
Improvement of On-Site Sensor for Simultaneous Determination of Phosphate, Silicic Acid, Nitrate plus Nitrite in Seawater


Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Standards Preparation
- -
- The acidic MoO42− reagent (R1) was prepared by dissolving 12.8 g ammonium molybdate tetrahydrate ((NH4)6Mo7O24 4H2O, Sigma Aldrich, Burlington, MA, USA) and 140 mL sulfuric acid (H2SO4, 98%, Merck, Kenilworth, NJ, USA) to obtain a concentration of 2.57 M (pH 0.6), 3.5 mL of a solution of potassium antimony (III) oxide tartrate trihydrate (PAT; C8H4K2O12Sb2 3H2O; Merck) (5.3 g/100 mL deionized water), and 1 mL of solution of sodium dodecyl sulfate (C₁₂H₂₅OSO₂ONa; Merck, Kenilworth, NJ, USA) (30 g/L) in 1000 mL deionized water.
- -
- Ascorbic acid reagent (R2) was prepared by dissolving 25 g of L(+)-ascorbic acid (C6H8O6; ≥99%, Carl Roth, Karlsruhe, Germany) in 1000 mL of deionized water.
- -
- The MoO42− reagent (R1) was prepared by dissolving 15 g of ammonium molybdate tetrahydrate, 5.4 mL of H2SO4, and 1 mL of sodium dodecyl sulfate solution in 1000 mL of deionized water.
- -
- The oxalic acid reagent (R2) was prepared by dissolving 50 g of oxalic acid dihydrate (C2H2O4.2H2O; ≥99%, Carl Roth, Karlsruhe, Germany) into 1000 mL of deionized water.
- -
- The ascorbic acid reagent (R3) was the same as that used for PO43− determination.
- -
