Abstract
New perylene monoimide (PMI) derivatives bearing a seven-membered heterocycle and 1,8-diaminosarcophagine (DiAmSar) or N,N-dimethylaminoethyl chelator fragments were synthesized, and their spectroscopic properties in the absence and presence of metal cations were determined to evaluate their potential applications as PET optical sensors for such analytes. DFT and TDDFT calculations were employed to rationalize the observed effects.
1. Introduction
Metal ions play a substantial role in the functioning of every living organism. On the one hand, their presence as macro or microelements is essential for almost any biological process, such as intra- and intercellular communications, electron and proton transfer processes, photosynthesis, the regulation of nerve impulses, gas transfers, etc. [1]. On the other hand, depending on the absorbed dose, they may be a major threat to the health and life of living systems [2]. There are plenty of scientifically proven environmental and health effects of metals, their cations, and their complexes, as well as several myths about their toxicity [3], but it is inarguable that their qualitative and quantitative detection is of utmost importance [4]. One of the most versatile and promising techniques for detecting and quantifying cations of interest are fluorescent sensors [5], which are capable of changing their emissions upon specific binding with the target analytes [6]. Such fluorometric probes provide excellent sensitivity, fast responses [7], and are widely applied in both optical imaging and analytical sensing [8,9].
The rational design of optical sensors incorporates a fluorophore as an active signaling unit, which is covalently tethered to a receptor fragment through a spacer [10]. While the chelating moiety provides the selectivity, an appropriate mechanism for translating the chemical changes to a modulated signal from the reporter should be employed. Photoinduced electron transfer (PET) is one of the most popular such techniques since it can selectively switch the fluorescence emission on or off in the presence of analytes [11,12]. Various classes of light-active organic compounds were successfully used as fluorophores in the design of PET sensors—1,8-naphthalimides, benzanthrone, benzofurazan and xanthene derivatives, etc. [13,14,15,16,17,18]. One of the emerging classes of fluorophores used as signal fragments are perylene monoimides (PMIs) and diimides (PDIs) [19,20,21]. Due to their outstanding chemical, thermal, and photostability, they are among the most promising functional dyes [22,23] and have numerous applications in organic electronics [24], photovoltaics [25], membrane labeling [26], and OLEDs [27]. Due to the constant search for receptor fragments providing the best possible match for any specific fluorophore, various open-chain and cyclic chelators have been tested. A very promising macrobicyclic hexamine cage ligand named sarcophagine (Sar = 3,6,10,13,16,19-hexaaza-bicyclo[6.6.6]-eicosane) is very well known and widely used for developing 64Cu complexes for radiolabeling [28,29,30], but, to the best of our knowledge, it has never been used as a receptor in PET optical sensors. Diamino-sarcophagine (DiAmSar) was chosen as the most promising Sar derivative for this purpose, as it is relatively easy to chemically attach to different organic structures [31] and has been proven to be able to quench the intrinsic fluorescence of albumins [32].
The aim of this work is to synthesize the first PMI derivatives with seven membered dioxepine rings annulated to the peri-positions, as well as having DiAmSar or N,N-dimethylaminoethyl chelating moiety, and to investigate their photophysical properties in the absence and presence of metal cations to determine the PET sensing potential of the new compounds. To better evaluate the proposed PET sensor design, a fluorophore without a receptor was also prepared, and all the structures were investigated via density-functional theory (DFT) and time-dependent density-functional theory (TDDFT) methods to gain a better understanding of the relevant photophysical processes.
2. Materials and Methods
UV/VIS absorption spectra were recorded on a “Cary 5000 UV/VIS/NIR” spectrophotometer (Varian, Mulgrave, Australia). Fluorescence investigations were performed on a “Cary Eclipse” spectrophotometer (Varian, Australia). In all spectroscopic measurements, 1 cm path length synthetic quartz glass cells were used. The organic solvents (spectroscopic grade) used in this study were dichloromethane (DCM) and methanol (MeOH).
The relative fluorescence quantum yield (RQY) was calculated based on the absorption and fluorescence spectra in the same solvent mixture (DCM/MeOH 1:1 v/v), assuming 100% emission quantum yield for the reference compound in the series. The RQYs were calculated as the Gradsample/Gradref ratio (%), where Gradsample and Gradref are the gradients (slopes) of the integrated emission band areas as a function of the absorbance (measured at five different concentrations) for the sample and reference compound, respectively. The RQYs are only used to compare the emissions of the studied compounds and should not be compared with other published fluorescence quantum yields.
To study the effect of the metal cations on the fluorescence intensity, a series of solutions were prepared in volumetric flasks by adding increasing amounts of stock metal cation solution (c = 10−2 mol l−1) to the same volume of the studied compound and then diluting the mixture to the same final volume. The following salts were used as sources of metal cations: AgNO3, BaCl2.2H2O, Cu(NO3)2.5H2O, FeCl3.6H2O, MgCl2.6H2O, Pb(NO3)2, SnCl2.2H2O, and Zn(NO3)2.4 H2O (Sigma-Aldrich, Darmstadt, Germany).
The NMR spectra were obtained using a Bruker DRX-250 spectrometer (Bruker, Karlsruhe, Germany) operating at 400 and 101 MHz for 1H and 13C, respectively, using a dual 5 mm probe head. DMSO-d6 and CDCl3 were used as solvents. Thin layer chromatographic (TLC) analysis was performed on silica gel plates (Macherey-Nagel F60 254 40 × 80; 0.2 mm, Macherey-Nagel, Duren, Germany) using the solvent system n-hexane/dichloromethane as an eluent, unless otherwise stated.
2.1. Synthesis
2.1.1. Synthesis of 1,8-Diamino-3,6,10,13,16,19-hexaazabicyclo-(6,6,6)eicosane (DiAmSar)
The bicyclic cage structure of DiAmSar was synthesized via four-step template synthesis following known procedures [33,34], and the synthetic steps are represented in the Supplementary Materials. Briefly, a Co(en)3Cl3 complex (en = ethylenediamine) was isolated first and used as a template in the following step, in which it was reacted with formaldehyde and nitromethane in an ice-cold basic medium. The template-directed condensation involves all six N-atoms in the octahedron’s vertices of the Co(en)3Cl3 complex, thereby forming the bicyclic cage structure of Co(diNO2Sar)Cl3 (diNO2Sar = dinitrosarcophagine, or 1,8-dinitro-3,6,10,13,16,19-hexaazabicyclo-(6,6,6)eicosane). The latter was precipitated via the addition of 6N HCl and isolated as orange crystals. Its reduction to the diamino-analogue Co(DiAmSar)Cl3 was performed under nitrogen with an excess of Zn powder and concentrated HCl. To avoid the isolation of the mixture of Co(III) and Co(II) complexes, of which the latter is not stable in air, the formed product was oxidized to Co(III) with H2O2. The resulting product mixture of differently protonated amines was separated by column chromatography with the cation exchange resin Dowex 50WX2 (Sigma-Aldrich, Darmstadt, Germany) to allow for the isolation of the doubly protonated Co(DiAmSarH2)Cl5 via elution with 3M HCl. Yellow crystals were isolated after the concentration of the eluate. The removal of the Co(III) ion from the bicyclic cage was performed with an excess of cobaltous cyanide (formed in situ) and sodium cyanide under nitrogen. The metal-free DiAmSar was extracted with boiling acetonitrile from the dry residue of the evaporated reaction mixture. White crystals were collected after recrystallization from a minimal amount of acetonitrile.
