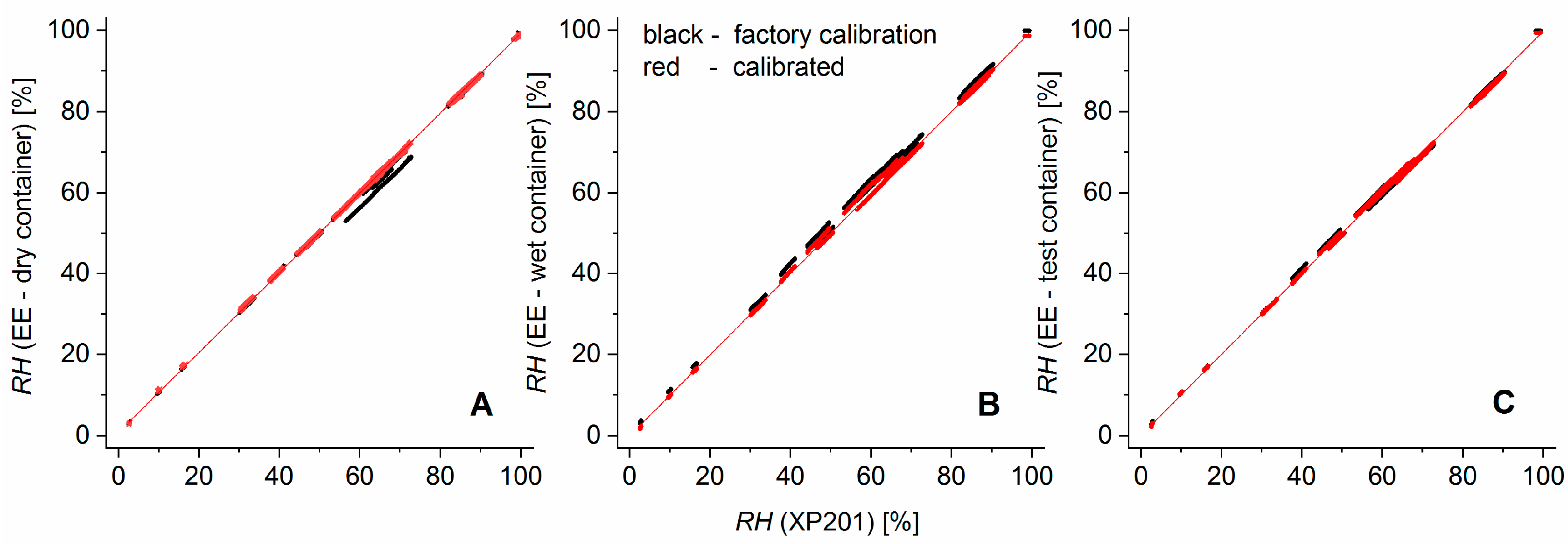
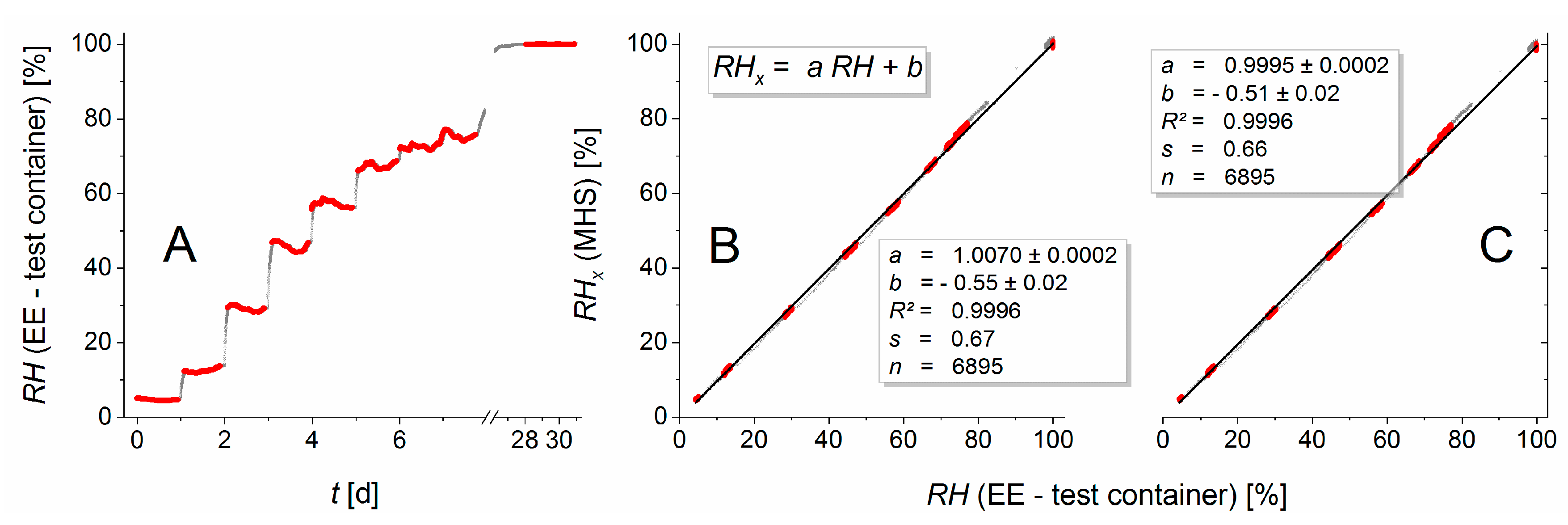
Figure 3 illustrates the improvement of the calibration over the factory calibration. The residuals of the EE humidity measurements versus calibration curve had a standard deviation of 0.4% (EE in the dry container), 0.6% (wet container), and 0.55% (test container). For the temperature, the standard deviations of the residuals were 0.019 °C (dry container), 0.013 °C (wet container), and 0.014 °C (test container).
The shift from the factory calibration (black dots) to the user-calibrated sensors (red dots, final calibration curve: red line) was achieved in a two-step calibration procedure: First the temperature dependence of the
RH signal was recalibrated over a temperature range from 10 to 35 °C by applying a quadratic regression polynomial on the
RH differences from EE sensors and the XP201 with respect to the previously calibrated EE sensor temperature. This additive correction was sensor-specific but behaved nearly independently of the relative humidity. It shifted measurement values parallel to the EE—axes of the sensor characteristics and, thereby, reduced the artefact of having parallel bands of relative humidity values (
Figure 3). In essence, the need for this elaborate recalibration arose from the very high sensitivity of the relative humidity reported by the sensors to the temperature. In the second step, a quadratic regression polynomial was fitted to calibrate the temperature-corrected EE humidity sensors against various humidity levels in a range from 3 to 100% for ambient temperature.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}