- The Griess reagent and VCl3 reducing agent reagent were prepared by dissolving 5 g of VCl3 (Sigma Aldrich, Burlington, MA, USA) in 200 mL of deionized water until the solution turned a dark brown color. Then, 15 mL of concentrated HCl (37%, trace-metal grade, Fisher Scientific, Waltham, MA, USA) was added. After a dark-turquoise color appeared, 10 g of sulfanilamide (H2NC6H4SO2NH2; Merck, USA) was added by dissolving 1 g of N-(1-naphthyl) ethylenediamine dihydrochloride (C10H7NHCH2CH2NH2.2HCl; Merck, Kenilworth, NJ, USA) and 1 mL of a solution of Triton x-100 50% (v/v) (50 mL Triton x-100 (Sigma Aldrich, Burlington, MA, USA): 50 mL isopropanol (Fisher Scientific, Waltham, MA, USA) in 1000 mL of deionized water.
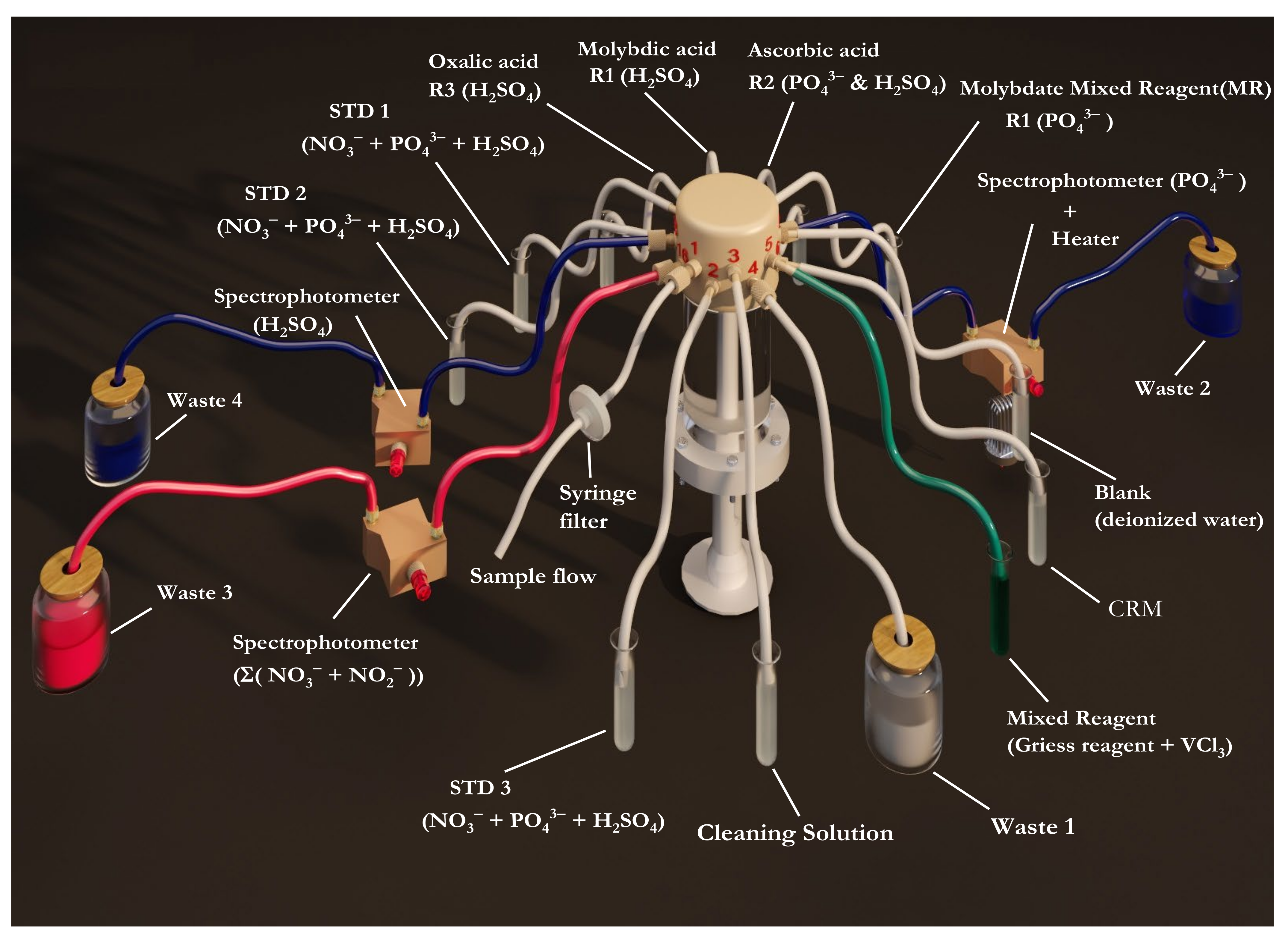
2.2. Multinutrient Analyzer Description
2.3. Chemical Methods
2.3.1. Phosphate Chemical Assay
2.3.2. Silicic Acid Chemical Assay
2.3.3. Nitrate and Nitrite Chemical Assay
2.4. Analytical Protocol
2.5. Data Processing
2.6. Field Deployment and Discrete Sampling
3. Results and Discussion
3.1. Optimization of Analytical Conditions
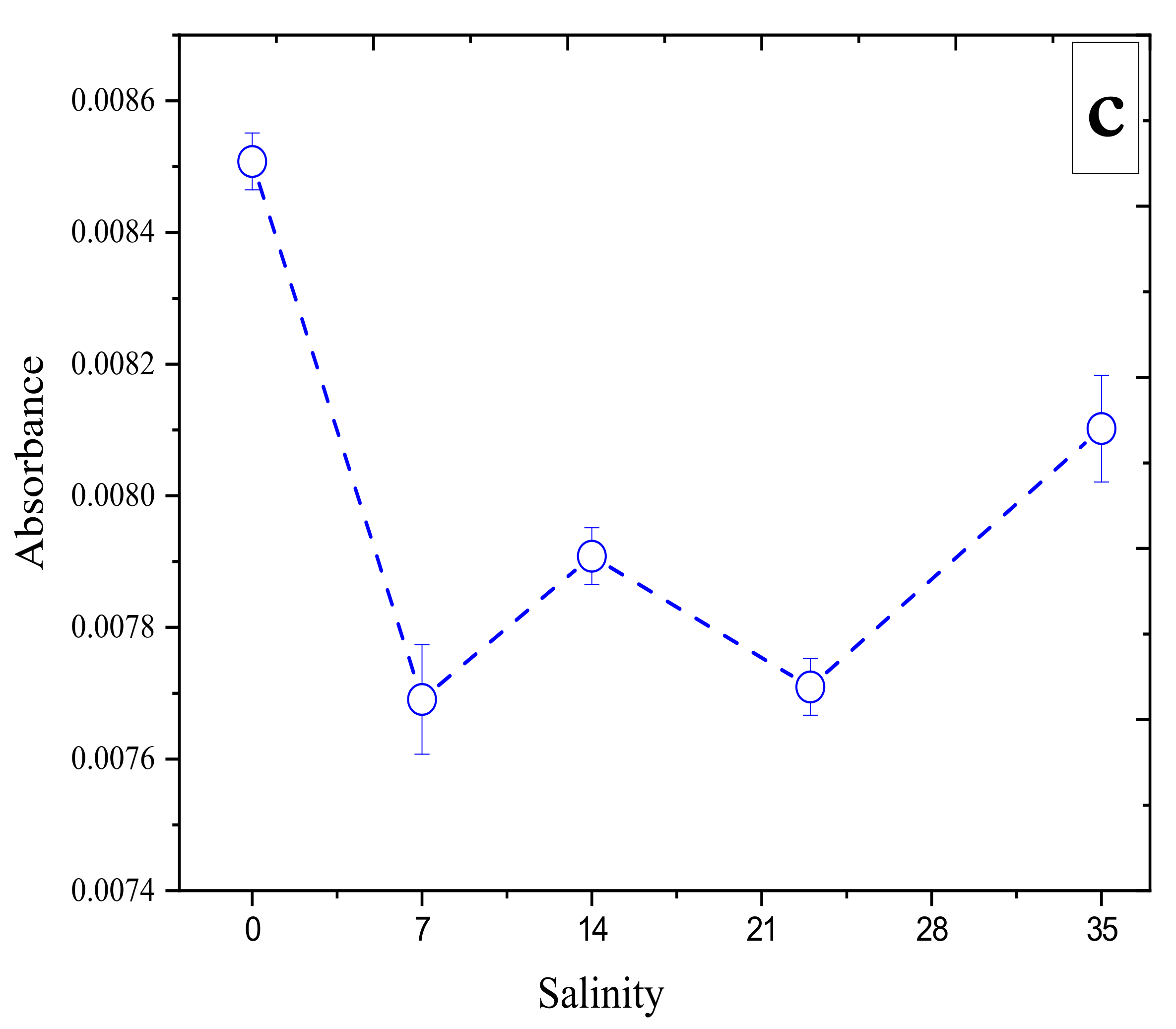
3.2. Effect of Salinity
3.3. Analytical Performance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyzer | Method | Linear Range (μM) | LOD (μM) | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PO43− | NO3− | NO2− | H4SiO4 | PO43− | NO3− | NO2− | H4SiO4 | |||
| WIZ | µLFA (a)/ wet chemistry | 0.19–32.2 | 0.28–71.4 | 0.15–19.2 | ------- | 0.19 | 0.28 | 0.15 | ------ | [25] |
| APNA, ChemFIN | CFIA (b)/ wet chemistry | 0.03–16 | 0.03–15 | 0.02–10 | 0.05–50 | 0.03 | 0.03 | 0.02 | 0.05 | [54] |
| Hydrocycle, Sea-Bird | FIA (c)/ wet chemistry | 0–10 | ------- | ------- | ------- | 0.075 | ------ | ------ | ------ | [66] |
