
A Portable Tunable Diode Laser Absorption Spectroscopy System for Dissolved CO2 Detection Using a High-Efficiency Headspace Equilibrator



Abstract
:1. Introduction
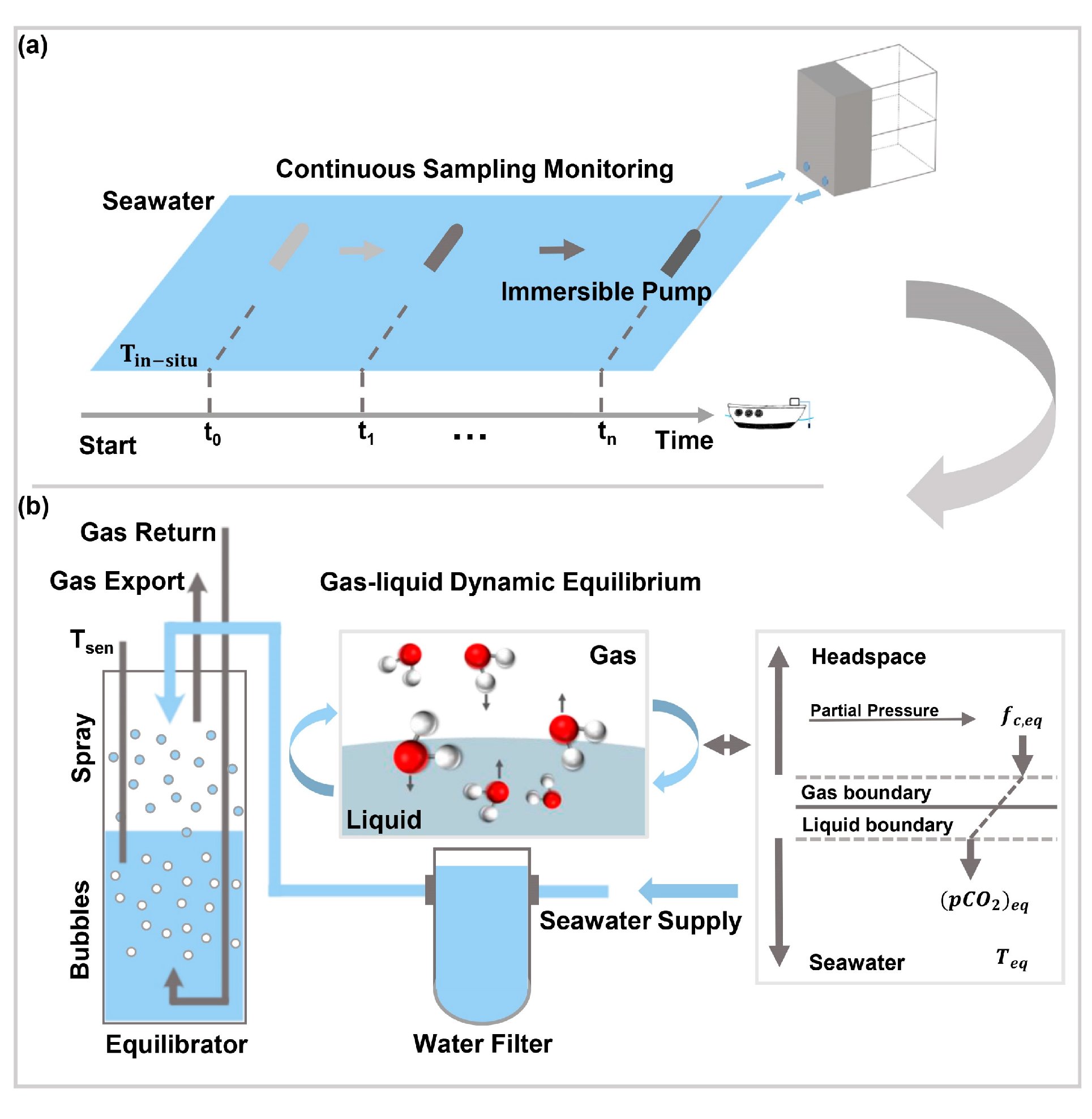
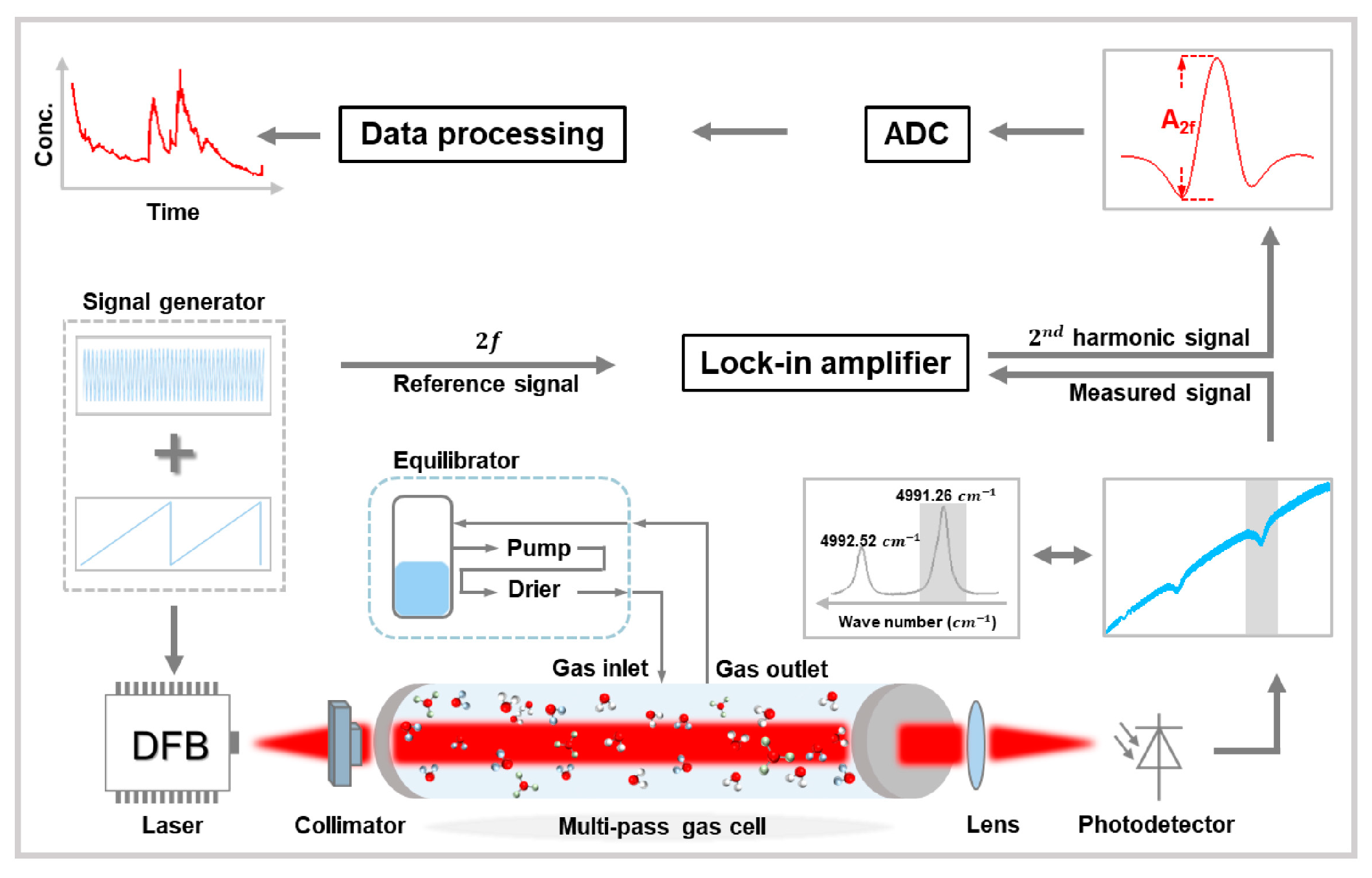
2. Apparatus and Methods
3. Results and Discussion
3.1. System Calibration
3.2. System Precision and Accuracy
3.3. System Response Time
3.4. Atmosphere Monitoring in the Laboratory
3.5. Field Application
- Surface Runoff & Human Activities
- Geographic Position
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Doney, S.C.; Busch, D.S.; Cooley, S.R.; Kroeker, K.J. The impacts of ocean acidification on marine ecosystems and reliant human communities. Annu. Rev. Environ. Resour. 2020, 45, 83–112. [Google Scholar] [CrossRef]
- Gingerich, P.D. Temporal scaling of carbon emission and accumulation rates: Modern anthropogenic emissions compared to estimates of PETM onset accumulation. Paleoceanogr. Paleoclimatol. 2019, 34, 329–335. [Google Scholar] [CrossRef]
- DeVries, T. The oceanic anthropogenic CO2 sink: Storage, air-sea fluxes, and transports over the industrial era. Glob. Biogeochem. Cycles 2014, 28, 631–647. [Google Scholar] [CrossRef] [Green Version]
- IPCC. Climate Change 2013—The Physical Science Basis: Working Group I Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press: Cambridge, UK, 2014; pp. 465–570. [Google Scholar]
- Wang, S.; Moore, J.; Primeau, F.; Khatiwala, S. Simulation of anthropogenic CO2 uptake in the CCSM3. 1 ocean circulation-biogeochemical model: Comparison with data-based estimates. Biogeosciences 2012, 9, 1321–1336. [Google Scholar] [CrossRef] [Green Version]
- Perez, F.F.; Fontela, M.; Garcia-Ibanez, M.I.; Mercier, H.; Velo, A.; Lherminier, P.; Zunino, P.; de la Paz, M.; Alonso-Perez, F.; Guallart, E.F.; et al. Meridional overturning circulation conveys fast acidification to the deep Atlantic Ocean. Nature 2018, 554, 515–518. [Google Scholar] [CrossRef] [PubMed]
- Bindoff, N.L.; Cheung, W.W.L.; Kairo, J.G.; Arístegui, J.; Guinder, V.A.; Hallberg, R.; Hilmi, N.; Jiao, N.; Karim, M.S.; Levin, L.; et al. Changing Ocean, Marine Ecosystems, and Dependent Communities. In IPCC Special Report on the Ocean and Cryosphere in a Changing Climate; Intergovernmental Panel on Climate Change: Geneva, Switzerland, 2019. [Google Scholar]
