
Rapid and Online Microvolume Flow-Through Dialysis Probe for Sample Preparation in Veterinary Drug Residue Analysis


Abstract
:1. Introduction
2. Materials and Methods
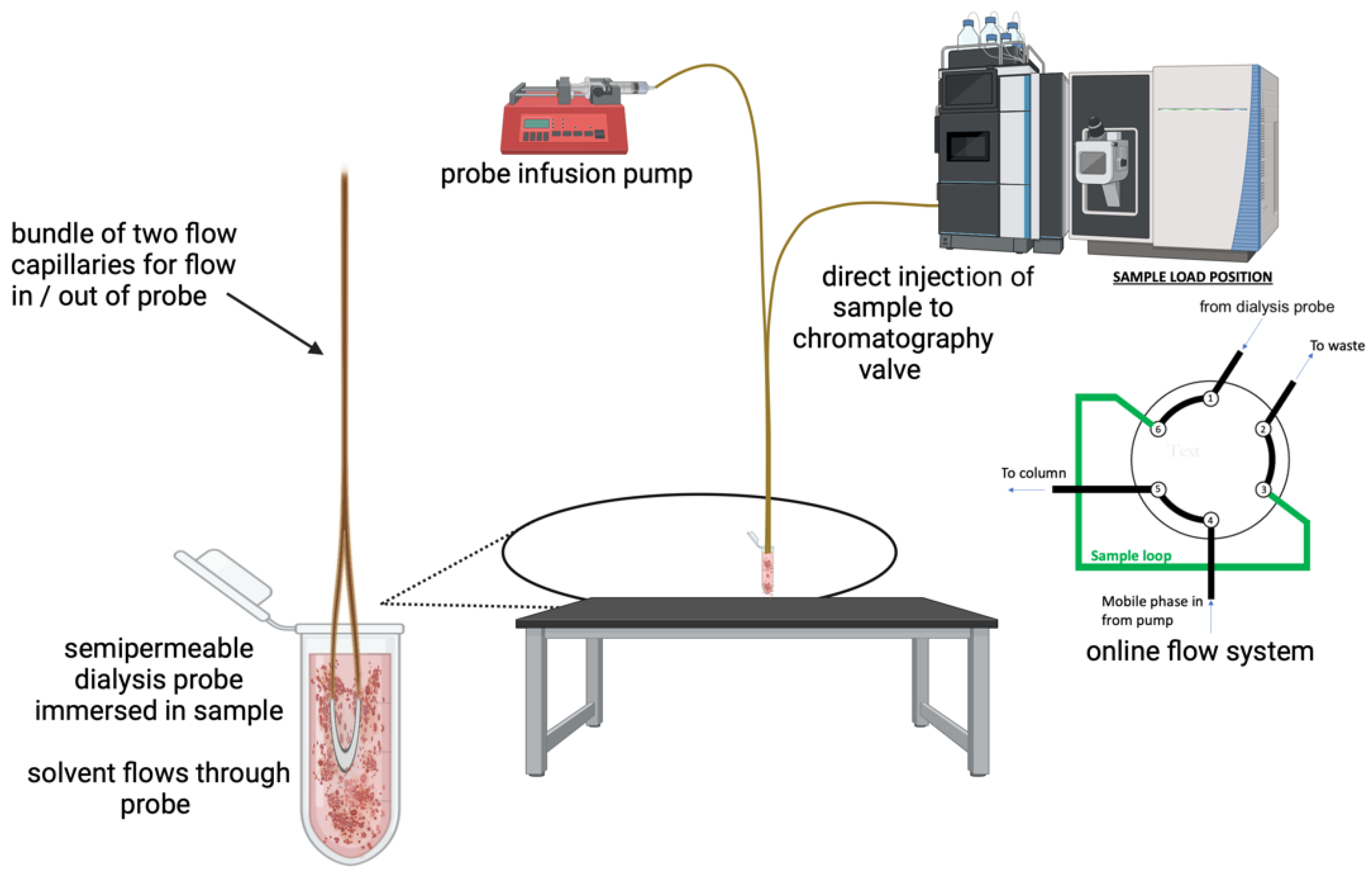
2.1. Microdialysis Probe Construction
2.2. Sample Preparation
2.3. LC-MS Conditions
3. Results
3.1. Chemical Analysis of Veterinary Drug Residues
3.1.1. Chromatography
3.1.2. Mass Spectrometry
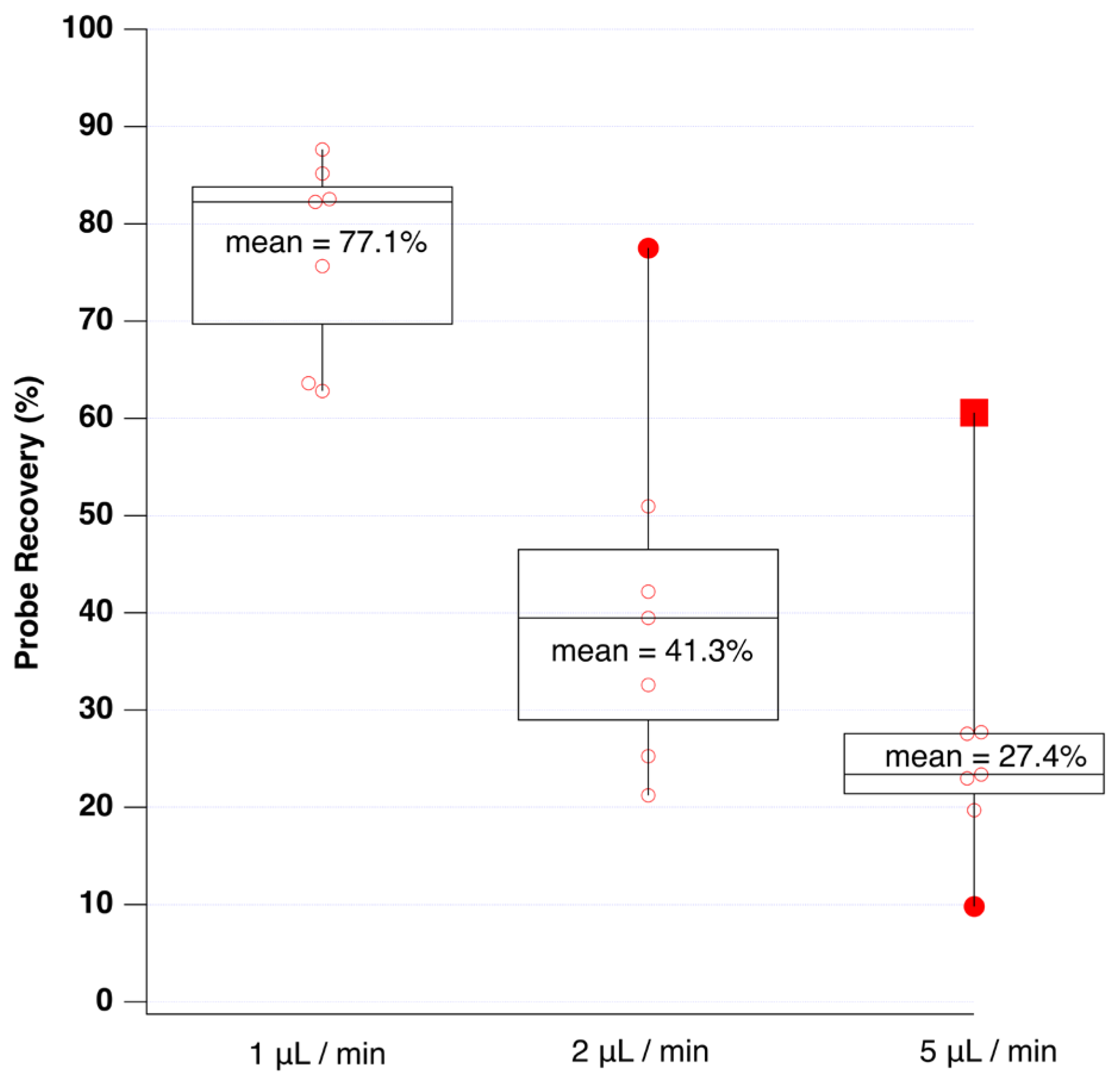
3.2. Microdialysis Probe Performance and Recovery
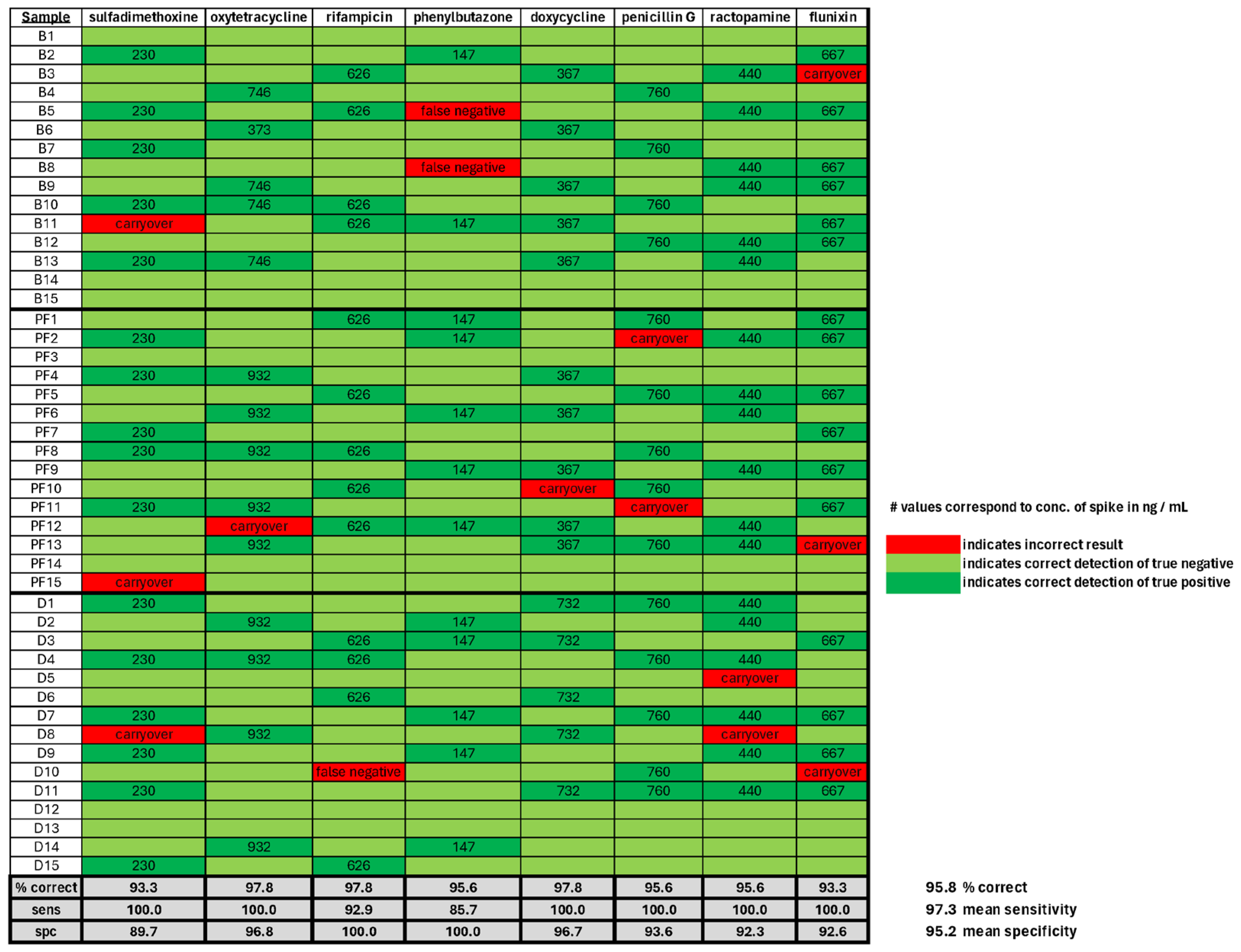
3.3. Veterinary Drug Residue Detection in Mock Samples
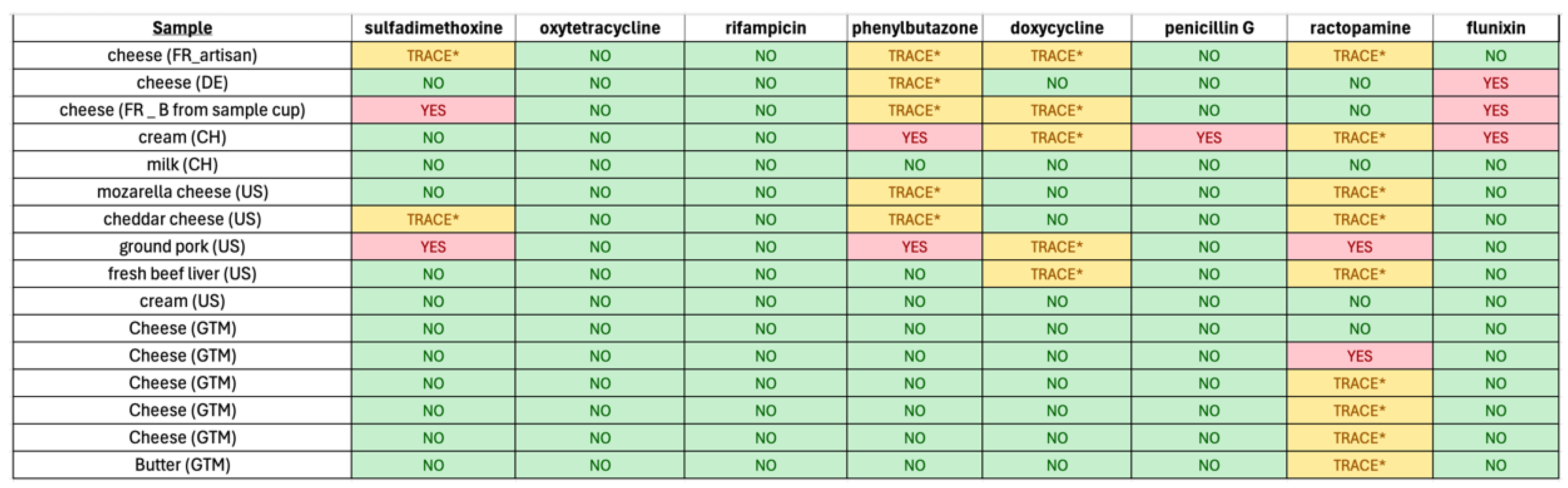
3.4. Veterinary Drug Residue Detection in Food Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- National Research Council. Expanding the Vision of Sensor Materials; The National Academies Press: Washington, DC, USA, 1995. [Google Scholar] [CrossRef]
- Bihar, E.; Wustoni, S.; Pappa, A.M.; Salama, K.N.; Baran, D.; Inal, S. A Fully Inkjet-Printed Disposable Glucose Sensor on Paper. npj Flex. Electron. 2018, 2, 30. [Google Scholar] [CrossRef]