| NAS3X | FIA/ wet chemistry | 0–6 | 0-300 | ------- | 0–60 | 0.06 | 0.05 | ------ | 0.06 | [67] |
| ANAIS | rFIA (d)/ wet chemistry | 0.1–5 | 0.1–40 | ------- | 0.5–150 | 0.1 | 0.1 | ------ | 0.5 | [26] |
| ALCHEMIST | FIA/ wet chemistry | ------- | 0–40 (e) | ------- | ------ | 0.5 | ------ | [23] | ||
| Lab-on-Chip | Micro-fluidics/ wet chemistry | ------- | 0.025–350 | 0–0.25 | ------- | ------ | 0.05 | 0.02 | ------ | [56] |
| 0.14–10 | ------- | ------- | ------- | 0.04 | ------ | ------ | ------ | [57,58] | ||
| ------- | ------- | ------- | 0–400 | ------ | ------ | ------ | 0.045 | [68] | ||
| NuLAB | FIA/ wet chemistry | 0.2–25 | 0.2–50 (e) | 0.15-35 | 0.3–60 | 0.2 | 0.2 | 0.15 | 0.3 | [55] |
| SUNA | UV-spectral | ------- | 2.4–4000 | ------- | ------- | ------ | 2 | ----- | ----- | [59] |
| OPUS | UV-spectral | ------- | 1–60 | ------- | ------- | ------ | 2 | ----- | ----- | [13] |
| SUV-6 | UV-spectral | ------- | 0–400 | ------- | ------- | ------ | 0.21 | ----- | ----- | [61] |
| ANESIS | Electro-chemistry | ------- | ------- | ------- | 1.63–132.8 | ------ | ------ | ----- | 0.32 | [69] |
| AutoLAB (modified) | FIA/wet chemistry | 0.2–100 | 0.5–100 | 0.4–100 | 0.2–100 | 0.18 | 0.45 | 0.35 | 0.15 | This work |
3.4. Field Deployment
4. Conclusions and Future Implications
- -
- The option to only determine a single nutrient by an analyzer.
- -
- The use of a cadmium column for nitrate reduction, which may degrade by organic matter in the water, and also regular regeneration is typically needed. Our VCl3 reduction approach therefore provides an important step forward.