1H-NMR (δ (ppm), DMSO-d6): 2.61 (s, 6H); 2.62 (s, 6H); 3.09 (bs, 10H, NH, NH2).
13C-NMR (δ (ppm), DMSO-d6): 50.00; 55.34; 58.64.
All intermediate products were characterized by IR (in KBr) and 1H NMR, and the spectra are given in the Supplementary Materials.
2.1.2. Synthesis of Dibutyl 9,10-Dibromo-1,6,7,12-tetrachloroperylene-3,4-dicarboxylate (B2)
To a suspension of crude 9,10-dibromo-1,6,7,12-tetrachloroperylene-3,4-anhydride [35] (B1) (5 mmol 3.09 g), potassium hydroxide (20 mmol, 1.32 g) in 100 mL of water and 0.5 mL of aliquat 336 was added. The mixture was stirred and refluxed for 30 min, and then butyl bromide (50 mmol, 5.40 mL) was added in one portion. The reaction mixture was refluxed for 2 h and was then cooled down to room temperature and extracted with dichloromethane. The crude product was purified via column chromatography using hexane/dichloromethane as the eluent on silica. It yielded 3.29 g (88%) as a yellow solid.
1H-NMR (δ (ppm), CDCl3): 1.00 (t, 6H, CH3, J = 7.4 Hz); 1.50 (m, 4H, CH2); 1.80 (m, 4H, CH2); 4.36 (m, 4H, CH2, J = 10.8 Hz, J = 6.8 Hz); 8.03 (s, 2H); 8.05 (s, 2H).
13C-NMR (δ (ppm), CDCl3): 13.92; 19.37; 30.71; 66.13; 121.44; 123.09; 125.01; 125.12; 127.55; 131.45; 130.28; 132.11; 132.70; 133.95; 134.21; 135.70; 136.86; 167.22.
2.1.3. Synthesis of Dibutyl 3,4,15,16-Tetrachloronaphtho[2,3-b]peryleno[3,4-ef][1,4]dioxepine-1,18-dicarboxylate (B3)
A mixture of dibutyl 9,10-dibromo-1,6,7,12-tetrachloroperylene-3,4-dicarboxylate (B2) (4.0 mmol, 2.99 g), 2,3-dihydroxynaphthalene (5.0 mmol, 0.80 g), and potassium carbonate (10 mmol, 1.38 g) in 20 mL NMP was stirred and heated to 140 °C for 2 h. The mixture was cooled down to room temperature and poured into ice containing 2 mL of concentrated hydrochloric acid. The precipitate was filtered, washed with water, and dried. The crude product was purified via column chromatography using hexane/dichloromethane as the eluent on silica. This yielded 2.72 g (91%) as an orange solid.
1H-NMR (δ (ppm), CDCl3): 1.00 (t, 6H, CH3, J = 7.4 Hz); 1.50 (m, 4H, CH2); 1.80 (m, 4H, CH2); 4.35 (qt, 4H, CH2, J = 10.8 Hz, J = 6.8 Hz); 7.48 (m, 2H); 7.61 (s, 2H); 7.81 (m, 2H); 7.83 (s, 2H); 8.05 (s, 2H).
13C-NMR (δ (ppm), CDCl3): 13.93; 19.38; 30.73; 65.98; 115.18; 119.08; 119.45; 121.29; 123.37; 126.59; 127.46; 127.91; 129.46; 131.45; 131.92; 132.02; 134.65; 135.37; 135.59; 148.82; 153.11; 167.41.
Elemental analysis (Anal.) Calculated (calcd.) for C40H28Cl4O6: C, 64.36; H, 3.78; Found: C, 64.51; H, 3.99.
2.1.4. Synthesis of Dibutyl 3,4,15,16-Tetrachloronaphtho[2,3-b]peryleno[3,4-ef][1,4]dioxepine-1,18-dicarboxylate (B4)
To a solution of B3 (3.5 mmol, 2.61 g) in 50 mL of glacial acetic acid, 10 mL sulfuric acid (95–97%) was added. The mixture was stirred at 110 °C for 2 h. The mixture was cooled down to room temperature and poured into ice. The precipitate was filtered, washed with water, and dried. This yielded 2.09 g (97%) as a purple solid. The product was pure according to the TLC analysis and was used in the next steps without further purification.
2.1.5. Synthesis of 5,6,17,18-Tetrachloro-2-(2,6-diisopropylphenyl)-1H-naphtho[2″,3″:2′,3′][1,4]dioxepino[5′,6′,7′:9,10]peryleno[3,4-cd]pyridine-1,3(2H)-dione (1a)
To a suspension of B4 (1.0 mmol, 0.62 g) in a mixture of 25 mL of NMP and 5 mL of glacial acetic acid, 2,6-diisopropylaniline (4.0 mmol, 0.71 g) was added. The mixture was stirred at 150 °C for 8 h. After cooling to room temperature, 30 mL water was added and the precipitate was filtered, washed with methanol, and dried. The product was purified via column chromatography using hexane/dichloromethane as the eluent on silica. This yielded 0.63 g (81%) as a purple solid.
1H-NMR (δ (ppm), CDCl3): 1.17 (d, 6H, CH3, J = 6.1 Hz); 1.19 (d, 6H, CH3, J = 6.0 Hz); 2.74 (hept, 2H, CH, J = 6.8 Hz); 7.35 (d, 2H, J = 7.8 Hz); 7.51 (m, 3H); 7.71 (s, 2H); 7.84 (m, 2H); 7.87 (s, 2H); 8.66 (s, 2H).
13C-NMR (δ (ppm), CDCl3): 24.02; 29.21; 115.39; 119.00; 119.87; 120.68; 121.19; 123.81; 124.17; 126.63; 127.38; 129.81; 130.31; 131.37; 132.30; 132.86; 133.12; 135.30; 136.49; 145.66; 148.42; 148.51; 153.92; 162.81.
Anal. calcd. C44H27Cl4NO4: C, 68.15; H, 3.51; N, 1.81; Found: C, 68.33; H, 3.38; N, 1.90.