- Martins, S.A.M.; Martins, V.C.; Cardoso, F.A.; Germano, J.; Rodrigues, M.; Duarte, C.; Bexiga, R.; Cardoso, S.; Freitas, P.P. Biosensors for On-Farm Diagnosis of Mastitis. Front. Bioeng. Biotechnol. 2019, 7, 186. [Google Scholar] [CrossRef]
- Davitt, K.; Song, Y.-K.; Patterson III, W.R.; Nurmikko, A.V.; Pan, Y.-L.; Chang, R.K.; Han, J.; Gherasimova, M.; Cobler, P.J.; Butler, P.D.; et al. Spectroscopic Sorting of Aerosols by a Compact Sensor Employing UV LEDs. Aerosol Sci. Technol. 2006, 40, 1047–1051. [Google Scholar] [CrossRef]
- Cao, T.; Thompson, J.E. Personal Monitoring of Ozone Exposure: A Fully Portable Device for under $150 USD Cost. Sens. Actuators B Chem. 2016, 224, 936–943. [Google Scholar] [CrossRef]
- Cao, T.; Thompson, J.E. Portable, Ambient PM2.5 Sensor for Human and/or Animal Exposure Studies. Analytical Letters 2017, 50, 712–723. [Google Scholar] [CrossRef]
- Thompson, J.E. Improved Measurement Performance for the Sharp Gp2y1010 Dust Sensor: Reduction of Noise. Atmosphere 2021, 12, 775. [Google Scholar] [CrossRef]
