
First-Principle Studies of the Vibrational Properties of Carbonates under Pressure


Abstract
:1. Introduction
2. Calculation Method
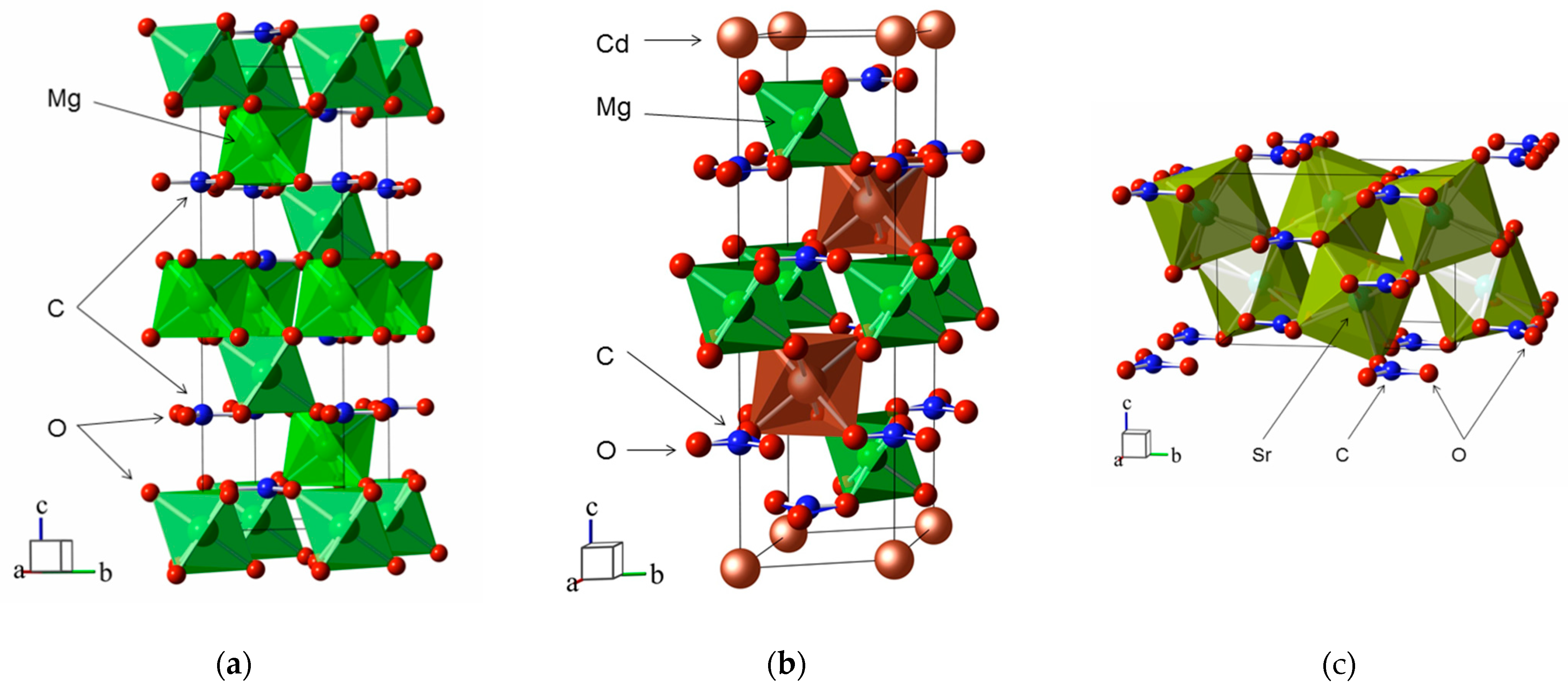
3. Crystal Structure and Pressure
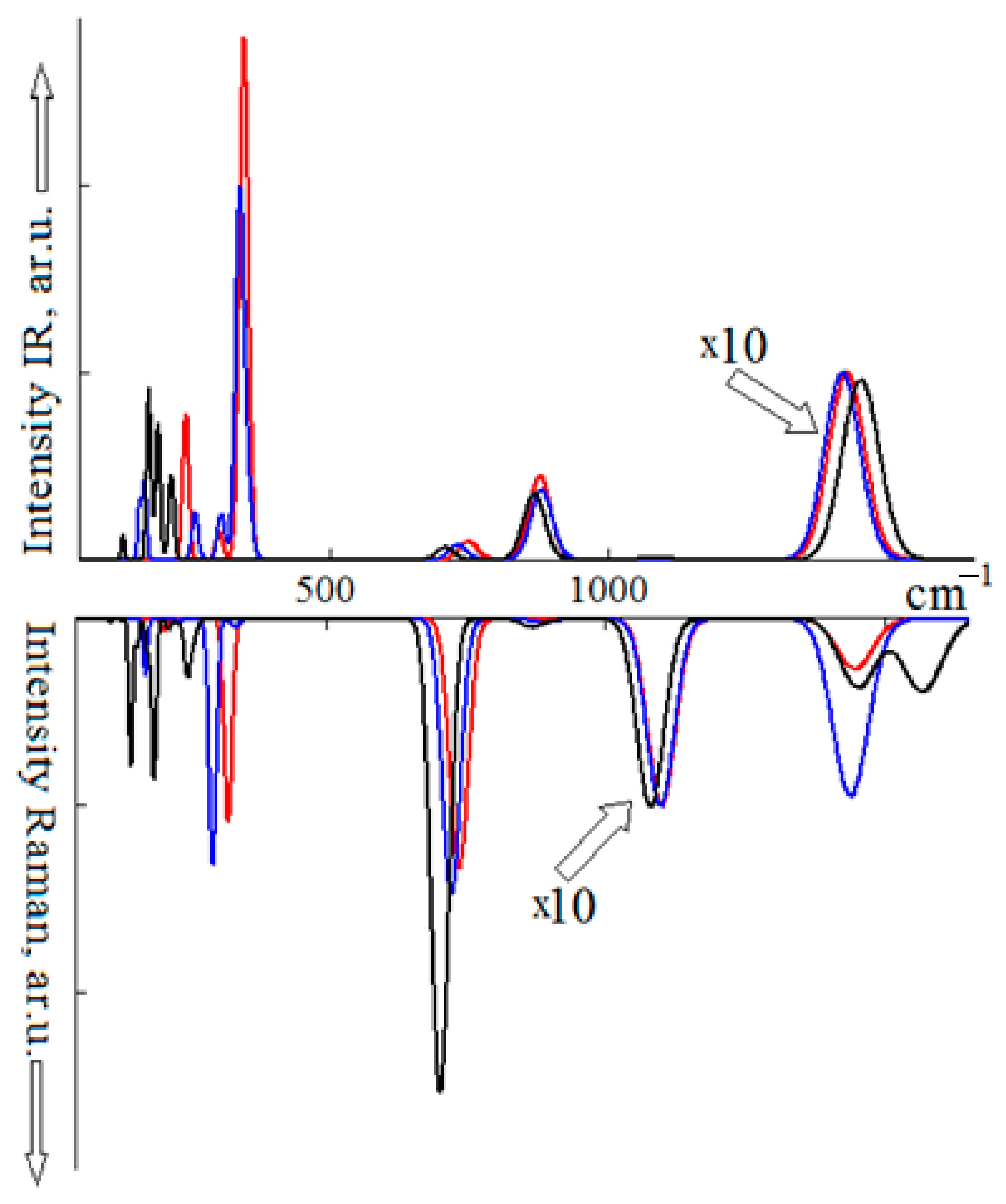
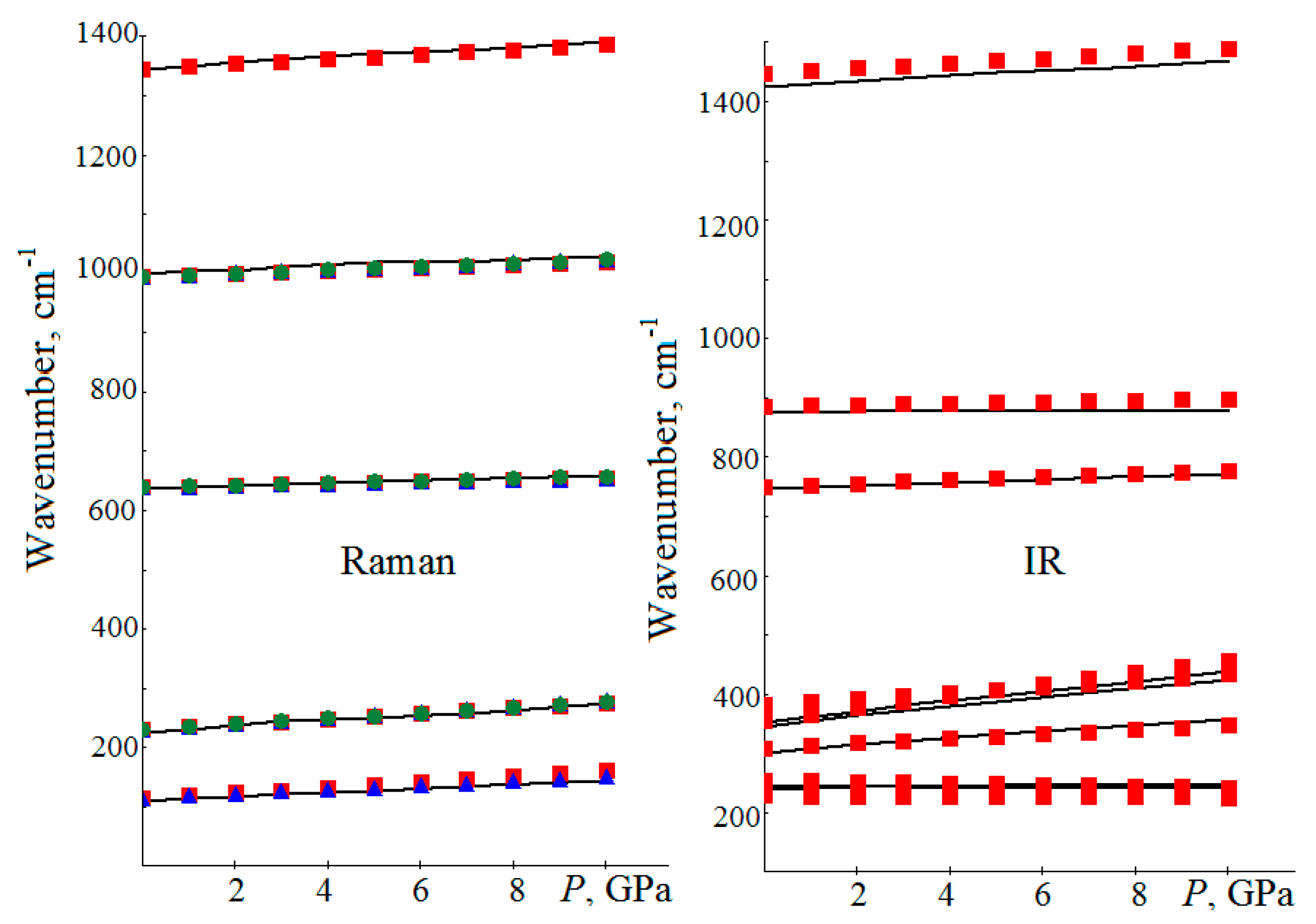
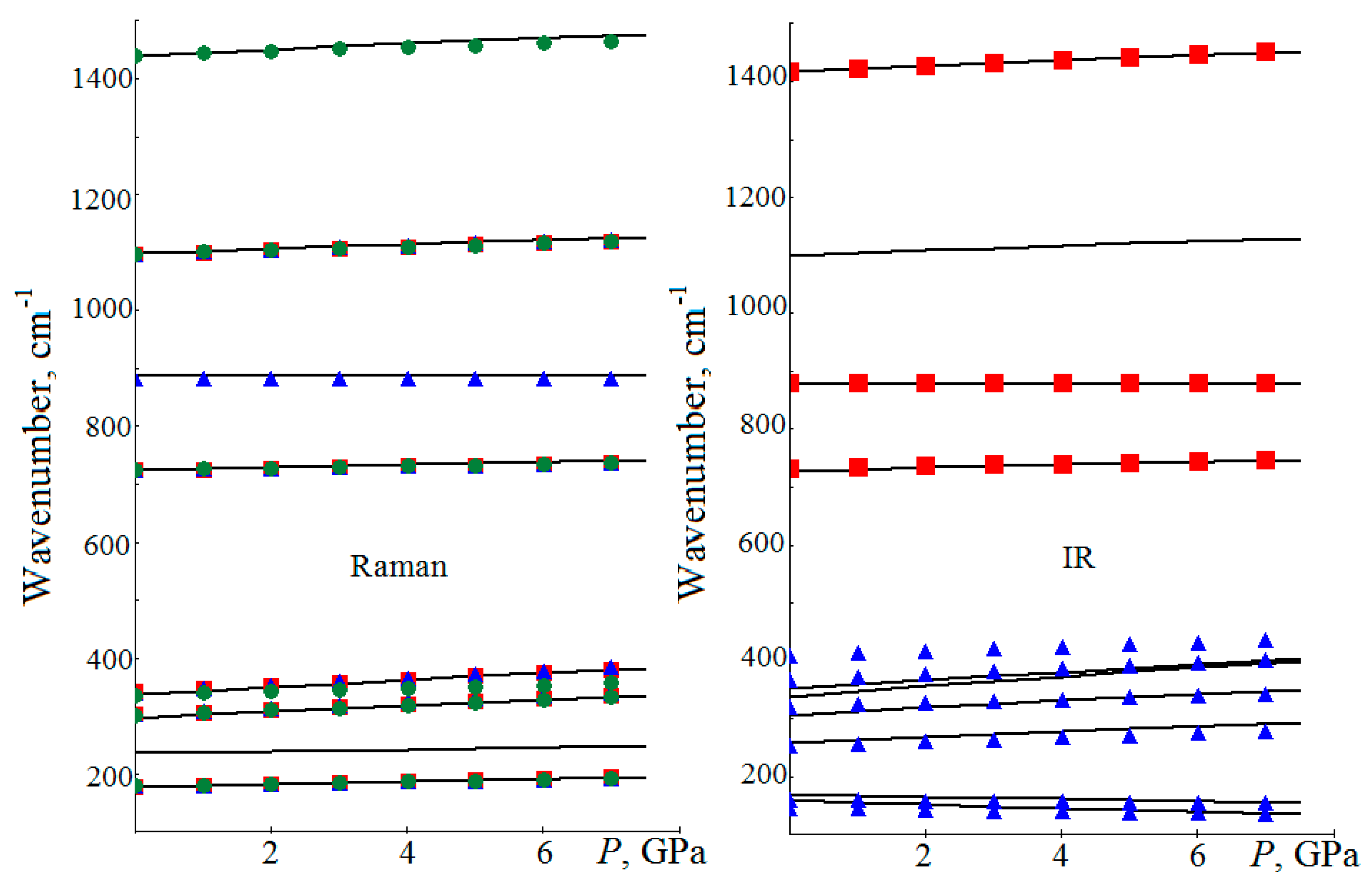
4. Vibrational Spectra under Pressure
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shatskiy, A.F.; Litasov, K.D.; Palyanov, Y.N. Phase relations in carbonate systems at pressures and temperatures of lithospheric mantle: Review of experimental data. Russ. Geol. Geophys. 2015, 56, 113–142. [Google Scholar] [CrossRef]
- Shena, J.; Lib, S.; Wang, S.; Teng, F.; LiLi, Q.; Liu, Y. Subducted Mg-rich carbonates into the deep mantle wedge. Earth Planet. Sci. Lett. 2018, 503, 118–130. [Google Scholar] [CrossRef]
- White, W.B. The Carbonate Minerals. In The Infrared Spectra of Minerals; Farmer, V.C., Ed.; Mineralogical Society: London, UK, 1974. [Google Scholar]
- Ferraro, J.R. Vibrational Spectroscopy at High External Pressures: The Diamond Anvil Cell; Academic Press: Orlando, FL, USA, 1984. [Google Scholar]
- Yuan, X.; Xiong, X.; Zhang, G.; Mayanovic, R.A. Application of calcite, Mg-calcite, and dolomite as Raman pressure sensors for high-pressure, high-temperature studies. J. Raman Spectrosc. 2020, 51, 1248–1259. [Google Scholar] [CrossRef]
- Wang, X.; Ye, Y.; Wu, X.; Smyth, J.R.; Yang, Y.; Zhang, Z.; Wang, Z. High-temperature Raman and FTIR study of aragonite-group carbonates. Phys. Chem. Miner. 2019, 46, 51–62. [Google Scholar] [CrossRef]
- Grzechnik, A.; Simon, P.; Gillet, P.; McMillan, P. An infrared study of MgCO3 at high pressure. Phys. B Condens. Matter 1999, 262, 67–73. [Google Scholar] [CrossRef]
- Clark, S.J.; Jouanna, P.; Haines, J.; Mainprice, D. Calculation of infrared and Raman vibration modes of magnesite at high pressure by density-functional perturbation theory and comparison with experiments. Phys. Chem. Miner. 2010, 38, 193–202. [Google Scholar] [CrossRef]
- Liang, W.; Li, Z.; Yin, Y.; Li, R.; Chen, L.; He, Y.; Dong, H.; Dai, L.; Li, H. Single crystal growth, characterization and high-pressure Raman spectroscopy of impurity-free magnesite (MgCO3). Phys. Chem. Miner. 2018, 45, 423–434. [Google Scholar] [CrossRef]
- Yao, C.; Wu, Z.; Zou, F.; Sun, W. Thermodynamic and elastic properties of magnesite at mantle conditions: First-principles calculations, Geochem. Geophys. Geosyst. 2018, 19, 2719–2731. [Google Scholar] [CrossRef]