2.1.6. Synthesis of 5,6,17,18-Tetrachloro-2-(2-(dimethylamino)ethyl)-1H-naphtho[2″,3″:2′,3′][1,4]dioxepino-[5′,6′,7′:9,10]peryleno[3,4-cd]pyridine-1,3(2H)-dione (1b)
To a suspension of B4 (1.0 mmol, 0.62 g) in a mixture of 25 mL of NMP and 5 mL of glacial acetic acid, N1,N1-dimethylethylenediamine (4.0 mmol, 0.35 g) was added. The mixture was stirred at 120 °C for 2 h. After cooling to room temperature, 30 mL of water was added and the precipitate was filtered, washed with methanol, and dried. This yielded 0.65 g (95%).
1H-NMR (δ (ppm), DMSO-d6): 2.35 (s, 6H, NCH3); 2.66 (m, 2H, CH2); 4.34 (m, 2H, CH2); 7.49 (m, 2H); 7.66 (s, 2H); 7.82 (m, 2H); 7.84 (s, 2H); 8.58 (s, 2H).
13C-NMR (δ (ppm), DMSO-d6): 38.55; 45.95; 57.16; 115.43; 119.10; 119.92; 120.81; 121.37; 123.36; 126.72; 127.49; 130.11; 131.48; 132.23; 132.83; 132.87; 135.32; 136.48; 148.64; 153.92; 162.93.
Anal. calcd. C36H20Cl4N2O4: C, 63.00; H, 2.94; N, 4.08; Found: C, 63.21; H, 3.07; N, 4.19.
2.1.7. Synthesis of 2-(8-Amino-3,6,10,13,16,19-hexaazabicyclo[6.6.6]icosan-1-yl)-5,6,17,18-tetrachloro-1H-naphtho[2″,3″:2′,3′][1,4]dioxepino[5′,6′,7′:9,10]peryleno[3,4-cd]pyridine-1,3(2H)-dione (1c)
To a suspension of B4 (1.0 mmol, 0.62 g) in a mixture of 25 mL of NMP and 5 mL of glacial acetic acid, DiAmSar (2 mmol, 0.63 g) was added. The mixture was stirred at 150 °C for 12 h. After cooling to room temperature, 30 mL of water was added and the precipitate was filtered, washed with water, and dried. The product was purified via column chromatography using dichloromethane/methanol as the eluent on silica. This yielded 0.30 g (33%) as a purple solid.
1H-NMR (δ (ppm), DMSO-d6): 1.25 (m, 6H, CH2); 1.82 (m, 6H, CH2); 2.34 (t, 6H, CH2, J = 7.3 Hz); 2.85 (t, 6H, CH2, J = 7.5 Hz); 7.22-7.56 (m, 6H); 7.98 (bs, 2H); 8.14 (bs, 2H); 7.84 (s, 2H); 9.03 (bs, 6H, NH); 12.23 (bs, 2H, NH2).
Anal. calcd. C46H42Cl4N8O4: C, 60.54; H, 4.64; N, 12.28; Found: C, 60.30; H, 4.81; N, 12.07.
2.2. Computational Details
The geometry optimization and photophysical properties of the compounds were modeled with the G16 software package [36]. The optimization of the ground and excited state geometry for 1a–c was performed within the DFT [37] and TDDFT [38] formalisms, respectively. The theoretical computations were carried out using B3LYP/6-31G(d,p) and PBE0/6-311+G(2d,p) levels of theory. The vibrational frequencies were evaluated for each structure at the same method/basis to verify that the structures are indeed a minimum of the potential energy surface, and no imaginary frequency was found.
The absorption wavelengths were determined by TDDFT calculations of vertical excitations. The absorption spectra of 1a–c were simulated by TDDFT using the same functionals and basis sets. Twelve excited singlet states were studied. The lowest energy transition with non-zero oscillator strength is considered for each molecule. To simulate the fluorescence, the optimization of the excited state corresponding to the transition of interest was performed at TDDFT, starting from the ground state geometry. Calculations of the vibrational frequencies and the absence of imaginary frequencies confirm that the excited state geometries are minimum on the potential energy surface. The fluorescence electronic transitions were calculated as vertical de-excitations based on the optimized geometries of the excited state. A detailed description of the procedure can be found elsewhere [39].
The solvent effects were examined at each step by means of PCM formalism [40]. All the computations were performed in methanol as the solvent.
3. Results and Discussion
3.1. Synthesis of Compounds
One of our synthetic goals was to obtain new PMI derivatives with an expanded heterocyclic system by annulating a new 7-membered 1,4-dioxepine ring at the peri position of the perylene core. A suitable building block was 9,10-dibromo-6,7,12,13-tetrachloroperylene monoanhydride (B1), which can be easily obtained in gram scale according to the procedure published by Zagranyarski et al. [41]. We intended to use 2,3-dihydroxynaphtalene to substitute both Br-atoms in basic media (NMP, K2CO3), but the anhydride was not reactive enough in such Nucleophilic Aromatic Substitution (SnAr) reactions. One possible approach to overcome this was to perform the imidization first, which is usually conducted with a bulky amine to prevent stacking, thus improving the solubility and making the corresponding imides easier to purify via column chromatography. In this way, we were able to obtain 1a in two steps with a 53% overall yield (Supplementary Materials, Figure S3), thus proving that ring extension via SnAr with suitable dihydroxy arenes is possible. Unfortunately, this “direct” path towards PMIs with benzo or naphtho-dioxepine rings fused at positions 9 and 10 (peri-annulated) has its limitations: (i) all our efforts to use an amine with chelating properties (vide infra) led to no reaction or complex reaction mixtures, from which the target compounds were impossible to isolate and purify; (ii) it is practically impossible to hydrolyze 1a to the corresponding anhydride (B4, Scheme 1), which would allow us to introduce the target receptor fragments. Therefore, we decided to employ the corresponding diester derivatives, which had the following advantages: (i) the opening of the anhydride cycle can be performed easily in basic or acidic media—our method of choice was saponification and then alkylation with 1-bromobutane; (ii) the 9,10-dibromodiester B2 is compatible with the key ring extension SnAr step, and we were able to substitute both peri-bromine atoms, thus forming the new seven-membered cycle with a very good yield; (iii) the resulting dioxepino-diester B3 is of good solubility, can be easily purified, and can be practically quantitatively converted back to the corresponding anhydride B4; (iv) once the extended heterocyclic system is formed, a variety of amines can be used for the imidization of B4, making it a valuable building block for rylene chemistry.
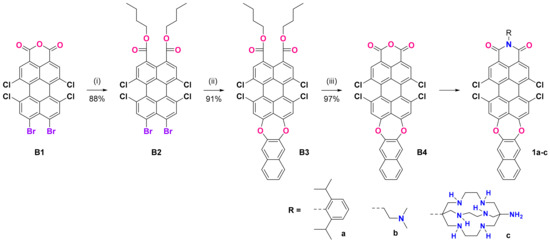
Scheme 1.