- Agrawaal, H.; Jones, C.; Thompson, J.E. Personal Exposure Estimates via Portable and Wireless Sensing and Reporting of Particulate Pollution. Int. J. Env. Res. Public. Health 2020, 17, 843. [Google Scholar] [CrossRef]
- Xu, M.; Wang, J.; Zhu, L. The Qualitative and Quantitative Assessment of Tea Quality Based on E-Nose, E-Tongue and E-Eye Combined with Chemometrics. Food Chem. 2019, 289, 482–489. [Google Scholar] [CrossRef]
- Ye, Z.; Liu, Y.; Li, Q. Recent Progress in Smart Electronic Nose Technologies Enabled with Machine Learning Methods. Sensors 2021, 21, 7620. [Google Scholar] [CrossRef]
- Nyein, H.Y.Y.; Tai, L.C.; Ngo, Q.P.; Chao, M.; Zhang, G.B.; Gao, W.; Bariya, M.; Bullock, J.; Kim, H.; Fahad, H.M.; et al. A Wearable Microfluidic Sensing Patch for Dynamic Sweat Secretion Analysis. ACS Sens. 2018, 3, 944–952. [Google Scholar] [CrossRef]
- Ates, H.C.; Brunauer, A.; von Stetten, F.; Urban, G.A.; Güder, F.; Merkoçi, A.; Früh, S.M.; Dincer, C. Integrated Devices for Non-Invasive Diagnostics. Adv. Funct. Mater. 2021, 31, 2010388. [Google Scholar] [CrossRef]