- Binck, J.; Bayarjargal, L.; Lobanov, S.S.; Morgenroth, W.; Luchitskaia, R.; Pickard, C.J.; Milman, V.; Refson, K.; Jochym, D.B.; Byrne, P.; et al. Phase stabilities of MgCO3 and MgCO3-II studied by Raman spectroscopy, X-ray diffraction, and density functional theory calculations. Phys. Rev. Mater. 2020, 4, 055001. [Google Scholar] [CrossRef]
- Liu, J.; Caracas, R.; Fan, D.; Bobocioiu, E.; Zhang, D.; Mao, W.L. High-pressure compressibility and vibrational properties of (Ca,Mn)CO3. Am. Miner. 2016, 101, 2723–2730. [Google Scholar] [CrossRef]
- Minch, R.; Seoung, D.-H.; Ehm, L.; Winkler, B.; Knorr, K.; Peters, L.; Borkowski, L.A.; Parise, J.B.; Lee, Y.; Dubrovinsky, L.; et al. High-pressure behavior of otavite (CdCO3). J. Alloys Compd. 2010, 508, 251–257. [Google Scholar] [CrossRef]
- Efthimiopoulos, I.; Jahn, S.; Kuras, A.; Schade, U.; Koch-Muller, M. Combined high-pressure and high-temperature vibrational studies of dolomite: Phase diagram and evidence of a new distorted modification. Phys. Chem. Miner. 2017, 44, 465–476. [Google Scholar] [CrossRef]
- Efthimiopoulos, I.; Germer, M.; Jahn, S.; Harms, M.; Reichmann, H.J.; Speziale, S.; Schade, U.; Sieber, M.; Koch-Müller, M. Effects of hydrostaticity on the structural stability of carbonates at lower mantle pressures: The case study of dolomite. High Press. Res. 2018, 39, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Catalli, K.; Santillan, J.; Williams, Q. A high pressure infrared spectroscopic study of PbCO3-cerussite: Constraints on the structure of the post-aragonite phase. Phys. Chem. Miner. 2005, 32, 412–417. [Google Scholar] [CrossRef]
- Chaney, J.; Santillán, J.D.; Knittle, E.; Williams, Q. High-pressure infrared and Raman spectroscopic study of BaCO3: The aragonite, trigonal and Pmmn structures. Phys. Chem. Miner. 2015, 42, 83–93. [Google Scholar] [CrossRef]
- Biedermann, N.; Speziale, S.; Winkler, B.; Reichmann, H.J.; Koch-Muller, M.; Heide, G. High-pressure phase behavior of SrCO3: An experimental and computational Raman scattering study. Phys. Chem. Miner. 2017, 44, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Efthimiopoulos, I.; Muller, J.; Winkler, B.; Otzen, C.; Harms, M.; Schade, U.; Koch-Muller, M. Vibrational response of strontianite at high pressures and high temperatures and construction of P–T phase diagram. Phys. Chem. Miner. 2019, 46, 27–35. [Google Scholar] [CrossRef]