Preparation of N-substituted dioxepino PMIs 1a–c: (i) KOH, aliquat 336, water, and then BuBr, reflux, 2 h; (ii) 2,3-dihydroxynaphthalene, NMP, K2CO3, 150 °C, 2 h; (iii) H2SO4/CH3COOH, 110 °C, 2 h.
The compounds 1a–c were synthesized with different substituents at the imide N atom. The one with 2,6-diisopropylphenyl (1a) had no chelating fragments and was prepared to investigate the intrinsic photophysical properties of the new fluorophore. Compound 1b bears a N,N-dimethylaminoethyl receptor fragment, which has been tested in our previous investigations with naphthalimides [42,43]. In contrast, in the case of 1c, we used the bulky bicyclic sarcophagine (DiAmSar), which is known as an excellent chelator for many metal ions. The DiAmSar was obtained in a four-step template synthesis, which employs the template effect of a metal ion to ensure a high yield and the isolation of the unique product with a bicyclic (cage-like) structure that would otherwise be difficult to obtain in a metal-free organic synthesis. Starting from a simple CoCl3 salt, a stable octahedral complex with ethylenediamine (en) can be readily obtained with a high yield and purity. Using it as a substrate in the following one-pot condensation reaction with formaldehyde and nitromethane leads to the formation of the bicyclic structure stabilized by the Co(III) present in its core, Co(diNO2Sar)Cl3. As the Co(III) ions are known for their inertness, the formed complex and its reduced analogue Co(diNO2Sar)Cl3 are very stable, even in a highly acidic medium. Therefore, the removal of the cobalt ions is the most elaborate step, but several procedures are available in the literature [33,34], of which we employed the use of an excess of cyanide that first reduces the Co(III) to the more labile Co(II) complex. The Co(II) ion can more easily be removed from the cage via the formation of stable cobaltous cyanide complexes. The unambiguous proof that the metal ion is removed from the cage is the color of the isolated sarcophagine, which is white, in contrast to the intensely colored cobalt complexes in the preceding steps, and the absence of Co complex-related signals in the NMR spectra. The isolated white crystals of DiAmSar were used in the synthesis of 1c.
3.2. Spectral Properties
Compounds 1a–c show broad absorption bands in the 425–600 nm region, with maxima around 520 nm and a visible shoulder at the shorter wavelength side (Figure 1a). This is typical for rylene imides with two electron donating groups at the peri-positions due to the two different polarization modes in the formed donor–π–acceptor system [44].
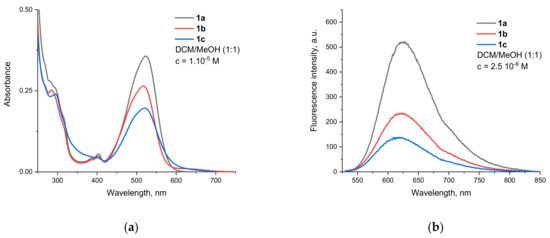
Figure 1.
Spectra of compounds 1a–c in DCM/MeOH 1:1 (v/v). (a) Absorption. (b) Fluorescence (excitation wavelength 520 nm).
Since the additional dioxepine cycle fused at positions 9 and 10 is not aromatic and does not lead to the extension of the perylene π-system, both oxygens should be considered auxochromes. Their donating capabilities are weakened due to their partial conjugation with the naphthalene unit in the opposite direction, but they still determine the intramolecular charge-transfer (ICT) nature of the observed transition. It should be noted that similar substitution patterns in which the two O-atoms are part of a seven membered cycle is not known from the literature. For similar structures, with two separate aryloxy substituents, Sahoo et al. reported a λmax of 540 and 590 nm for absorption and emission, respectively [45]. As Table 1 shows, the absorption bands of the compounds investigated in this work are slightly hypsochromically shifted, which can be attributed to the combined influence of several factors (vide infra).

Table 1.
Photophysical characteristics of the investigated compounds in mixed DCM/MeOH (1:1) solvent.
The fluorescence emission spectra of compounds 1a–c are shown on Figure 1b. All of them emit in the orange-red part of the spectrum and have maxima of around 620 nm. The bands are bathochromically shifted in comparison to the aforementioned diaryloxy-substituted PMIs, leading to Stokes shifts almost twice as big at around 3000 cm−1. This is an indication that substantial conformational changes take place upon the relaxation of the Franck–Condon to the locally excited (LE) emissive state.
To clarify the nature of the electronic transitions and the influence of the N-substituents in the PMIs on the geometry and spectral properties of the studied compounds, DFT and TDDFT calculations were performed. The B3LYP/6-31G(d,p) optimized structures of 1a–c in solvent methanol are presented in Figure 2. The gas-phase optimized molecular geometry of unsubstituted PMI is planar [40]. The bulky chlorine substituents in the bay positions result in the twisting of the molecular geometry to avoid steric hindrance. Therefore, the geometry is not planar, and the π–π interaction is weaker. The calculated (B3LYP/6-31G(d,p)) dihedral angles between the two naphthalene rings in the perylene moiety are rather high (37.5° for 1a and 37.4° and 37.8° for 1b and 1c, respectively) and are consistent with the X-ray data for tetra-substituted PMIs [41].
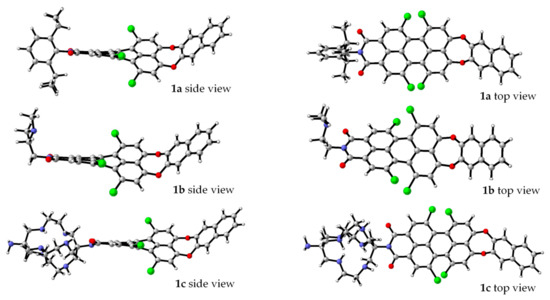
Figure 2.
B3LYP/6-31G(d,p) optimized geometries of 1a–c in methanol [46].
It is known that electron-donor (EDG) substituents at the bay position cause a bathochromic shift of the longest-wavelength electronic transition in the compounds, while electron-withdrawing substituents (EWG) lead to a hypsochromic shift [47]. It can be concluded that the combination of the core twisting, fixed bended conformation at the donor part and the EWG at the bay position define the observed absorption maxima and are in line with the hypsochromic shift.
The theoretical results for the absorption transition energies obtained with the two functionals (B3LYP, and PBE0) and two basis sets (6-31G(d,p) and 6-311+G(2d,p)) in methanol solution for the three substituted PMIs are shown in Table 2. Substitution at the imide nitrogen atom usually improves the solubility of the compounds and modulates their aggregation properties in the solid state but has a very weak effect on their spectral properties, as can be seen from the experimental and theoretical data. Both combinations of functional and basis sets used for the spectral calculations proved to be suitable for the studied substituted PMIs. The transitions are simulated with good accuracy by the theoretical calculations. PBE0 gives better results, and the difference between the calculated and measured transitions is 0.08 eV at most.