- Kim, J.; Campbell, A.S.; de Ávila, B.E.F.; Wang, J. Wearable Biosensors for Healthcare Monitoring. Nat. Biotechnol. 2019, 37, 389–406. [Google Scholar] [CrossRef]
- Guo, S.; Wu, K.; Li, C.; Wang, H.; Sun, Z.; Xi, D.; Zhang, S.; Ding, W.; Zaghloul, M.E.; Wang, C.; et al. Integrated Contact Lens Sensor System Based on Multifunctional Ultrathin MoS2 Transistors. Matter 2020, 4, 969–985. [Google Scholar] [CrossRef]
- Ates, H.C.; Nguyen, P.Q.; Gonzalez-Macia, L.; Morales-Narváez, E.; Güder, F.; Collins, J.J.; Dincer, C. End-to-End Design of Wearable Sensors. Nat. Rev. Mater. 2022, 7, 887–907. [Google Scholar] [CrossRef]
- McCracken, K.E.; Yoon, J.Y. Recent Approaches for Optical Smartphone Sensing in Resource-Limited Settings: A Brief Review. Anal. Methods 2016, 8, 6591–6601. [Google Scholar] [CrossRef]
- Xiao, M.; Liu, Z.; Xu, N.; Jiang, L.; Yang, M.; Yi, C. A Smartphone-Based Sensing System for On-Site Quantitation of Multiple Heavy Metal Ions Using Fluorescent Carbon Nanodots-Based Microarrays. ACS Sens. 2020, 5, 870–878. [Google Scholar] [CrossRef]