- Gillet, P.; Biellmann, C.; Reynard, B.; McMillan, P. Raman Spectroscopic studies of carbonates. Part I: High-pressure and high-temperature behavior of Calcite, Magnesite, Dolomite and Aragonite. Phys. Chem. Miner. 1993, 20, 1–18. [Google Scholar] [CrossRef]
- Matas, J.; Gillet, P.; Ricard, Y.; Meartinez, I. Thermodynamic properties of carbonates at high pressures from vibrational modelling. Eur. J. Miner. 2000, 12, 703–720. [Google Scholar] [CrossRef]
- Farsang, S.; Facq, S.; Redfern, S.A.T. Raman modes of carbonate minerals as pressure and temperature gauges up to 6 GPa and 500 °C. Am. Miner. 2018, 103, 1988–1998. [Google Scholar] [CrossRef]
- Zhuravlev, Y.N.; Korabel’nikov, D.V. An ab initio study of the vibrational properties of alkaline-earth metal nitrates and their crystallohydrates. Opt. Spectr. 2017, 122, 929–936. [Google Scholar] [CrossRef]
- Atuchin, V.V.; Liang, F.; Grazhdannikov, S.; Isaenko, L.I.; Krinitsin, P.G.; Molokeev, M.S.; Prosvirin, I.P.; Jiang, X.; Lin, Z. Negative thermal expansion and electronic structure variation of chalcopyrite type LiGaTe2. RSC Adv. 2018, 8, 9946–9955. [Google Scholar] [CrossRef] [Green Version]
- Plazyan, T.L.; Zhuravlev, Y.N. The first-principle studies of the elastic, electronic, and vibrational properties of L-alanine. Struct. Chem. 2019, 30, 1243–1255. [Google Scholar] [CrossRef]
- Vinnik, D.A.; Prosvirin, I.P.; Zhivulin, V.E.; Wang, N.; Jiang, X.; Trofimov, E.A.; Zaitseva, O.V.; Gudkova, S.A.; Nemrava, S.; Zherebtsov, D.A.; et al. Crystal growth, structural characteristics and electronic structure of Ba1-xPbxFe12O19 (x = 0.23 − 0.80) hexaferrites. J. Alloys Compd. 2020, 844, 156036. [Google Scholar] [CrossRef]
- Korabel’nikov, D.V.; Zhuravlev, Y.N. Semi-empirical calculations for crystals under pressure at fixed temperatures: The case of guanidinium perchlorate. RSC Adv. 2020, 10, 42204–42211. [Google Scholar] [CrossRef]
- Denisenko, Y.G.; Atuchin, V.V.; Molokeev, M.S.; Wang, N.; Jiang, X.; Aleksandrovsky, A.S.; Krylov, A.S.; Oreshonkov, A.S.; Sedykh, A.E.; Volkova, S.S.; et al. Klaus Müller-Buschbaum, Negative thermal expansion in one-dimension of a new double sulfate AgHo(SO4)2 with isolated SO4 tetrahedra. J. Mater. Sci. Technol. 2021, 76, 111–121. [Google Scholar] [CrossRef]