- -
- An absence of reports on long-term use or field testing in natural waters for some promising analyzers.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Watson, A.J.; Lenton, T.M.; Mills, B.J. Ocean deoxygenation, the global phosphorus cycle and the possibility of human-caused large-scale ocean anoxia. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2017, 375, 20160318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altieri, K.E.; Fawcett, S.E.; Hastings, M.G. Reactive nitrogen cycling in the atmosphere and ocean. Annu. Rev. Earth Planet. Sci. 2021, 49, 523–550. [Google Scholar] [CrossRef]
- Ragueneau, O.; Tréguer, P.; Leynaert, A.; Anderson, R.; Brzezinski, M.; DeMaster, D.; Dugdale, R.; Dymond, J.; Fischer, G.; Francois, R. A review of the Si cycle in the modern ocean: Recent progress and missing gaps in the application of biogenic opal as a paleoproductivity proxy. Glob. Planet. Change 2000, 26, 317–365. [Google Scholar] [CrossRef]
- Field, C.B.; Behrenfeld, M.J.; Randerson, J.T.; Falkowski, P. Primary production of the biosphere: Integrating terrestrial and oceanic components. Science 1998, 281, 237–240. [Google Scholar] [CrossRef] [Green Version]
- Rabalais, N.N.; Cai, W.-J.; Carstensen, J.; Conley, D.J.; Fry, B.; Hu, X.; Quinones-Rivera, Z.; Rosenberg, R.; Slomp, C.P.; Turner, R.E. Eutrophication-driven deoxygenation in the coastal ocean. Oceanography 2014, 27, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Deutsch, C.; Sarmiento, J.L.; Sigman, D.M.; Gruber, N.; Dunne, J.P. Spatial coupling of nitrogen inputs and losses in the ocean. Nature 2007, 445, 163–167. [Google Scholar] [CrossRef]
- Krause, J.W.; Schulz, I.K.; Rowe, K.A.; Dobbins, W.; Winding, M.H.; Sejr, M.K.; Duarte, C.M.; Agustí, S. Silicic acid limitation drives bloom termination and potential carbon sequestration in an Arctic bloom. Sci. Rep. 2019, 9, 8149. [Google Scholar] [CrossRef]
- Blaen, P.J.; Khamis, K.; Lloyd, C.E.; Bradley, C.; Hannah, D.; Krause, S. Real-time monitoring of nutrients and dissolved organic matter in rivers: Capturing event dynamics, technological opportunities and future directions. Sci. Total Environ. 2016, 569, 647–660. [Google Scholar] [CrossRef] [Green Version]
- Rode, M.; Wade, A.J.; Cohen, M.J.; Hensley, R.T.; Bowes, M.J.; Kirchner, J.W.; Arhonditsis, G.B.; Jordan, P.; Kronvang, B.; Halliday, S.J. Sensors in the Stream: The High-Frequency Wave of the Present; ACS Publications: Washington, WA, USA, 2016. [Google Scholar]
- Moscetta, P.; Sanfilippo, L.; Savino, E.; Moscetta, P.; Allabashi, R.; Gunatilaka, A. Instrumentation for continuous monitoring in marine environments. In Proceedings of the OCEANS 2009, Biloxi, MS, USA, 26–29 October 2009; pp. 1–10. [Google Scholar]
- Mills, D.; Greenwood, N.; Kröger, S.; Devlin, M.; Sivyer, D.; Pearce, D.; Cutchey, S.; Malcolm, S. New approaches to improve the detection of eutrophication in UK coastal waters. In Proceedings of the USA-Baltic Internation Symposium, Klaipeda, Lithuania, 15–17 June 2004; pp. 1–7. [Google Scholar]
- Meyer, D.; Prien, R.D.; Rautmann, L.; Pallentin, M.; Waniek, J.J.; Schulz-Bull, D.E. In situ determination of nitrate and hydrogen sulfide in the Baltic Sea using an ultraviolet spectrophotometer. Front. Mar. Sci. 2018, 5, 431. [Google Scholar] [CrossRef]
- Nehir, M.; Esposito, M.; Begler, C.; Frank, C.; Zielinski, O.; Achterberg, E.P. Improved calibration and data processing procedures of OPUS optical sensor for high-resolution in situ monitoring of nitrate in seawater. Front. Mar. Sci. 2021, 8, 663800. [Google Scholar] [CrossRef]
- Daniel, A.; Laës-Huon, A.; Barus, C.; Beaton, A.D.; Blandfort, D.; Guigues, N.; Knockaert, M.; Munaron, D.; Salter, I.; Woodward, E.M.S. Toward a harmonization for using in situ nutrient sensors in the marine environment. Front. Mar. Sci. 2020, 6, 773. [Google Scholar] [CrossRef] [Green Version]
- Pellerin, B.A.; Bergamaschi, B.A.; Downing, B.D.; Saraceno, J.F.; Garrett, J.D.; Olsen, L.D. Optical Techniques for the Determination of Nitrate in Environmental Waters: Guidelines for Instrument Selection, Operation, Deployment, Maintenance, Quality Assurance, and Data Reporting; 1-D5: Reston, VA, USA, 2013; p. 48. [Google Scholar]