Table 2.
Theoretically calculated absorption maxima (in eV), values of oscillator strength (f), and molecular orbital contributions to electronic transition in methanol for 1a–c compared with experimental absorption maxima.
The frontier molecular orbitals for 1b and 1c in methanol are represented in Figure 3. The two functionals applied describe the orbitals’ shape in an analogous way.
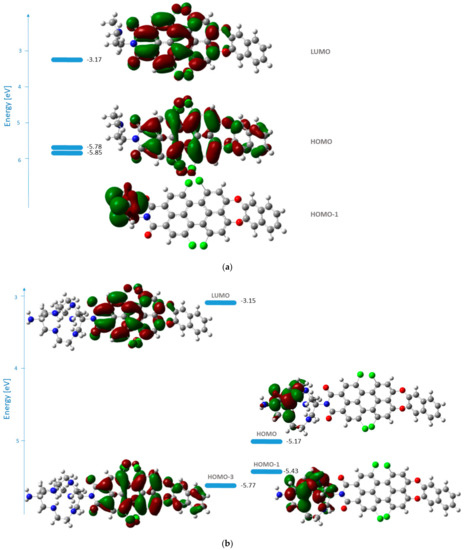
Figure 3.
Energies and shape representations of the ground state orbitals in methanol from TDB3LYP/6-31G(d,p) computations: (a) HOMO−1, HOMO, and LUMO of 1b; (b) HOMO−3, HOMO−1, HOMO, and LUMO of 1c.
The HOMO of 1b is delocalized over the perylene core and the naphthodioxepine moiety (the donor), while LUMO covers the acceptor part of the molecule. The orbitals’ shape indicates that an effective intramolecular charge transfer (ICT) occurs from the donor group toward the imide moiety. Hence, the HOMO→LUMO transition for 1b can be classified as an ICT transition with contributions from the aryloxy substituents. In the case of 1c, the transition with the highest oscillator strength that corresponds to the one in the experimental spectrum arises from HOMO−3→LUMO. In this case, the nature of the transition is also ICT.
The excited state geometries for 1b and 1c were optimized at the PBE0/6-311+G(2d,p) level. For 1b, the predicted fluorescence maximum in methanol is 1.85 eV (671 nm and oscillator strength f = 1.12), which is in good agreement with the experimental value (1.99 eV). The geometry of the excited state of 1b is shown in Figure 4. The main difference in the optimized geometry for the ground S0 and the excited S1 states concerns the dioxepine substituent at the peri position. The donor part of the molecule becomes planar in its excited state.

Figure 4.
TDPBE0/6-311+G(2d,p)-optimized excited state geometry (S1) of 1b in methanol. (a) Side view. (b) Top view.
3.3. PET Design Evaluation
Comparison of the fluorescence intensity of the sensor fragment containing compounds 1b,c with that of the reference compound 1a shows a decrease in the signal due to a PET from the nitrogen atoms that are two carbon atoms away from the imide nitrogen. This can best be illustrated by the spectra of compounds 1a and 1b, which are shown in Figure 5 and clearly show that the weaker fluorescence is not only due to the lower molar absorbance at the excitation wavelength.
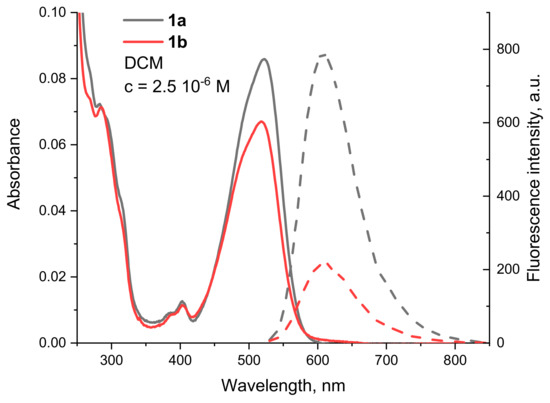
Figure 5.
Absorption (solid line) and fluorescence (dashed line) spectra of 1a (black) and 1b (red) in DCM, c = 2.5 10−6 M.
From the relative quantum yields calculated in Table 1, the effect can be determined quantitatively. The most efficient fluorescence quenching was observed for 1c, where the RQY was half that of 1a. The emission of 1b also clearly decreases due to the presence of the tertiary nitrogen of the dimethylaminoethyl group, with an RQY of 61%. These results suggest that the best match between the HOMO energies of the unbound donor and the fluorophore (condition for reductive PET) is found in 1c. This was also confirmed by the calculated frontier molecular orbitals. For 1c (Figure 3b), both HOMO and HOMO−1 are located at the DiAmSar fragment and can transfer an electron towards the HOMO−3, which is responsible for the ICT transition and is lower by 0.6 and 0.33 eV, respectively. This is consistent with a reductive PET process occurring for 1c. For 1b, the electron transfer should be from HOMO−1, which is located at the chelator, towards the HOMO, from where the electron is excited during the ICT transition (Figure 3a). The energy of the donor was found to be slightly lower than the acceptor, but the difference is only 0.07 eV, suggesting that PET is possible, although less favorable compared to 1c. In general, it can be concluded that the design of the PET sensing system was successful, but the efficiency of the PET is not particularly great.
3.4. Influence of Metal Cations
The presence of analytes in the solution affects the emission of compound 1b. While the absorption spectra remain the same when increasing the metal cations’ concentration, fluorescence intensity changes were observed. As is expected in PET sensing systems, blocking the electron transfer “turns on” the fluorescence without changing its wavelength. Fluorescence titration experiments were performed by varying the metal ion concentration from 0 to at least 2 times the concentration of the free ligand. Typical results are shown in Figure 6. The signal enhancement with increasing concentration of silver and copper ions is demonstrated. An interesting result is registered for Fe3+, where the intensity decreases (far right). A similar effect, but less pronounced, is also observed with Ba2+.

Figure 6.
Illustrative examples of the observed changes in the fluorescence spectra of 1b upon increasing the respective metal cation concentration.
The PET sensors’ sensitivity can be quantified by the fluorescence enhancement (FE) factor, which is defined as the ratio of the maximum intensity achieved (when the signal reaches a plateau and the emission does not change with a further increase in the analyte’s concentration) to the initial intensity of the sample in the absence of analyte.
The FE factors calculated for compound 1b are presented in Figure 7. As can be seen, the sensor has the greatest sensitivity to Ag+ and Cu2+. The values close to unity (the red line for FE = 1 shows no change whatsoever) for the remaining cations suggest some selectivity, most probably due to lower rates of complex formation in their presence. Titrations of 1c did not lead to a change in the fluorescence intensity, from which it can be concluded that either there is no complex formation between the sarcophagine and the studied metal ions under these conditions (not very likely) or that the introduction of the analyte into the chelator cavity does not sufficiently engage all electron pairs at the nitrogen atoms in it and fails to block the PET.