- Moroz, T.N.; Edwards, H.G.M.; Zhmodik, S.M. Detection of carbonate, phosphate minerals and cyanobacteria in rock from the Tomtor deposit, Russia, by Raman spectroscopy. Spectrochim. Acta Part Mol. Biomol. Spectrosc. 2021, 250, 119372. [Google Scholar] [CrossRef]
- Gao, J.; Yuan, X.; Chen, B.; Liu, Z.; Su, W. High-pressure phase transformation of carbonate malachite Cu2(CO3) (OH)2 driven by [CuO6] regularization and [CO3] rotation. Geosci. Front. 2021, 12, 965–973. [Google Scholar] [CrossRef]
- Hollis, J.R.; Ireland, S.; Abbey, W.; Bhartia, R.; Beegle, L.W. Deep-ultraviolet Raman spectra of Mars-relevant evaporite minerals under 248.6 nm excitation. Icarus 2021, 357, 114067. [Google Scholar] [CrossRef]
- Erba, A.; Baima, J.; Bush, I.; Orlando, R.; Dovesi, R. Large-scale condensed matter DFT simulations: Performance and capabilities of the CRYSTAL code. J. Chem. Theory Comput. 2017, 13, 5019–5027. [Google Scholar] [CrossRef]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenzano, L.; Torres, F.J.; Doll, K.; Pascale, F.; Zicovich-Wilson, C.M.; Dovesi, R. Ab Initio study of the vibrational spectrum and related properties of crystalline compounds; the case of CaCO3 calcite. Z. Phys. Chem. 2006, 220, 893–912. [Google Scholar] [CrossRef]
- Laun, J.; Oliveira, D.V.; Bredow, T. Consistent gaussian basis sets of double- and triple-zeta valence with polarization quality of the fifth period for solid-state calculations. J. Comput. Chem. 2018, 39, 1285–1290. [Google Scholar] [CrossRef] [PubMed]
- Piskunov, S.; Heifets, E.; Eglitis, R.I.; Borstel, G. Bulk properties and electronic structure of SrTiO3, BaTiO3, PbTiO3 perovskites: An ab initio HF/DFT study. Comp. Mat. Sci. 2004, 29, 165–178. [Google Scholar] [CrossRef]
- Sophia, G.; Baranek, P.; Sarrazin, C.; Rerat, M.; Dovesi, R. First-principles study of the mechanisms of the pressure-induced dielectric anomalies in ferroelectric perovskites. Phase Trans. Multinat. J. 2013, 86, 1069–1084. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pascale, F.; Zicovich-Wilson, C.M.; Lopez, F.; Civalleri, B.; Orlando, R.; Dovesi, R. The calculation of the vibration frequencies of crystalline compounds and its implementation in the CRYSTAL code. J. Comput. Chem. 2004, 25, 888–897. [Google Scholar] [CrossRef]
- Zicovich-Wilson, C.M.; Pascale, F.; Roetti, C.; Saunders, V.R.; Orlando, R.; Dovesi, R. The calculation of the vibration frequencies of alpha-quartz: The efect of Hamiltonian and basis set. J. Comput. Chem. 2004, 25, 1873–1881. [Google Scholar] [CrossRef]