- Lacombe, M.; Garçon, V.; Thouron, D.; Le Bris, N.; Comtat, M. Silicate electrochemical measurements in seawater: Chemical and analytical aspects towards a reagentless sensor. Talanta 2008, 77, 744–750. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, D.; Barus, C.; Giraud, W.; Calas, E.; Vanhove, E.; Laborde, A.; Launay, J.; Temple-Boyer, P.; Striebig, N.; Armengaud, M. Silicon-based electrochemical microdevices for silicate detection in seawater. Sens. Actuators B Chem. 2015, 211, 116–124. [Google Scholar] [CrossRef] [Green Version]
- Barus, C.; Chen Legrand, D.; Striebig, N.; Jugeau, B.; David, A.; Valladares, M.; Munoz Parra, P.; Ramos, M.E.; Dewitte, B.; Garçon, V. First deployment and validation of in situ silicate electrochemical sensor in seawater. Front. Mar. Sci. 2018, 5, 60. [Google Scholar] [CrossRef] [Green Version]
- Jońca, J.; Giraud, W.; Barus, C.; Comtat, M.; Striebig, N.; Thouron, D.; Garçon, V. Reagentless and silicate interference free electrochemical phosphate determination in seawater. Electrochim. Acta 2013, 88, 165–169. [Google Scholar] [CrossRef] [Green Version]
- Barus, C.; Romanytsia, I.; Striebig, N.; Garçon, V. Toward an in situ phosphate sensor in seawater using Square Wave Voltammetry. Talanta 2016, 160, 417–424. [Google Scholar] [CrossRef]
- Beaton, A.D.; Sieben, V.J.; Floquet, C.F.; Waugh, E.M.; Bey, S.A.K.; Ogilvie, I.R.; Mowlem, M.C.; Morgan, H. An automated microfluidic colourimetric sensor applied in situ to determine nitrite concentration. Sens. Actuators B Chem. 2011, 156, 1009–1014. [Google Scholar] [CrossRef]
- Beaton, A.D.; Wadham, J.L.; Hawkings, J.; Bagshaw, E.A.; Lamarche-Gagnon, G.; Mowlem, M.C.; Tranter, M. High-resolution in situ measurement of nitrate in runoff from the Greenland ice sheet. Environ. Sci. Technol. 2017, 51, 12518–12527. [Google Scholar] [CrossRef] [Green Version]
- Le Bris, N.; Sarradin, P.-M.; Birot, D.; Alayse-Danet, A.-M. A new chemical analyzer for in situ measurement of nitrate and total sulfide over hydrothermal vent biological communities. Mar. Chem. 2000, 72, 1–15. [Google Scholar] [CrossRef]
- Copetti, D.; Valsecchi, L.; Capodaglio, A.; Tartari, G. Direct measurement of nutrient concentrations in freshwaters with a miniaturized analytical probe: Evaluation and validation. Environ. Monit. Assess. 2017, 189, 144. [Google Scholar] [CrossRef]
- Bodini, S.; Sanfilippo, L.; Savino, E.; Moscetta, P. Automated micro Loop Flow Reactor technology to measure nutrients in coastal water: State of the art and field application. In Proceedings of the OCEANS 2015-Genova, Genova, Italy, 18–21 May 2015; pp. 1–7. [Google Scholar]
- Thouron, D.; Vuillemin, R.; Philippon, X.; Lourenço, A.; Provost, C.; Cruzado, A.; Garçon, V. An autonomous nutrient analyzer for oceanic long-term in situ biogeochemical monitoring. Anal. Chem. 2003, 75, 2601–2609. [Google Scholar] [CrossRef] [PubMed]
- Racicot, J.M.; Mako, T.L.; Olivelli, A.; Levine, M. A paper-based device for ultrasensitive, colorimetric phosphate detection in seawater. Sensors 2020, 20, 2766. [Google Scholar] [CrossRef] [PubMed]
- Charbaji, A.; Heidari-Bafroui, H.; Anagnostopoulos, C.; Faghri, M. A new paper-based microfluidic device for improved detection of nitrate in water. Sensors 2021, 21, 102. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Li, P.; Fang, T.; Jiang, Y.; Chen, J.; Chen, N.; Yuan, D.; Ma, J. Automated determination of dissolved reactive phosphorus at nanomolar to micromolar levels in natural waters using a portable flow analyzer. Anal. Chem. 2020, 92, 4379–4386. [Google Scholar] [CrossRef] [PubMed]
- FIALab Instruments, INC. Available online: https://www.flowinjection.com/hardware/sia-analyzers (accessed on 21 April 2022).
- Hansen, E.H.; Ruzicka, J.; Chocholous, P. Advances in Flow Injection Analysis. Available online: https://www.flowinjectiontutorial.com/index.html (accessed on 21 April 2022).