- Clarke, J.S.; Achterberg, E.P.; Connelly, D.P.; Schuster, U.; Mowlem, M. Developments in marine pCO2 measurement technology; towards sustained in situ observations. TrAC Trends Anal. Chem. 2017, 88, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Doney, S.C.; Fabry, V.J.; Feely, R.A.; Kleypas, J.A. Ocean acidification: The other CO2 problem. Annu. Rev. Mar. Sci. 2009, 1, 169–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arruda, R.; Atamanchuk, D.; Cronin, M.; Steinhoff, T.; Wallace, D.W.R. At-sea intercomparison of three underway pCO2 systems: Intercomparison of pCO2 systems. Limnol. Oceanogr. Methods 2019, 18, 63–76. [Google Scholar] [CrossRef]
- Takahashi, T.; Sutherland, S.; Sweeney, C.; Poisson, A.; Metzl, N.; Tilbrook, B.; Bates, N.; Wanninkhof, R.; Feely, R.; Chris, S.; et al. Global sea-air CO2 flux based on climatological surface ocean pCO2, and seasonal biological and temperature effects. Deep Sea Res. Part II Top. Stud. Oceanogr. 2002, 49, 1601–1622. [Google Scholar] [CrossRef]
- Friedrich, T.; Oschlies, A. Neural network-based estimates of North Atlantic surface pCO2 from satellite data: A methodological study. J. Geophys. Res. Ocean. 2009, 114, 3020. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Zheng, C.; Zhang, T.; Li, Y.; Ren, Q.; Chen, C.; Ye, W.; Zhang, Y.; Wang, Y.; Tittel, F.K. Midinfrared Sensor System Based on Tunable Laser Absorption Spectroscopy for Dissolved Carbon Dioxide Analysis in the South China Sea: System-Level Integration and Deployment. Anal. Chem. 2020, 92, 8178–8185. [Google Scholar] [CrossRef]
- Sepulveda-Jauregui, A.; Martinez-Cruz, K.; Strohm, A.; Walter Anthony, K.M.; Thalasso, F. A new method for field measurement of dissolved methane in water using infrared tunable diode laser absorption spectroscopy. Limnol. Oceanogr. Methods 2012, 10, 560–567. [Google Scholar] [CrossRef]
- You, O.-R.; Son, S.K.; Baker, E.T.; Son, J.; Kim, M.J.; Barcelona, M.J.; Kim, M. Bathymetric influence on dissolved methane in hydrothermal plumes revealed by concentration and stable carbon isotope measurements at newly discovered venting sites on the Central Indian Ridge (11–13°S). Deep Sea Res. Part I Oceanogr. Res. Pap. 2014, 91, 17–26. [Google Scholar] [CrossRef]