- Baima, J.; Ferrabone, M.; Orlando, R.; Erba, A.; Dovesi, R. Thermodynamics and phonon dispersion of pyrope and grossular silicate garnets from ab initio simulations. Phys. Chem. Miner. 2016, 43, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Birch, F. Finite strain isotherm and velocities for single-crystal and polycrystalline NaCl at high pressures and 300 °K. J. Geophys. Res. 1978, 83, 1257–1268. [Google Scholar] [CrossRef]
- Gao, J.; Zhu, F.; Lai, X.; Huang, R.; Qin, S.; Chen, D.; Liu, J.; Zheng, L.; Wu, X. Compressibility of a natural smithsonite ZnCO3 up to 50 GPa. High Press. Res. 2014, 34, 89–99. [Google Scholar] [CrossRef]
- Bromiley, F.A.; Ballaran, T.B.; Langenhorst, F.; Seifert, F. Order and miscibility in the otavite-magnesite solid solution locality: Synthetic. Am. Miner. 2007, 92, 829–836. [Google Scholar] [CrossRef]
- Zucchini, A.; Comodi, P.; Nazzareni, S.; Hanfland, M. The effect of cation ordering and temperature on the high-pressure behaviour of dolomite. Phys. Chem. Miner. 2014, 41, 783–793. [Google Scholar] [CrossRef]
- Garavelli, C.G.; Vurro, F.; Fioravanti, G.C. Minrecordite, a new mineral from Tsumeb. Mineral. Rec. 1982, 13, 131–136. [Google Scholar]
- Graf, D.L. Crystallographic tables for the rhombohedral carbonates. Am. Miner. 1961, 46, 1283–1316. [Google Scholar]
- Ye, Y.; Smyth, J.R.; Boni, P. Crystal structure and thermal expansion of aragonite-group carbonates by single-crystal X-ray diffraction. Am. Miner. 2012, 97, 707–712. [Google Scholar] [CrossRef]
- Zhuravlev, Y.N.; Atuchin, V.V. Comprehensive density functional theory studies of vibrational spectra of carbonates. Nanomaterials 2020, 10, 2275. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Cryst. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Fiquet, G.; Reynard, B. High-pressure equation of state of magnesite: New data and a reappraisal. Am. Miner. 1999, 84, 856–860. [Google Scholar] [CrossRef]
- Holl, C.M.; Smyth, J.R.; Lausten, H.M.S.; Jacobsen, S.D.; Downs, R.T. Compression of witherite to 8 GPa and the crystal structure of BaCO3 II. Phys. Chem. Miner. 2000, 27, 467–473. [Google Scholar] [CrossRef]
- Dovesi, R.; Saunders, V.R.; Roetti, C.; Orlando, R.; Zicovich-Wilson, C.M.; Pascale, F.; Civalleri, B.; Doll, K.; Harrison, N.M.; Bush, I.J.; et al. CRYSTAL17 User’s Manual, Università di Torino, Torino. 2017. Available online: http://www.crystal.unito.it/news.php (accessed on 19 May 2021).