- Ma, J.; Li, P.; Chen, Z.; Lin, K.; Chen, N.; Jiang, Y.; Chen, J.; Huang, B.; Yuan, D. Development of an integrated syringe-pump-based environmental-water analyzer (i SEA) and application of it for fully automated real-time determination of ammonium in fresh water. Anal. Chem. 2018, 90, 6431–6435. [Google Scholar] [CrossRef]
- Griess, P. Griess reagent: A solution of sulphanilic acid and α-naphthylamine in acetic acid which gives a pink colour on reaction with the solution obtained after decomposition of nitrosyl complexes. Chem. Ber 1879, 12, 427. [Google Scholar]
- Murphy, J.; Riley, J. Colorimetric method for determination of P in soil solution. Anal. Chim. Acta 1962, 27, 31–36. [Google Scholar]
- Mullin, J.; Riley, J. The colorimetric determination of silicate with special reference to sea and natural waters. Anal. Chim. Acta 1955, 12, 162–176. [Google Scholar] [CrossRef]
- Wang, S.; Lin, K.; Chen, N.; Yuan, D.; Ma, J. Automated determination of nitrate plus nitrite in aqueous samples with flow injection analysis using vanadium (III) chloride as reductant. Talanta 2016, 146, 744–748. [Google Scholar] [CrossRef]
- Fang, T.; Li, P.; Lin, K.; Chen, N.; Jiang, Y.; Chen, J.; Yuan, D.; Ma, J. Simultaneous underway analysis of nitrate and nitrite in estuarine and coastal waters using an automated integrated syringe-pump-based environmental-water analyzer. Anal. Chim. Acta 2019, 1076, 100–109. [Google Scholar] [CrossRef]
- Nightingale, A.M.; Hassan, S.-U.; Warren, B.M.; Makris, K.; Evans, G.W.; Papadopoulou, E.; Coleman, S.; Niu, X. A droplet microfluidic-based sensor for simultaneous in situ monitoring of nitrate and nitrite in natural waters. Environ. Sci. Technol. 2019, 53, 9677–9685. [Google Scholar] [CrossRef]
- Ellis, P.S.; Shabani, A.M.H.; Gentle, B.S.; McKelvie, I.D. Field measurement of nitrate in marine and estuarine waters with a flow analysis system utilizing on-line zinc reduction. Talanta 2011, 84, 98–103. [Google Scholar] [CrossRef]
- Jońca, J.; Comtat, M.; Garçon, V. In situ phosphate monitoring in seawater: Today and tomorrow. In Smart Sensors for Real-Time Water Quality Monitoring; Springer: Berlin/Heidelberg, Germany, 2013; pp. 25–44. [Google Scholar]
- Directive, W.F. Water framework directive. J. Ref. OJL 2000, 327, 1–73. [Google Scholar]
- Altahan, M.; Esposito, M.; Achterberg, E.P. Video S1.mp4. In Figshare: 2022. Available online: https://doi.org/10.6084/m9.figshare.19608597.v1 (accessed on 17 April 2022).
- Meteorological Station GEOMAR & Kiel Lighthouse. Available online: https://www.geomar.de/service/wetter (accessed on 13 July 2021).
- García-Robledo, E.; Corzo, A.; Papaspyrou, S. A fast and direct spectrophotometric method for the sequential determination of nitrate and nitrite at low concentrations in small volumes. Mar. Chem. 2014, 162, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.; Li, P.; Ma, J.; Yuan, D. An automatic reserve flow injection method using vanadium (III) reduction for simultaneous determination of nitrite and nitrate in estuarine and coastal waters. Talanta 2019, 195, 613–618. [Google Scholar] [CrossRef]
- Worsfold, P.J.; Clough, R.; Lohan, M.C.; Monbet, P.; Ellis, P.S.; Quétel, C.R.; Floor, G.H.; McKelvie, I.D. Flow injection analysis as a tool for enhancing oceanographic nutrient measurements—A review. Anal. Chim. Acta 2013, 803, 15–40. [Google Scholar] [CrossRef]
- Grasshoff, K.; Kremling, K.; Ehrhardt, M. Methods of Seawater Analysis; John and Wiley and Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Murphy, J.; Riley, J.P. A modified single solution method for the determination of phosphate in natural waters. Anal. Chim. Acta 1962, 27, 31–36. [Google Scholar] [CrossRef]
- Nagul, E.A.; McKelvie, I.D.; Worsfold, P.; Kolev, S.D. The molybdenum blue reaction for the determination of orthophosphate revisited: Opening the black box. Anal. Chim. Acta 2015, 890, 60–82. [Google Scholar] [CrossRef] [Green Version]
- Dias, A.C.B.; Borges, E.P.; Zagatto, E.A.G.; Worsfold, P.J. A critical examination of the components of the Schlieren effect in flow analysis. Talanta 2006, 68, 1076–1082. [Google Scholar] [CrossRef]
- Arar, E.J. Method 366.0 Determination of Dissolved Silicate in Estuarine and Coastal Waters by Gas Segmented Continuous Flow Colorimetric Analysis; United States Environmental Protection Agency: Durham, NC, USA, 1997.
