
Fast and Economic Microarray-Based Detection of Species-, Resistance-, and Virulence-Associated Genes in Clinical Strains of Vancomycin-Resistant Enterococci (VRE)
 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bioinformatic Analysis: Target Gene Selection, and Oligonucleotide Sequence Design
2.2. Strains/Isolates
2.3. Genome Sequencing
2.4. Genomic DNA Isolation from Enterococci
2.5. Microarray Production
2.6. Labeling and Hybridization
2.7. Data Analysis
2.8. Analyzing Similar Hybridization Profiles for Typing Purposes
3. Results
3.1. Optimization of DNA Isolation Protocol
3.2. Comparison between Microarray Results and Sequence-Based Predictions
3.3. Detection of Resistance and Virulence-Associated Genes Alongside Species Markers
3.4. Generation of Genetic Fingerprints for Typing Purposes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miller, W.R.; Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance in enterococci. Expert Rev. Anti-Infect. Ther. 2014, 12, 1221–1236. [Google Scholar] [CrossRef]
- Long, D.R.; Bryson-Cahn, C.; Waalkes, A.; Holmes, E.A.; Penewit, K.; Tavolaro, C.; Bellabarba, C.; Zhang, F.; Chan, J.D.; Fang, F.C.; et al. Contribution of the patient microbiome to surgical site infection and antibiotic prophylaxis failure in spine surgery. Sci. Transl. Med. 2014, 16, eadk8222. [Google Scholar] [CrossRef]
- Hammerum, A.M.; Karstensen, K.T.; Roer, L.; Kaya, H.; Lindegaard, M.; Porsbo, L.J.; Kjerulf, A.; Pinholt, M.; Holzknecht, B.J.; Worning, P.; et al. Surveillance of vancomycin-resistant enterococci reveals shift in dominating clusters from vanA to vanB Enterococcus faecium clusters, Denmark, 2015 to 2022. Eurosurveillance 2024, 29, 2300633. [Google Scholar] [CrossRef]
- Di Cagno, R.; De Angelis, M.; Calasso, M.; Gobbetti, M. Proteomics of the bacterial cross-talk by quorum sensing. J. Proteom. 2011, 74, 19–34. [Google Scholar] [CrossRef]
- Kayaoglu, G.; Ørstavik, D. Virulence Factors ofEnterococcus faecalis: Relationship to Endodontic Disease. Crit. Rev. Oral Biol. Med. 2004, 15, 308–320. [Google Scholar] [CrossRef]
- Geraldes, C.; Tavares, L.; Gil, S.; Oliveira, M. Enterococcus Virulence and Resistant Traits Associated with Its Permanence in the Hospital Environment. Antibiotics 2022, 11, 857. [Google Scholar] [CrossRef]
- Wagner, T.M.; Janice, J.; Paganelli, F.L.; Willems, R.J.; Askarian, F.; Pedersen, T.; Top, J.; de Haas, C.; van Strijp, J.A.; Johannessen, M.; et al. Enterococcus faecium TIR-Domain Genes Are Part of a Gene Cluster Which Promotes Bacterial Survival in Blood. Int. J. Microbiol. 2018, 2018, 1435820. [Google Scholar] [CrossRef]
- Rice, L.B.; Bellais, S.; Carias, L.L.; Hutton-Thomas, R.; Bonomo, R.A.; Caspers, P.; Page, M.G.P.; Gutmann, L. Impact of specific pbp5 mutations on expression of beta-lactam resistance in Enterococcus faecium. Antimicrob. Agents Chemother. 2004, 48, 3028–3032. [Google Scholar] [CrossRef]
- Huycke, M.M.; Sahm, D.F.; Gilmore, M.S. Multiple-drug resistant enterococci: The nature of the problem and an agenda for the future. Emerg. Infect. Dis. 1998, 4, 239–249. [Google Scholar] [CrossRef]