- Gao, J.; Liu, Y.; Wu, X.; Yuan, X.; Liu, Y.; Su, W. Structural modifications of single-crystal aragonite CaCO3 beginning at ~15 GPa: In situ vibrational spectroscopy and X-ray difraction evidence. Minerals 2020, 10, 924. [Google Scholar] [CrossRef]
- Minch, R.; Dubrovinsky, L.; Kurnosov, A.; Ehm, L.; Knorr, K.; Depmeier, W. Raman spectroscopic study of PbCO3 at high pressures and temperatures. Phys. Chem. Miner. 2010, 37, 45–56. [Google Scholar] [CrossRef]
- Gao, J.; Wu, X.; Qin, S.; Li, Y.C. Pressure-induced phase transformations of PbCO3 by X-ray diffraction and Raman spectroscopy. High Press. Res. 2016, 36, 1–15. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carbonate | a(b), Å | c, Å | RM-O, Å | RC-O, Å |
|---|---|---|---|---|
| MgCO3 | 4.662 | 15.189 | 2.123 | 1.286 |
| ZnCO3 | 4.709 | 15.130 | 2.134 | 1.297 |
| CdCO3 | 4.982 | 16.616 | 2.331 | 1.287 |
| CdMg(CO3)2 | 4.814 | 15.863 | 2.345, 2.106 | 1.286 |
| CaMg(CO3)2 | 4.838 | 16.256 | 2.409, 2.105 | 1.286 |
| CaZn(CO3)2 | 4.856 | 16.296 | 2.393, 2.136 | 1.287 |
| SrCO3 | 5.147, 8.442 | 6.195 | 2.684 | 1.288 |
| PbCO3 | 5.245, 8.572 | 6.373 | 2.745 | 1.290 |
| BaCO3 | 5.367, 8.933 | 6.685 | 2.857 | 1.291 |
| Carbonate | V0/Z, Å3 | B0, GPa | B1 | Ba (Bb), GPa | Bc, GPa | BM1-O (BM2-O), GPa | BC-O, GPa |
|---|---|---|---|---|---|---|---|
| MgCO3 | 47.64 | 107.11 | 4.28 | 539 | 238 | 326 | 1749 |
| ZnCO3 | 48.41 | 123.0 | 4.91 | 692 | 248 | 384 | 1768 |
| CdCO3 | 59.53 | 97.84 | 3.52 | 894 | 159 | 319 | 1508 |
| CdMg(CO3)2 | 53.06 | 100.38 | 4.86 | 601 | 189 | 340 (286) | 1589 |
| CaMg(CO3)2 | 54.92 | 88.82 | 4.33 | 522 | 169 | 269 (276) | 1508 |
| CaZn(CO3)2 | 55.47 | 93.39 | 4.89 | 587 | 189 | 287 (325) | 1577 |
| SrCO3 | 67.29 | 63.27 | 3.45 | 397 (328) | 125 | 268 | 1422 |
| PbCO3 | 71.63 | 56.21 | 4.01 | 403 (496) | 100 | 394 | 1675 |
| BaCO3 | 80.13 | 54.39 | 2.74 | 477 (434) | 88 | 208 | 1740 |
| Carbonate | E0, kJ/mol | dE0/dP, kJ/(mol·GPa) | d2E0/dP2, kJ/(mol·GPa2) |
|---|---|---|---|
| MgCO3 | 51.201 | 0.359 | −0.006 |
| ZnCO3 | 49.356 | 0.255 | −0.002 |
| CdCO3 | 46.209 | 0.243 | −0.003 |
| CaMg(CO3)2 | 49.109 | 0.308 | −0.004 |
| CdMg(CO3)2 | 48.759 | 0.305 | −0.005 |
| CaZn(CO3)2 | 47.613 | 0.314 | −0.007 |
| SrCO3 | 46.346 | 0.240 | 0.017 |
| BaCO3 | 45.211 | 0.322 | −0.003 |
| PbCO3 | 42.774 | 0.386 | −0.008 |
| Mode | Lattice Modes | Internal Modes | ||||||
|---|---|---|---|---|---|---|---|---|
| Symmetry | Eg | Eg | Eg(ν4) | A1g(ν1) | Eg(ν3) | |||
| MgCO3, RS | ||||||||
| ν0 dν/dP d2ν/dP2 d2P/dν2 × 10−3 dv/da | 208 | 323 | 737 | 1099 | 1444 | |||
| 4.214 | 5.541 | 2.90 | 4.378 | 5.567 | ||||
| −0.069 | −0.078 | −0.072 | −0.143 | −0.11 | ||||
| 1.47 | 0.606 | 6.34 | 4.42 | 1.06 | ||||
| −416 | −600 | −261 | −360 | −530 | ||||
| ZnCO3, RS | ||||||||
| ν0 dν/dP d2ν/dP2 d2P/dν2 × 10−3 dv/da | 208 | 306 | 723 | 1105 | 1457 | |||
| 3.153 | 4.611 | 1.203 | 2.704 | 5.426 | ||||
| 0.006 | −0.018 | 0.042 | 0.001 | −0.114 | ||||
| −0.448 | −3.706 | −15.73 | 0.977 | 12.7 | ||||
| −470 | −653 | −231 | −398 | −649 | ||||
| CdCO3, RS | ||||||||
| ν0 dν/dP d2ν/dP2 d2P/dν2 × 10−3 dv/da | 156 | 259 | 713 | 1093 | 1395 | |||
| 2.648 | 4.945 | 1.938 | 3.759 | 6.925 | ||||
| 0.017 | −0.047 | −0.005 | −0.004 | −0.149 | ||||