- Dickson, A.G.; Sabine, C.L.; Christian, J.R. Guide to Best Practices for Ocean CO2 Measurements; North Pacific Marine Science Organization: Sidney, BC, USA, 2007. [Google Scholar]
- Grilli, R.; Triest, J.; Chappellaz, J.; Calzas, M.; Desbois, T.; Jansson, P.; Guillerm, C.; Ferré, B.; Lechevallier, L.; Ledoux, V.; et al. Sub-Ocean: Subsea Dissolved Methane Measurements Using an Embedded Laser Spectrometer Technology. Environ. Sci. Technol. 2018, 52, 10543–10551. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Olafsson, J.; Goddard, J.G.; Chipman, D.W.; Sutherland, S.C. Seasonal variation of CO2 and nutrients in the high-latitude surface oceans: A comparative study. Glob. Biogeochem. Cycles 1993, 7, 843–878. [Google Scholar] [CrossRef]
- Liu, Y.; Sharma, K.R.; Fluggen, M.; O’Halloran, K.; Murthy, S.; Yuan, Z. Online dissolved methane and total dissolved sulfide measurement in sewers. Water Res. 2015, 68, 109–118. [Google Scholar] [CrossRef]
- Vautier, C.; Abhervé, R.; Labasque, T.; Laverman, A.M.; Guillou, A.; Chatton, E.; Dupont, P.; Aquilina, L.; de Dreuzy, J.-R. Mapping gas exchanges in headwater streams with membrane inlet mass spectrometry. J. Hydrol. 2020, 581, 124398. [Google Scholar] [CrossRef]
- Douchi, D.; Liang, F.; Cano, M.; Xiong, W.; Wang, B.; Maness, P.C.; Lindblad, P.; Yu, J. Membrane-Inlet Mass Spectrometry Enables a Quantitative Understanding of Inorganic Carbon Uptake Flux and Carbon Concentrating Mechanisms in Metabolically Engineered Cyanobacteria. Front. Microbiol. 2019, 10, 1356. [Google Scholar] [CrossRef] [PubMed]
- Prytherch, J.; Yelland, M.J.; Pascal, R.W.; Moat, B.I.; Skjelvan, I.; Neill, C.C. Direct measurements of the CO2 flux over the ocean: Development of a novel method. Geophys. Res. Lett. 2010, 37. [Google Scholar] [CrossRef]
- Sutton, A.J.; Sabine, C.L.; Maenner-Jones, S.; Lawrence-Slavas, N.; Meinig, C.; Feely, R.; Mathis, J.; Musielewicz, S.; Bott, R.; McLain, P. A high-frequency atmospheric and seawater pCO2 data set from 14 open-ocean sites using a moored autonomous system. Earth Syst. Sci. Data 2014, 6, 353–366. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Ye, W.; Liu, Q.; Qi, F.; Cheng, K.; Yang, D.; Zheng, R. Development of a compact deep-sea Raman spectroscopy system and direct bicarbonate detection in sea trials. Appl. Opt. 2019, 58, 2630–2634. [Google Scholar] [CrossRef]