- Maharjan, S.M.; Poudyal, A.; van Heerden, A.; Byanjankar, P.; Thapa, A.; Islam, C.; Kohrt, B.A.; Hagaman, A. Passive Sensing on Mobile Devices to Improve Mental Health Services with Adolescent and Young Mothers in Low-Resource Settings: The Role of Families in Feasibility and Acceptability. BMC Med. Inf. Decis. Mak. 2021, 21, 117. [Google Scholar] [CrossRef]
- Majumder, S.; Deen, M.J. Smartphone Sensors for Health Monitoring and Diagnosis. Sensors 2019, 19, 2164. [Google Scholar] [CrossRef]
- Fu, Q.; Wu, Z.; Li, X.; Yao, C.; Yu, S.; Xiao, W.; Tang, Y. Novel Versatile Smart Phone Based Microplate Readers for On-Site Diagnoses. Biosens. Bioelectron. 2016, 81, 524–531. [Google Scholar] [CrossRef]
- Cao, T.; Thompson, J.E. Remote Sensing of Atmospheric Optical Depth Using a Smartphone Sun Photometer. PLoS ONE 2014, 9, e84119. [Google Scholar] [CrossRef]
- Cao, T.; Zhang, Q.; Thompson, J.E. Designing, Constructing, and Using an Inexpensive Electronic Buret. J. Chem. Educ. 2014, 92, 106–109. [Google Scholar] [CrossRef]
- Pal, A.; Kaswan, K.; Barman, S.R.; Lin, Y.Z.; Chung, J.H.; Sharma, M.K.; Liu, K.L.; Chen, B.H.; Wu, C.C.; Lee, S.; et al. Microfluidic Nanodevices for Drug Sensing and Screening Applications. Biosens. Bioelectron. 2023, 219, 114783. [Google Scholar] [CrossRef]
- Amreen, K.; Goel, S. Microfluidic-Based Sensors. In Microfluidics and Multi Organs on Chip; Springer: Singapore, 2022; pp. 163–184. [Google Scholar] [CrossRef]
- Chen, S.; Qiao, Z.; Niu, Y.; Yeo, J.C.; Liu, Y.; Qi, J.; Fan, S.; Liu, X.; Lee, J.Y.; Lim, C.T. Wearable Flexible Microfluidic Sensing Technologies. Nat. Rev. Bioeng. 2023, 1, 950–971. [Google Scholar] [CrossRef]
- Gunawardhana, S.M.; Bulgakova, G.A.; Barybin, A.M.; Thomas, S.R.; Lunte, S.M. Progress toward the Development of a Microchip Electrophoresis Separation-Based Sensor with Electrochemical Detection for on-Line in Vivo Monitoring of Catecholamines. Analyst 2020, 145, 1768. [Google Scholar] [CrossRef]
- Microfluidic-Based Chemical Sensors for Analytical Chemistry and Diagnostics|Frontiers Research Topic. Available online: https://www.frontiersin.org/research-topics/60123/microfluidic-based-chemical-sensors-for-analytical-chemistry-and-diagnostics (accessed on 22 April 2024).