| −7.308 | 5.111 | 10.77 | 1.892 | 9.039 | ||||
| −452 | −736 | −306 | −603 | −918 | ||||
| Symmetry | Eu (T) | Eu (T) | A2u (T) | Eu (ν4) | Eu(ν3) | |||
| MgCO3, IRS | ||||||||
| ν0 dν/dP d2ν/dP2 d2P/dν2 × 10−3 dv/da | 301 | 344 | 351 | 746 | 1424 | |||
| 6.628 | 9.235 | 9.738 | 3.128 | 5.126 | ||||
| −0.104 | −0.144 | −0.116 | −0.049 | −0.087 | ||||
| 0.556 | 0.282 | 0.171 | 2.64 | 1.023 | ||||
| −659 | −919 | −1005 | −311 | −503 | ||||
| ZnCO3, IRS | ||||||||
| ν0 dν/dP d2ν/dP2 d2P/dν2 × 10−3 dv/da | 212 | 287 | 348 | 735 | 1447 | |||
| 1.352 | 5.761 | 4.36 | 1.725 | 4.95 | ||||
| 0.036 | −0.04 | 0.078 | 0.04 | −0.09 | ||||
| −6.443 | −0.262 | −0.475 | −2.666 | −1.215 | ||||
| −245 | −794 | −740 | −305 | −610 | ||||
| CdCO3, IRS | ||||||||
| ν0 dν/dP d2ν/dP2 d2P/dν2 × 10−3 dv/da | 158 | 267 | 303 | 722 | 1388 | |||
| 1.616 | 6.274 | 6.339 | 2.295 | 5.326 | ||||
| 0.035 | −0.095 | −0.075 | 0.003 | −0.041 | ||||
| −4.869 | 0.590 | 0.422 | −0.048 | 0.339 | ||||
| −310 | −887 | −925 | −375 | −807 | ||||
| Mode | Lattice Modes | Internal Modes | |||||
|---|---|---|---|---|---|---|---|
| Symmetry | Eg | Ag | Eg | Ag | Eg(ν4) | Ag(ν1) | Eg(ν3) |
| CaMg(CO3)2, RS | |||||||
| ν0 dν/dP d2ν/dP2 d2P/dν2 ×10−3 dv/da | 175 | 235 | 296 | 336 | 723 | 1097 | 1437 |
| 2.719 | 1.402 | 5.397 | 6.789 | 2.73 | 4.596 | 6.01 | |
| −0.02 | 0.032 | −0.041 | −0.09 | −0.05 | −0.099 | −0.109 | |
| −1.114 | −7.874 | −0.30 | 0.366 | 3.473 | 1.607 | 0.740 | |
| −279 | −171 | −552 | −670 | −260 | −260 | −573 | |
| CdMg(CO3)2, RS | |||||||
| ν0 dν/dP d2ν/dP2 γ d2P/dν2 × 10−3 dv/da | 183 | 258 | 286 | 365 | 725 | 1098 | 1419 |
| 3.516 | 2.431 | 5.562 | 6.094 | 2.258 | 3.793 | 5.437 | |
| −0.071 | −0.011 | −0.08 | −0.04 | −0.042 | −0.045 | −0.07 | |
| 1.93 | 0.94 | 1.95 | 1.68 | 0.31 | 0.35 | 0.38 | |
| 2.43 | 0.91 | −0.014 | 0.20 | 6.685 | 1.202 | 0.577 | |
| −383 | −293 | −629 | −726 | −249 | −437 | −622 | |
| CaZn(CO3)2, RS | |||||||
| ν0 dν/dP d2ν/dP2 d2P/dν2 × 10−3 dv/da | 165 | 233 | 283 | 350 | 719 | 1095 | 1416 |
| 3.429 | 1.796 | 5.566 | 7.743 | 2.622 | 3.992 | 5.209 | |
| −0.088 | −0.041 | −0.072 | −0.204 | −0.069 | −0.059 | −0.007 | |
| 4.661 | 0.016 | 0.602 | 0.925 | 8.81 | 1.5 | 0.043 | |
| −320 | −173 | −592 | −717 | −243 | −417 | −618 | |
| Symmetry | Eu | Au | Eu | Au | Eu(ν4) | Au(ν1) | Eu(ν3) |
| CaMg(CO3)2, IRS | |||||||
| ν0 dν/dP d2ν/dP2γ d2P/dν2 × 10−3 dv/da | 258 | 304 | 337 | 352 | 727 | 1098 | 1416 |
| 5.26 | 7.298 | 9.316 | 6.647 | 2.972 | 4.538 | 5.6109 | |
| −0.078 | −0.168 | −0.162 | 0.022 | −0.041 | −0.076 | −0.112 | |
| 0.733 | 0.702 | 0.288 | −0.069 | 2.003 | 1.143 | 0.966 | |
| −513 | −673 | −893 | −727 | −292 | −437 | −528 | |
| CdMg(CO3)2, IRS | |||||||
| ν0 dν/dP d2ν/dP2 d2P/dν2 × 10−3 dv/da | 236 | 285 | 338 | 354 | 733 | 1098 | 1407 |
| 2.315 | 2.876 | 8.981 | 8.119 | 2.627 | 3.964 | 5.061 | |
| −0.017 | −0.064 | −0.181 | −0.091 | −0.022 | −0.06 | −0.046 | |
| −9.999 | 4.62 | 0.378 | 0.232 | 2.316 | 1.401 | 0.457 | |
| −274 | −308 | −977 | −938 | −309 | −446 | −593 | |
| CaZn(CO3)2, IRS | |||||||
| ν0 dν/dP d2ν/dP2 d2P/dν2 × 10−3 dv/da | 189 | 242 | 290 | 310 | 724 | 1094 | 1400.9 |
| 4.978 | 3.453 | 8.139 | 9.431 | 2.948 | 4.089 | 4.67 | |