- Humborg, C.; Geibel, M.; Sun, X.; McCrackin, M.; Mörth, C.-M.; Stranne, C.; Jakobsson, M.; Gustafsson, B.; Sokolov, A.; Norkko, A.; et al. High Emissions of Carbon Dioxide and Methane From the Coastal Baltic Sea at the End of a Summer Heat Wave. Front. Mar. Sci. 2019, 6, 493. [Google Scholar] [CrossRef] [Green Version]
- Grefe, I.; Kaiser, J. Equilibrator-based measurements of dissolved nitrous oxide in the surface ocean using an integrated cavity output laser absorption spectrometer. Ocean Sci. 2014, 10, 501. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Zhu, F.; Zhang, S.; Kolomenskii, A.; Schuessler, H. A ppb level sensitive sensor for atmospheric methane detection. Infrared Phys. Technol. 2017, 86, 194. [Google Scholar] [CrossRef]
- Ren, Q.; Chen, C.; Wang, Y.; Li, C.; Wang, Y. A prototype of ppbv-level mid-infrared CO2 sensor for potential application in deep-sea natural-gas-hydrate exploration. IEEE Trans. Instrum. Meas. 2020, 69, 7200–7208. [Google Scholar] [CrossRef]
- Gordon, I.E.; Rothman, L.S.; Hill, C.; Kochanov, R.V.; Tan, Y.; Bernath, P.F.; Birk, M.; Boudon, V.; Campargue, A.; Chance, K.V.; et al. The HITRAN2016 molecular spectroscopic database. J. Quant. Spectrosc. Radiat. Transf. 2017, 203, 3–69. [Google Scholar] [CrossRef]
- Šimečková, M.; Jacquemart, D.; Rothman, L.S.; Gamache, R.R.; Goldman, A. Einstein A-coefficients and statistical weights for molecular absorption transitions in the HITRAN database. J. Quant. Spectrosc. Radiat. Transf. 2006, 98, 130–155. [Google Scholar] [CrossRef]
- Webb, J.R.; Maher, D.T.; Santos, I.R.J.L.; Methods, O. Automated, in situ measurements of dissolved CO2, CH4, and δ13C values using cavity enhanced laser absorption spectrometry: Comparing response times of air-water equilibrators. Limnol. Oceanogr. Methods 2016, 14, 323–337. [Google Scholar] [CrossRef] [Green Version]
- Frankignoulle, M.; Borges, A.; Biondo, R. A new design of equilibrator to monitor carbon dioxide in highly dynamic and turbid environments. Water Res. 2001, 35, 1344–1347. [Google Scholar] [CrossRef] [Green Version]