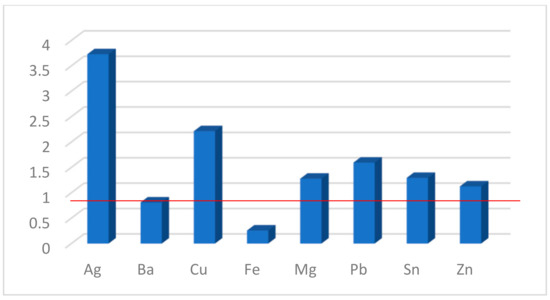
Figure 7.
Fluorescence enhancement (FE) factors for 1b in the presence of metal salts.
To gain a better understanding of the observed PET, even in the presence of a metal cation at the chelator, the geometry of 1c complex with Cu2+ ions was studied theoretically at the B3LYP/6-31G(d,p) level of theory, and its structure is given in Figure 8.
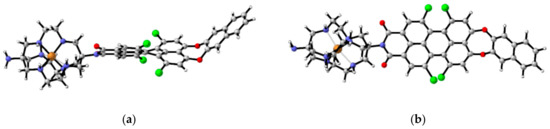
Figure 8.
Optimized structure of the complex of 1c with Cu2+ ions from B3LYP/6-31G(d,p) computations. (a) Side view. (b) Top view.
The S1-excited state of the complex was optimized at the same level of theory, and the energies and shape representation of the frontier molecular orbitals are given in Figure 9. The S1-excited state geometry optimization is necessary as the consideration of the MO in the ground state only is not sufficient to reliably predict the photoinduced electron transfer [14,48].
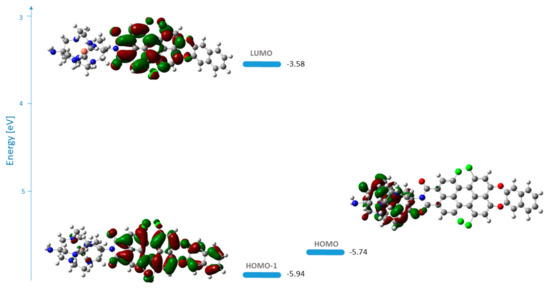
Figure 9.
Energies and shape representation of HOMO−1, HOMO, and LUMO in the excited state (S1) of the complex of 1c with Cu2+ ions in methanol from the TDB3LYP/6-31G(d,p) computations.
Fluorescence enhancement will only take place if the sarcophagine orbitals in the bound state lie lower than the fluorophore orbitals and the reductive PET process cannot occur. In the Cu2+ complex of 1c, the intense transition localized on the fluorophore corresponds to excitation from the HOMO−1→LUMO transition (Figure 9). HOMO is located at the DiAmSar fragment, and an electron can be transferred towards HOMO−1, explaining its inability to block the PET and why 1c cannot be used as a PET sensor.
We also optimized the geometry of the complex between 1b and hydrated Cu2+ ions at the B3LYP/6-31G(d,p) level of theory (Figure S12 in Supplementary Materials). As can be seen, in this case the metal ion is coordinated with one of the N atoms and one O from the imide moiety of the dye, effectively engaging the electron lone pair at the tertiary nitrogen and blocking the PET.
4. Conclusions
Three new perylene monoimide (PMI) derivatives bearing a seven-membered 1,4-dioxepine ring fused at positions 9 and 10 (peri-annulated) were synthesized. An improved synthetic protocol utilizing diester derivatives was employed to maintain compatibility with the SnAr reaction while allowing for the easy purification and introduction of the target receptor fragments. Two Photoinduced Electron Transfer (PET) optical sensors bearing DiAmSar and N,N-dimethylaminoethyl chelator moieties were prepared along with a reference compound with classical 2,6-diisopropylphenyl substituents at the imide nitrogen. The intramolecular charge transfer (ICT) character of the transitions in the recorded absorption spectra was shown, and its efficiency was explained in terms of the molecular geometry and the overlapping of the corresponding molecular orbitals. Both receptor fragments were found to quench the intrinsic emission of the fluorophore, proving the successful design of the PET systems. The DiAmSar derivative 1c showed a better match in the energies of the participating frontier molecular orbitals. Unfortunately, the complex formation does not engage the lone electron pairs in the chelator strongly enough to block the PET and the fluorescence signal does not change in the presence of metal cations. Another reason from a practical point of view is that cyclic chelators usually need additional time and energy to initiate complex formation. These demands are lower in the case of non-cyclic chelators. Therefore, a reasonable compromise between the kinetics of the complexation and the stability of the formed complexes is another concern in the design of a successful optical sensor for metal ions. Compound 1b showed a fluorescence enhancement upon the addition of analytes, with selectivity towards Ag+ and Cu2+ with FE factors of 3.7 and 2.3, respectively.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/s23062902/s1, Figure S1: Synthesis of DiAmSar.; Figure S2: Synthetic scheme for “direct” preparation of 1a.; Figure S3. 1H NMR (a) and IR (b) spectra of Co(en)3Cl3.; Figure S4. 1H NMR (a), 13C NMR (b) and IR (c) spectra of [Co(diNO2Sar)]Cl3.; Figure S5. 1H NMR (a), 13C NMR (b) and IR (c) spectra of [Co(diAmSarH2)]Cl5.; Figure S6. 1H NMR (a) and 13C NMR (b) spectra of DiAmSar.; Figure S7. 1H NMR (a) and 13C NMR (b) spectra of B2.; Figure S8. 1H NMR (a) and 13C NMR (b) spectra of B3.; Figure S9. 1H NMR (a) and 13C NMR (b) spectra of 1a; Figure S10. 1H NMR (a) and 13C NMR (b) spectra of 1b.; Figure S11. 1H NMR spectrum of 1c.; Figure S12. Optimized structure for the complex of 1b with hydrated Cu2+ ion from B3LYP/6-31G(d,p) computations.; Table S1. Cartesian coordinates, electronic energies in Hartree, and the number of imaginary frequencies for the B3LYP/6-31G(d,p) fully-optimized geometries of the ground state for dyes 1a–c in methanol and complexes of dyes 1b,c with Cu2+ ions.; Table S2. Cartesian coordinates, electronic energies in Hartree, and the number of imaginary frequencies for the B3LYP/6-31G(d,p) fully optimized geometry of the excited state for the complex of dye 1c with Cu2+ ions.; Table S3. Cartesian coordinates, electronic energies in Hartree, and the number of imaginary frequencies for the optimized geometry of the excited state for dye 1b in methanol at the PBE1PBE/6-311+G(2d,p) level of theory.