| −0.118 | −0.084 | −0.18 | −0.171 | −0.066 | −0.064 | 0.013 | |
| 1.895 | 4.171 | 0.622 | 0.344 | 5.179 | 1.543 | −0.128 | |
| −475 | −327 | −790 | −954 | −285 | −424 | −573 | |
| Mode | ν4 | ν1 | ν3 | ||||
|---|---|---|---|---|---|---|---|
| Symmetry | Ag | B1g | B3g | Ag | Ag | B3g | B2g |
| SrCO3, RS | |||||||
| ν0 dν/dP d2ν/dP2 d2P/dν2 × 10−3 dv/da | 704 | 704 | 708 | 1080 | 1453 | 1453 | 1567 |
| 4.693 | 1.546 | 1.844 | 3.632 | 3.649 | 4.123 | 4.479 | |
| −0.447 | 0.012 | −0.006 | −0.103 | −0.008 | 0.027 | 0.11 | |
| - | 0.968 | 4.234 | 2.909 | −0.959 | −0.236 | −0.886 | |
| - | −124 | −139 | −234 | −276 | −328 | −391 | |
| BaCO3, RS | |||||||
| ν0 dν/dP d2ν/dP2 d2P/dν2 × 10−3 dv/da | 694 | 696 | 700 | 1066 | 1426 | 1430 | 1528 |
| 2.264 | 1.92 | 2.275 | 3.672 | 4.742 | 4.739 | 6.239 | |
| 0.039 | 0.031 | 0.04 | −0.043 | −0.349 | −0.067 | −0.24 | |
| −3.024 | −4.593 | −3.192 | 0.768 | 8.081 | 0.822 | 2.967 | |
| −217 | −183 | −218 | −300 | −244 | −382 | −429 | |
| PbCO3, RS | |||||||
| ν0 dν/dP d2ν/dP2 d2P/dν2 × 10−3 dv/da | 680 | 678 | 688 | 1068 | 1384 | 1394 | 1487 |
| 1.006 | 1.067 | 2.025 | 3.261 | 5.308 | 2.405 | 8.416 | |
| 0.012 | −0.054 | −0.067 | −0.1 | −0.248 | 0.198 | −0.37 | |
| −2.255 | 112 | 016 | 5.402 | 4.669 | −0.698 | 1.654 | |
| −80.5 | −55.9 | −122 | −200 | −288 | −266 | −467 | |
| Symmetry | B2u | B3u | B1u | B1u | B2u | B1u | B3u |
| SrCO3, IRS | |||||||
| ν0 dν/dP d2ν/dP2 d2P/dν2 × 10−3 dv/da | 701 | 708 | 718 | 1079 | 1441 | 1448 | 1457 |
| 1.754 | 2.176 | 2.418 | 3.063 | 4.427 | 4.162 | 4.306 | |
| 0.00 | 0.029 | 0.01 | −0.017 | 0.075 | 0.067 | 0.063 | |
| 3.915 | −1.672 | 0.340 | 0.094 | −0.584 | −0.889 | −0.717 | |
| −134 | −180 | −91 | −229 | −372 | −349 | −358 | |
| BaCO3, IRS | |||||||
| ν0 dν/dP d2ν/dP2 d2P/dν2 × 10−3 dv/da | 695 | 697 | 707 | 1066 | 1426 | 1420 | 1431 |
| 2.298 | 2.471 | 2.791 | 3.647 | 5.441 | 5.789 | 5.92 | |
| 0.04 | 0.031 | 0.037 | −0.049 | −0.077 | −0.33 | −0.233 | |
| 3.1 | −2.038 | −1.549 | 0.912 | 0.613 | 4.294 | 2.27 | |
| −221 | −231 | −262 | −295 | −439 | −345 | −404 | |
| PbCO3, IRS | |||||||
| ν0 dν/dP d2ν/dP2 d2P/dν2 × 10−3 dv/da | 684 | 682 | 702 | 1066 | 1402 | 1390 | 1395 |
| 1.826 | 1.104 | 2.196 | 3.033 | 2.733 | 6.143 | 7.152 | |
| −0.063 | 0.01 | 0.013 | −0.105 | 0.208 | −0.237 | −0.294 | |
| 21.0 | −0.641 | −0.804 | 7.777 | −0.604 | 2.375 | 1.999 | |
| −109 | −87.0 | −170 | −180 | −295 | −355 | −405 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhuravlev, Y.N.; Atuchin, V.V. First-Principle Studies of the Vibrational Properties of Carbonates under Pressure. Sensors 2021, 21, 3644. https://doi.org/10.3390/s21113644
Zhuravlev YN, Atuchin VV. First-Principle Studies of the Vibrational Properties of Carbonates under Pressure. Sensors. 2021; 21(11):3644. https://doi.org/10.3390/s21113644
Chicago/Turabian StyleZhuravlev, Yurii N., and Victor V. Atuchin. 2021. "First-Principle Studies of the Vibrational Properties of Carbonates under Pressure" Sensors 21, no. 11: 3644. https://doi.org/10.3390/s21113644
APA StyleZhuravlev, Y. N., & Atuchin, V. V. (2021). First-Principle Studies of the Vibrational Properties of Carbonates under Pressure. Sensors, 21(11), 3644. https://doi.org/10.3390/s21113644