- Liss, P.S.; Slater, P.G. Flux of Gases across the Air-Sea Interface. Nature 1974, 247, 181–184. [Google Scholar] [CrossRef]
- Takahashi, T.; Sutherland, S.C.; Wanninkhof, R.; Sweeney, C.; Feely, R.A.; Chipman, D.W.; Hales, B.; Friederich, G.; Chavez, F.; Sabine, C.; et al. Climatological mean and decadal change in surface ocean pCO2, and net sea–air CO2 flux over the global oceans. Deep Sea Res. Part II Top. Stud. Oceanogr. 2009, 56, 554–577. [Google Scholar] [CrossRef]
- Weiss, R.F.; Price, B.A. Nitrous oxide solubility in water and seawater. Mar. Chem. 1980, 8, 347–359. [Google Scholar] [CrossRef]
- Wu, Y.; Dai, M.; Guo, X.; Chen, J.; Xu, Y.; Xu, D.; Dai, J.; Zhang, Z. High-frequency time-series autonomous observations of sea surface pCO2 and pH. Limnol. Oceanogr. 2020. [Google Scholar] [CrossRef]
- Li, J.; Luo, G.; Du, Z.; Ma, Y. Hollow waveguide enhanced dimethyl sulfide sensor based on a 3.3μm interband cascade laser. Sens. Actuators B Chem. 2018, 255, 3550–3557. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | Mean |
| Value | 804.2 | 806.3 | 805.9 | 805.1 | 807.0 | 808.9 | 807.4 | 806.4 | 807.0 | 806.9 | 806.5 |
| Tidal Time (hh:mm) | 08:00 | 09:00 | 10:00 | 11:00 | 12:00 | 13:00 | 13:30 | 14:00 | 15:00 | 16:00 | 17:00 |
| Tidal height (cm) | 364 | 304 | 257 | 208 | 146 | 98 | 90 | 98 | 150 | 236 | 330 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; Li, M.; Guo, J.; Du, B.; Zheng, R. A Portable Tunable Diode Laser Absorption Spectroscopy System for Dissolved CO2 Detection Using a High-Efficiency Headspace Equilibrator. Sensors 2021, 21, 1723. https://doi.org/10.3390/s21051723
Zhang Z, Li M, Guo J, Du B, Zheng R. A Portable Tunable Diode Laser Absorption Spectroscopy System for Dissolved CO2 Detection Using a High-Efficiency Headspace Equilibrator. Sensors. 2021; 21(5):1723. https://doi.org/10.3390/s21051723
Chicago/Turabian StyleZhang, Zhihao, Meng Li, Jinjia Guo, Baolu Du, and Ronger Zheng. 2021. "A Portable Tunable Diode Laser Absorption Spectroscopy System for Dissolved CO2 Detection Using a High-Efficiency Headspace Equilibrator" Sensors 21, no. 5: 1723. https://doi.org/10.3390/s21051723