- Long, G.L.; Winefordner, J.D. Limit of detection. A closer look at the IUPAC definition. Anal. Chem. 1983, 55, 712A–724A. [Google Scholar]
- Belter, M.; Sajnóg, A.; Barałkiewicz, D. Over a century of detection and quantification capabilities in analytical chemistry—Historical overview and trends. Talanta 2014, 129, 606–616. [Google Scholar] [CrossRef]
- Egli, P.J.; Veitch, S.P.; Hanson, A.K. Sustained, autonomous coastal nutrient observations aboard moorings and vertical profilers. In Proceedings of the OCEANS 2009, Biloxi, MS, USA, 26–29 October 2009; pp. 1–9. [Google Scholar]
- Green Eyes, LLC. Available online: http://gescience.com/wp-content/uploads/2017/02/NuLAB-4.pdf (accessed on 13 November 2021).
- Beaton, A.D.; Cardwell, C.L.; Thomas, R.S.; Sieben, V.J.; Legiret, F.-E.; Waugh, E.M.; Statham, P.J.; Mowlem, M.C.; Morgan, H. Lab-on-chip measurement of nitrate and nitrite for in situ analysis of natural waters. Environ. Sci. Technol. 2012, 46, 9548–9556. [Google Scholar] [CrossRef]
- Grand, M.M.; Clinton-Bailey, G.S.; Beaton, A.D.; Schaap, A.M.; Johengen, T.H.; Tamburri, M.N.; Connelly, D.P.; Mowlem, M.C.; Achterberg, E.P. A lab-on-chip phosphate analyzer for long-term in situ monitoring at fixed observatories: Optimization and performance evaluation in estuarine and oligotrophic coastal waters. Front. Mar. Sci. 2017, 4, 255. [Google Scholar] [CrossRef] [Green Version]
- Clinton-Bailey, G.S.; Grand, M.M.; Beaton, A.D.; Nightingale, A.M.; Owsianka, D.R.; Slavik, G.J.; Connelly, D.P.; Cardwell, C.L.; Mowlem, M.C. A lab-on-chip analyzer for in situ measurement of soluble reactive phosphate: Improved phosphate blue assay and application to fluvial monitoring. Environ. Sci. Technol. 2017, 51, 9989–9995. [Google Scholar] [CrossRef]
- Sea-Bird Scientific. Available online: https://www.seabird.com/nutrient-sensors/suna-v2-nitrate-sensor/family-downloads?productCategoryId=54627869922/datasheet(SUNAV2.pdf) (accessed on 13 November 2021).
- Trios. Available online: https://www.trios.de/en/opus.html (accessed on 13 November 2021).
- Finch, M.S.; Hydes, D.J.; Clayson, C.H.; Weigl, B.; Dakin, J.; Gwilliam, P. A low power ultra violet spectrophotometer for measurement of nitrate in seawater: Introduction, calibration and initial sea trials. Anal. Chim. Acta 1998, 377, 167–177. [Google Scholar] [CrossRef]
- Mansour, F.R.; Danielson, N.D. Reverse flow-injection analysis. TrAC Trends Anal. Chem. 2012, 40, 1–14. [Google Scholar] [CrossRef]
- Cerdà, V.; Avivar, J.; Cerdà, A. Laboratory automation based on flow techniques. Pure Appl. Chem. 2012, 84, 1983–1998. [Google Scholar] [CrossRef] [Green Version]
- Monte-Filho, S.S.; Lima, M.B.; Andrade, S.I.; Harding, D.P.; Fagundes, Y.N.; Santos, S.R.; Lemos, S.G.; Araújo, M.C. Flow–batch miniaturization. Talanta 2011, 86, 208–213. [Google Scholar] [CrossRef] [Green Version]
- Schnetger, B.; Lehners, C. Determination of nitrate plus nitrite in small volume marine water samples using vanadium (III) chloride as a reduction agent. Mar. Chem. 2014, 160, 91–98. [Google Scholar] [CrossRef]
- Snazelle, T.T. Laboratory Evaluation of the Sea-Bird Scientific HydroCycle-PO4 Phosphate Sensor; United States Geological Survey: Reston, VA, USA, 2018; p. 20.
- Envirotech Instruments LLC. Available online: https://www.bodc.ac.uk/data/documents/nodb/pdf/envirotech_nas_nutrient_analyser.pdf (accessed on 13 November 2021).
- Cao, X.; Zhang, S.; Chu, D.; Wu, N.; Ma, H.; Liu, Y. A design of spectrophotometric microfluidic chip sensor for analyzing silicate in seawater. In Proceedings of the IOP Conference Series: Earth and Environmental Science, Qingdao, China, 26–29 June 2017; p. 012080. [Google Scholar]
- Legrand, D.C.; Mas, S.; Jugeau, B.; David, A.; Barus, C. Silicate marine electrochemical sensor. Sens. Actuators B Chem. 2021, 335, 129705. [Google Scholar] [CrossRef]
- Gibbons, R.D.; Coleman, D.E. Statistical Methods for Detection and Quantification of Environmental Contamination; John and Wiley and Sons: Hoboken, NJ, USA, 2001. [Google Scholar]
- Schories, D.; Selig, U.; Schubert, H.; Jegzentis, K.; Mertens, M.; Schubert, M.; Kaminski, T. Küstengewässer-Klassifizierung Deutsche Ostsee nach EU-WRRL. Teil A Äußere Küstengewässer Forsch. 2006. Available online: http://www.biologie.uni-rostock.de/oekologie/literature/RMB/Heft%2014/RMB-14-Schories-et-al-135-150.pdf (accessed on 9 September 2021).