- Li, S.; Zhang, Q.; Chen, M.; Zhang, X.; Liu, P. Determination of Veterinary Drug Residues in Food of Animal Origin: Sample Preparation Methods and Analytical Techniques. J. Liq. Chromatogr. Relat. Technol. 2020, 43, 701–724. [Google Scholar] [CrossRef]
- Pratiwi, R.; Ramadhanti, S.P.; Amatulloh, A.; Megantara, S.; Subra, L. Recent Advances in the Determination of Veterinary Drug Residues in Food. Foods 2023, 12, 3422. [Google Scholar] [CrossRef]
- Ochiai, N.; Sasamoto, K.; David, F.; Sandra, P. Recent Developments of Stir Bar Sorptive Extraction for Food Applications: Extension to Polar Solutes. J. Agric. Food Chem. 2018, 66, 7249–7255. [Google Scholar] [CrossRef]
- Yu, C.; Hu, B. C18-Coated Stir Bar Sorptive Extraction Combined with High Performance Liquid Chromatography–Electrospray Tandem Mass Spectrometry for the Analysis of Sulfonamides in Milk and Milk Powder. Talanta 2012, 90, 77–84. [Google Scholar] [CrossRef]
- Ibarra, I.S.; Miranda, J.M.; Rodriguez, J.A.; Nebot, C.; Cepeda, A. Magnetic Solid Phase Extraction Followed by High-Performance Liquid Chromatography for the Determination of Sulphonamides in Milk Samples. Food Chem. 2014, 157, 511–517. [Google Scholar] [CrossRef]
- Ye, Z.; Huang, Y.; Luo, Q.; Wang, L.; Huang, X. Preparation of Highly Fluorinated and Boron-Rich Adsorbent for Magnetic Solid-Phase Extraction of Fluoroquinolones in Water and Milk Samples. J. Chromatogr. A 2019, 1601, 86–94. [Google Scholar] [CrossRef]
- Anastassiades, M.; Lehotay, S.J.; Štajnbaher, D.; Schenck, F.J. Fast and Easy Multiresidue Method Employing Acetonitrile Extraction/Partitioning and “Dispersive Solid-Phase Extraction” for the Determination of Pesticide Residues in Produce. J. AOAC Int. 2003, 86, 412–431. [Google Scholar] [CrossRef]
- Mondal, S.; Xu, J.; Chen, G.; Huang, S.; Huang, C.; Yin, L.; Ouyang, G. Solid-Phase Microextraction of Antibiotics from Fish Muscle by Using MIL-101(Cr)NH2-Polyacrylonitrile Fiber and Their Identification by Liquid Chromatography-Tandem Mass Spectrometry. Anal. Chim. Acta 2019, 1047, 62–70. [Google Scholar] [CrossRef]
- Tang, Y.; Xu, J.; Chen, L.; Qiu, J.; Liu, Y.; Ouyang, G. Rapid in Vivo Determination of Fluoroquinolones in Cultured Puffer Fish (Takifugu Obscurus) Muscle by Solid-Phase Microextraction Coupled with Liquid Chromatography-Tandem Mass Spectrometry. Talanta 2017, 175, 550–556. [Google Scholar] [CrossRef]
- Ares, A.M.; Valverde, S.; Bernal, J.L.; Toribio, L.; Nozal, M.J.; Bernal, J. Determination of Flubendiamide in Honey at Trace Levels by Using Solid Phase Extraction and Liquid Chromatography Coupled to Quadrupole Time-of-Flight Mass Spectrometry. Food Chem. 2017, 232, 169–176. [Google Scholar] [CrossRef]
- Ha, J.; Song, G.; Ai, L.F.; Li, J. chen Determination of Six Polyether Antibiotic Residues in Foods of Animal Origin by Solid Phase Extraction Combined with Liquid Chromatography–Tandem Mass Spectrometry. J. Chromatogr. B 2016, 1017–1018, 187–194. [Google Scholar] [CrossRef]
- Casado, N.; Pérez-Quintanilla, D.; Morante-Zarcero, S.; Sierra, I. Evaluation of Bi-Functionalized Mesoporous Silicas as Reversed Phase/Cation-Exchange Mixed-Mode Sorbents for Multi-Residue Solid Phase Extraction of Veterinary Drug Residues in Meat Samples. Talanta 2017, 165, 223–230. [Google Scholar] [CrossRef]
- Guo, F.; Li, Q.X.; Alcantara-Licudine, J.P. Na(4)EDTA-Assisted Sub-/Supercritical Fluid Extraction Procedure for Quantitative Recovery of Polar Analytes in Soil. Anal. Chem. 1999, 71, 1309–1315. [Google Scholar] [CrossRef]