Author Contributions
Conceptualization, S.S. and Y.Z.; methodology, S.S., Y.Z. and D.V.C.; validation, A.A., M.M. and D.V.C.; formal analysis, Y.Z. and D.V.C.; investigation, M.M, A.A. and D.V.C.; writing—original draft preparation, S.S. and D.V.C.; writing—review and editing, S.S.; supervision, S.S.; project administration, S.S.; funding acquisition, S.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Bulgarian National Science Fund, grant number KP 06-N29/12.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Moustakas, M. The role of metal ions in biology, biochemistry and medicine. Materials. 2021, 14, 549. [Google Scholar] [CrossRef] [PubMed]
- Jaishankar, M.; Tseten, T.; Anbalagan, N.; Mathew, B.B.; Beeregowda, K.N. Toxicity, mechanism and health effects of some heavy metals. Interdiscip. Toxicol. 2014, 7, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Egorova, K.S.; Ananikov, V.P. Toxicity of metal compounds: Knowledge and myths. Organometallics 2017, 36, 4071–4090. [Google Scholar] [CrossRef]
- Lvova, L. Chemical sensors for heavy metals/toxin detection. Chemosensors 2020, 8, 14. [Google Scholar] [CrossRef]
- Li, L.; Wang, J.; Xu, S.; Li, C.; Dong, B. Recent progress in fluorescent probes for metal Ion detection. Front. Chem. 2022, 10, 1–15. [Google Scholar] [CrossRef]
- Fernandes, R.S.; Shetty, N.S.; Mahesha, P.; Gaonkar, S.L. A Comprehensive Review on Thiophene Based Chemosensors; Springer: New York, NY, USA, 2022; Volume 32, ISBN 0123456789. [Google Scholar]
- Park, S.H.; Kwon, N.; Lee, J.H.; Yoon, J.; Shin, I. Synthetic ratiometric fluorescent probes for detection of ions. Chem. Soc. Rev. 2020, 49, 143–179. [Google Scholar] [CrossRef]
- Wu, D.; Sedgwick, A.C.; Gunnlaugsson, T.; Akkaya, E.U.; Yoon, J.; James, T.D. Fluorescent chemosensors: The past, present and future. Chem. Soc. Rev. 2017, 46, 7105–7123. [Google Scholar] [CrossRef]
- You, L.; Zha, D.; Anslyn, E.V. Recent advances in supramolecular analytical chemistry using optical sensing. Chem. Rev. 2015, 115, 7840–7892. [Google Scholar] [CrossRef]
- Park, J.-K.; Shin, J.; Jang, S.; Seol, M.-L.; Kang, J.; Choi, S.; Eom, H.; Kwon, O.; Park, S.; Noh, D.-Y.; et al. Rational design of fluorescent/colorimetric chemosensors for detecting transition metal ions by varying functional groups. Inorganics 2022, 10, 189. [Google Scholar] [CrossRef]
- Daly, B.; Ling, J.; De Silva, A.P. Current developments in fluorescent PET (photoinduced electron transfer) sensors and switches. Chem. Soc. Rev. 2015, 44, 4203–4211. [Google Scholar] [CrossRef]
- De Silva, A.P.; Moody, T.S.; Wright, G.D. Fluorescent PET (Photoinduced Electron Transfer) sensors as potent analytical tools. Analyst 2009, 134, 2385–2393. [Google Scholar] [CrossRef]
- Georgiev, N.I.; Dimitrova, M.D.; Asiri, A.M.; Alamry, K.A.; Bojinov, V.B. Synthesis, sensor activity and logic behaviour of a novel bichromophoric system based on rhodamine 6G and 1,8-naphthalimide. Dye. Pigment. 2015, 115, 172–180. [Google Scholar] [CrossRef]
- Yordanova-Tomova, S.; Cheshmedzhieva, D.; Stoyanov, S.; Dudev, T.; Grabchev, I. Synthesis, photophysical characterization, and sensor activity of new 1,8-naphthalimide derivatives. Sensors 2020, 20, 3892. [Google Scholar] [CrossRef]
- Staneva, D.; Vasileva-Tonkova, E.; Grabchev, I. pH sensor potential and antimicrobial activity of a new PPA dendrimer modified with benzanthrone fluorophores in solution and on viscose fabric. J. Photochem. Photobiol. A Chem. 2019, 375, 24–29. [Google Scholar] [CrossRef]
- Abebe, F.; Perkins, P.; Shaw, R.; Tadesse, S. A rhodamine-based fluorescent sensor for selective detection of Cu2+ in aqueous media: Synthesis and spectroscopic properties. J. Mol. Struct. 2020, 1205, 127594. [Google Scholar] [CrossRef]
- Staneva, D.; Manov, H.; Yordanova, S.; Vasileva-Tonkova, E.; Stoyanov, S.; Grabchev, I. Synthesis, spectral properties and antimicrobial activity of a new cationic water-soluble pH-dependent poly(propylene imine) dendrimer modified with 1,8-naphthalimides. Luminescence 2020, 35, 947–954. [Google Scholar] [CrossRef]
- Staneva, D.; Yordanova, S.; Vasileva-Tonkova, E.; Stoyanov, S.; Grabchev, I. Synthesis of a new fluorescent poly(propylene imine) dendrimer modified with 4-nitrobenzofurazan. Sensor and antimicrobial activity. J. Photochem. Photobiol. A Chem. 2020, 395, 112506. [Google Scholar] [CrossRef]
- Chen, S.; Xue, Z.; Gao, N.; Yang, X.; Zang, L. Perylene diimide-based fluorescent and colorimetric sensors for environmental detection. Sensors. 2020, 20, 917. [Google Scholar] [CrossRef]
- Roy, R.; Khan, A.; Chatterjee, O.; Bhunia, S.; Koner, A.L. Perylene monoimide as a versatile fluoroprobe: The past, present, and future. Org. Mater. 2021, 3, 417–454. [Google Scholar] [CrossRef]
- Georgiev, N.I.; Said, A.I.; Toshkova, R.A.; Tzoneva, R.D.; Bojinov, V.B. A novel water-soluble perylenetetracarboxylic diimide as a fluorescent pH probe: Chemosensing, biocompatibility and cell imaging. Dye. Pigment. 2019, 160, 28–36. [Google Scholar] [CrossRef]
- Herrmann, A.; Müllen, K. From industrial colorants to single photon sources and biolabels: The fascination and function of rylene dyes. Chem. Lett. 2006, 35, 978–985. [Google Scholar] [CrossRef]
- Würthner, F. Perylene bisimide dyes as versatile building blocks for functional supramolecular architectures. Chem. Commun. 2004, 4, 1564–1579. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Facchetti, A.; Barlow, S.; Marks, T.J.; Ratner, M.A.; Wasielewski, M.R.; Marder, S.R. Rylene and related diimides for organic electronics. Adv. Mater. 2011, 23, 268–284. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wonneberger, H. Perylene imides for organic photovoltaics: Yesterday, today, and tomorrow. Adv. Mater. 2012, 24, 613–636. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.; Jung, C.; Wallis, P.; Schnitzler, T.; Li, C.; Müllen, K.; Bräuchle, C. Photophysics of new photostable rylene derivatives: Applications in single-molecule studies and membrane labelling. Chem. Phys. Chem. 2011, 12, 1588–1595. [Google Scholar] [CrossRef]