- Javidpour, J.; Molinero, J.C.; Peschutter, J.; Sommer, U. Seasonal changes and population dynamics of the ctenophore Mnemiopsis leidyi after its first year of invasion in the Kiel Fjord, Western Baltic Sea. Biol. Invasions 2009, 11, 873–882. [Google Scholar] [CrossRef] [Green Version]
- Schröder, K.; Kossel, E.; Lenz, M. Microplastic abundance in beach sediments of the Kiel Fjord, Western Baltic Sea. Environ. Sci. Pollut. Res. 2021, 28, 26515–26528. [Google Scholar] [CrossRef] [PubMed]
- Healy, T.; Wang, Y.; Healy, J.-A. Muddy Coasts of the World: Processes, Deposits and Function; Elsevier: Amsterdam, The Netherlands, 2002. [Google Scholar]
- WSV-Wasserstraßen- und Schifffahrtsverwaltung des Bundes Pegel Kiel-Holtenau. Available online: https://www.pegelonline.wsv.de/gast/stammdaten?pegelnr=9610066 (accessed on 15 January 2022).
- Lai, C.-Z.; DeGrandpre, M.D.; Darlington, R.C. Autonomous optofluidic chemical analyzers for marine applications: Insights from the Submersible Autonomous Moored Instruments (SAMI) for pH and pCO2. Front. Mar. Sci. 2018, 4, 438. [Google Scholar] [CrossRef]
- Fischer, M.; Friedrichs, G.; Lachnit, T. Fluorescence-based quasicontinuous and in situ monitoring of biofilm formation dynamics in natural marine environments. Appl. Environ. Microbiol. 2014, 80, 3721–3728. [Google Scholar] [CrossRef] [Green Version]
- Wasmund, N.; Dutz, J.; Pollehne, F.; Siegel, H.; Zettler, M.L. Biological assessment of the Baltic Sea. Marine Science Reports No 98 2015. Available online: https://doi.org/10.12754/msr-2015-0098 (accessed on 9 September 2021).
- Nedwell, D.B.; Jickells, T.D.; Trimmer, M.; Sanders, R. Nutrients in estuaries. In Advances in Ecological Research; Nedwell, D.B., Raffaelli, D.G., Eds.; Academic Press: Cambridge, MA, USA, 1999; Volume 29, pp. 43–92. [Google Scholar]
- Statham, P.J. Nutrients in estuaries—An overview and the potential impacts of climate change. Sci. Total Environ. 2012, 434, 213–227. [Google Scholar] [CrossRef]
- Marion, G. The geochemistry of natural waters: Surface and groundwater environments. J. Environ. Qual. 1998, 27, 245. [Google Scholar] [CrossRef]
- Burdige, D.J. Geochemistry of Marine Sediments; Princeton University Press: Princeton, NJ, USA, 2021. [Google Scholar]
- Conley, D.J.; Björck, S.; Bonsdorff, E.; Carstensen, J.; Destouni, G.; Gustafsson, B.G.; Hietanen, S.; Kortekaas, M.; Kuosa, H.; Markus Meier, H.E.; et al. Hypoxia-related processes in the Baltic Sea. Environ. Sci. Technol. 2009, 43, 3412–3420. [Google Scholar] [CrossRef] [Green Version]
- Latimer, J.S.; Charpentier, M.A. Nitrogen inputs to seventy-four southern New England estuaries: Application of a watershed nitrogen loading model. Estuar. Coast. Shelf Sci. 2010, 89, 125–136. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Altahan, M.F.; Esposito, M.; Achterberg, E.P. Improvement of On-Site Sensor for Simultaneous Determination of Phosphate, Silicic Acid, Nitrate plus Nitrite in Seawater. Sensors 2022, 22, 3479. https://doi.org/10.3390/s22093479
Altahan MF, Esposito M, Achterberg EP. Improvement of On-Site Sensor for Simultaneous Determination of Phosphate, Silicic Acid, Nitrate plus Nitrite in Seawater. Sensors. 2022; 22(9):3479. https://doi.org/10.3390/s22093479
Chicago/Turabian StyleAltahan, Mahmoud Fatehy, Mario Esposito, and Eric P. Achterberg. 2022. "Improvement of On-Site Sensor for Simultaneous Determination of Phosphate, Silicic Acid, Nitrate plus Nitrite in Seawater" Sensors 22, no. 9: 3479. https://doi.org/10.3390/s22093479