- Choi, J.H.; Mamun, M.I.R.; El-Aty, A.M.A.; Kim, K.T.; Koh, H.B.; Shin, H.C.; Kim, J.S.; Lee, K.B.; Shim, J.H. Inert Matrix and Na4EDTA Improve the Supercritical Fluid Extraction Efficiency of Fluoroquinolones for HPLC Determination in Pig Tissues. Talanta 2009, 78, 348–357. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, C.; Yang, L.; Zhang, W.; Lin, J.; Li, C. Determination of Eight Quinolones in Milk Using Immunoaffinity Microextraction in a Packed Syringe and Liquid Chromatography with Fluorescence Detection. J. Chromatogr. B 2017, 1064, 68–74. [Google Scholar] [CrossRef]
- Yamada, R.; Kozono, M.; Ohmori, T.; Morimatsu, F.; Kitayama, M. Simultaneous Determination of Residual Veterinary Drugs in Bovine, Porcine, and Chicken Muscle Using Liquid Chromatography Coupled with Electrospray Ionization Tandem Mass Spectrometry. Biosci. Biotechnol. Biochem. 2006, 70, 54–65. [Google Scholar] [CrossRef]
- Hajrulai-Musliu, Z.; Uzunov, R.; Jovanov, S.; Jankuloski, D.; Stojkovski, V.; Pendovski, L.; Sasanya, J.J. A New LC–MS/MS Method for Multiple Residues/Contaminants in Bovine Meat. BMC Chem. 2021, 15, 62. [Google Scholar] [CrossRef]
- Thompson, J.; Everhart Nunn, S.L.; Sarkar, S.; Clayton, B. Diagnostic Screening of Bovine Mastitis Using MALDI-TOF MS Direct-Spotting of Milk and Machine Learning. Vet. Sci. 2023, 10, 101. [Google Scholar] [CrossRef]
- Wittenberg, J.B.; Simon, K.A.; Wong, J.W. Targeted Multiresidue Analysis of Veterinary Drugs in Milk-Based Powders Using Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS). J. Agric. Food Chem. 2017, 65, 7288–7293. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Tret (min) | N (Theoretical Plates) * |
|---|---|---|
| sulfadimethoxine | 10.82 | 22,950 |
| oxytetracycline | 8.96 | 6980 |
| rifampicin | 10.74 | 10,100 |
| phenylbutazone | 12.96 | 33,000 |
| doxycycline | 9.80 | 8400 |
| penicillin G | 9.60 | 18,200 |
| ractopamine | 9.14 | 7200 |
| flunixin | 11.60 | 11,750 |
| Compound | MS (m/z) | MS/MS (m/z) |
|---|---|---|
| sulfadimethoxine | 311.078 | 92.0495, 108.0442, 156.0759, 218.0218, 245.1019 |
| oxytetracycline | 461.152 | 154.049, 226.0699, 337.0689, 381.0587, 426.1159 |
| rifampicin | 823.405 | n.a. * |
| phenylbutazone | 309.157 | 94.065, 120.044, 146.0592, 160.111, 188.106, 211.085, 253.0956 |
| doxycycline | 445.152 | 126.054, 154.049, 267.0636, 321.0743, 339.0849, 392.110, 410.1214, 428.1319 |
| penicillin G | 335.104 | 128.0523, 160.0418, 176.0697, 289.0987 |
| ractopamine | 302.1727 | 91.054, 107.049, 121.0644, 136.075, 164.1063, 284.1632 |
| flunixin | 297.0831 | 279.0728 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diab, H.; Calle, A.; Thompson, J. Rapid and Online Microvolume Flow-Through Dialysis Probe for Sample Preparation in Veterinary Drug Residue Analysis. Sensors 2024, 24, 3971. https://doi.org/10.3390/s24123971
Diab H, Calle A, Thompson J. Rapid and Online Microvolume Flow-Through Dialysis Probe for Sample Preparation in Veterinary Drug Residue Analysis. Sensors. 2024; 24(12):3971. https://doi.org/10.3390/s24123971
Chicago/Turabian StyleDiab, Hanin, Alexandra Calle, and Jonathan Thompson. 2024. "Rapid and Online Microvolume Flow-Through Dialysis Probe for Sample Preparation in Veterinary Drug Residue Analysis" Sensors 24, no. 12: 3971. https://doi.org/10.3390/s24123971