- Kozma, E.; Mróz, W.; Villafiorita-Monteleone, F.; Galeotti, F.; Andicsová-Eckstein, A.; Catellani, M.; Botta, C. Perylene diimide derivatives as red and deep red-emitters for fully solution processable OLEDs. RSC Adv. 2016, 6, 61175–61179. [Google Scholar] [CrossRef]
- Paterson, B.M.; Buncic, G.; McInnes, L.E.; Roselt, P.; Cullinane, C.; Binns, D.S.; Jeffery, C.M.; Price, R.I.; Hicks, R.J.; Donnelly, P.S. Bifunctional 64Cu-labelled macrobicyclic cage amine isothiocyanates for immuno-positron emission tomography. Dalt. Trans. 2015, 44, 4901–4909. [Google Scholar] [CrossRef]
- Liu, S.; Li, Z.; Conti, P. Development of multi-functional chelators based on sarcophagine cages. Molecules 2014, 19, 4246–4255. [Google Scholar] [CrossRef]
- Cooper, M.S.; Ma, M.T.; Sunassee, K.; Shaw, K.P.; Williams, J.D.; Paul, R.L.; Donnelly, P.S.; Blower, P.J. Comparison of 64cu-complexing bifunctional chelators for radioimmunoconjugation: Labeling efficiency, specific activity, and in vitro/in vivo stability. Bioconjug. Chem. 2012, 23, 1029–1039. [Google Scholar] [CrossRef]
- Brand, C.; Abdel-Atti, D.; Zhang, Y.; Carlin, S.; Clardy, S.M.; Keliher, E.J.; Weber, W.A.; Lewis, J.S.; Reiner, T. In vivo imaging of GLP-1R with a targeted bimodal PET/fluorescence imaging agent. Bioconjug. Chem. 2014, 25, 1323–1330. [Google Scholar] [CrossRef]
- Hooshyar, Z.; Rezanejade Bardajee, G.; Kakavand, N.; Khanjari, M.; Dianatnejad, N. Investigations on the interactions of DiAmsar with serum albumins: Insights from spectroscopic and molecular docking techniques. Luminescence 2015, 30, 538–548. [Google Scholar] [CrossRef]
- Bottomley, G.A.; Clark, I.J.; Creaser, I.I.; Engelhardt, L.M.; Geue, R.J.; Hagen, K.S.; Harrowfield, J.M.; Lawrance, G.A.; Lay, P.A.; Sargeson, A.M.; et al. The synthesis and structure of encapsulating ligands: Properties of bicyclic hexamines. Aust. J. Chem. 1994, 47, 143–179. [Google Scholar] [CrossRef]
- Cai, H.; Fissekis, J.; Conti, P.S. Synthesis of a novel bifunctional chelator AmBaSar based on sarcophagine for peptide conjugation and 64Cu radiolabelling. Dalt. Trans. 2009, 27, 5395–5400. [Google Scholar] [CrossRef]
- Zagranyarski, Y.; Chen, L.; Zhao, Y.; Wonneberger, H.; Li, C.; Müllen, K. Facile transformation of perylene tetracarboxylic acid dianhydride into strong donor-acceptor chromophores. Org. Lett. 2012, 14, 5444–5447. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision 16.A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Labanowski, J.K.; Andzelm, J.W. (Eds.) Density Functional Methods in Chemistry; Springer: New York, NY, USA, 1991. [Google Scholar]
- Gross, E.K.U.; Dobson, J.F.; Petersilka, M. Density functional theory of time-dependent phenomena. In Density Functional Theory II; Springer: Berlin/Heidelberg, Germany, 1996; pp. 81–172. [Google Scholar]
- Cheshmedzhieva, D.; Ivanova, P.; Stoyanov, S.; Tasheva, D.; Dimitrova, M.; Ivanov, I.; Ilieva, S. Experimental and theoretical study on the absorption and fluorescence properties of substituted aryl hydrazones of 1,8-naphthalimide. Phys. Chem. Chem. Phys. 2011, 13, 18530–18538. [Google Scholar] [CrossRef]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Zagranyarski, Y.; Chen, L.; Jänsch, D.; Gessner, T.; Li, C.; Müllen, K. Toward perylene dyes by the hundsdiecker reaction. Org. Lett. 2014, 16, 2814–2817. [Google Scholar] [CrossRef]
- Manov, H.; Staneva, D.; Vasileva-Tonkova, E.; Kukeva, R.; Stoyanov, S.; Grabchev, I.; Alexandrova, R.; Stoyanova, R. A new Cu(II) complex of PAMAM dendrimer modified with 1,8-naphthalimide: Antibacterial and anticancer activity. Biointerface Res. Appl. Chem. 2021, 12, 5534–5547. [Google Scholar] [CrossRef]
- Yordanova, S.; Grabchev, I.; Stoyanov, S.; Petkov, I. New detectors for metal cations and protons based on PAMAM dendrimers modified with 1,8-naphthalimide units. J. Photochem. Photobiol. A Chem. 2014, 283, 1–7. [Google Scholar] [CrossRef]
- Costabel, D.; Skabeev, A.; Nabiyan, A.; Luo, Y.; Max, J.B.; Rajagopal, A.; Kowalczyk, D.; Dietzek, B.; Wächtler, M.; Görls, H.; et al. 1,7,9,10-tetrasubstituted PMIs accessible through decarboxylative bromination: Synthesis, characterization, photophysical studies, and hydrogen evolution catalysis. Chem. A Eur. J. 2021, 27, 4081–4088. [Google Scholar] [CrossRef]
- Sahoo, D.; Sharma, V.; Roy, R.; Varghese, N.; Mohanta, K.; Koner, A.L. Synthesis of highly-soluble push-pull perylenemonoimide derivatives by regioselective peri-functionalization for switchable memory applications. Chem. Commun. 2019, 55, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Legault, C.Y. CYLview, 1.0b; Université de Sherbrooke: Sherbrooke, QC, Canada, 2009; Available online: https://www.cylview.org (accessed on 26 February 2023).
- Chen, Z.; Baumeister, U.; Tschierske, C.; Würthner, F. Effect of core twisting on self-assembly and optical properties of perylene bisimide dyes in solution and columnar liquid crystalline phases. Chem. A Eur. J. 2007, 13, 450–465. [Google Scholar] [CrossRef] [PubMed]
- Briggs, E.A.; Besley, N.A. Density functional theory based analysis of photoinduced electron transfer in a triazacryptand based K + sensor. J. Phys. Chem. A 2015, 119, 2902–2907. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).