- Arvanitidou, M.; Katsouyannopoulos, V.; Tsakris, A. Antibiotic resistance patterns of enterococci isolated from coastal bathing waters. J. Med. Microbiol. 2001, 50, 1001–1005. [Google Scholar] [CrossRef]
- Costa, Y.; Galimand, M.; Leclercq, R.; Duval, J.; Courvalin, P. Characterization of the chromosomal aac(6′)-Ii gene specific for Enterococcus faecium. Antimicrob. Agents Chemother. 1993, 37, 1896–1903. [Google Scholar] [CrossRef]
- Ramirez, M.S.; Nikolaidis, N.; Tolmasky, M. Rise and dissemination of aminoglycoside resistance: The aac(6′)-Ib paradigm. Front. Microbiol. 2013, 4, 121. [Google Scholar] [CrossRef]
- Portillo, A.; Ruiz-Larrea, F.; Zarazaga, M.; Alonso, A.; Martinez, J.L.; Torres, C. Macrolide resistance genes in Enterococcus spp. Antimicrob. Agents Chemother. 2000, 44, 967–971. [Google Scholar] [CrossRef]
- Arthur, M.; Reynolds, P.; Courvalin, P. Glycopeptide resistance in enterococci. Trends Microbiol. 1996, 4, 401–407. [Google Scholar] [CrossRef]
- Gorrie, C.; Higgs, C.; Carter, G.; Stinear, T.P.; Howden, B. Genomics of vancomycin-resistant Enterococcus faecium. Microb. Genom. 2019, 5, e000283. [Google Scholar] [CrossRef]
- Evers, S.; Courvalin, P. Regulation of VanB-type vancomycin resistance gene expression by the VanS(B)-VanR (B) two-component regulatory system in Enterococcus faecalis V583. J. Bacteriol. 1996, 178, 1302–1309. [Google Scholar] [CrossRef]
- Kerschner, H.; Cabal, A.; Hartl, R.; Machherndl-Spandl, S.; Allerberger, F.; Ruppitsch, W.; Apfalter, P. Hospital outbreak caused by linezolid resistant Enterococcus faecium in Upper Austria. Antimicrob. Resist. Infect. Control 2019, 8, 150. [Google Scholar] [CrossRef]
- Zeng, W.; Feng, L.; Qian, C.; Chen, T.; Wang, S.; Zhang, Y.; Zheng, X.; Wang, L.; Liu, S.; Zhou, T.; et al. Acquisition of Daptomycin Resistance by Enterococcus faecium Confers Collateral Sensitivity to Glycopeptides. Front. Microbiol. 2022, 13, 815600. [Google Scholar] [CrossRef]
- Chadi, Z.D.; Dib, L.; Zeroual, F.; Benakhla, A. Usefulness of molecular typing methods for epidemiological and evolutionary studies of Staphylococcus aureus isolated from bovine intramammary infections. Saudi J. Biol. Sci. 2022, 29, 103338. [Google Scholar]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef]
- Maiden, M.C.; Bygraves, J.A.; Feil, E.; Morelli, G.; Russell, J.E.; Urwin, R.; Zhang, Q.; Zhou, J.; Zurth, K.; Caugant, D.A.; et al. Multilocus sequence typing: A portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. USA 1998, 95, 3140–3145. [Google Scholar] [CrossRef]
- Yan, S.; Jiang, Z.; Zhang, W.; Liu, Z.; Dong, X.; Li, D.; Liu, Z.; Li, C.; Liu, X.; Zhu, L. Genomes-based MLST, cgMLST, wgMLST and SNP analysis of Salmonella Typhimurium from animals and humans. Comp. Immunol. Microbiol. Infect. Dis. 2023, 96, 101973. [Google Scholar] [CrossRef]
- Homan, W.L.; Tribe, D.; Poznanski, S.; Li, M.; Hogg, G.; Spalburg, E.; van Embden, J.D.A.; Willems, R.J.L. Multilocus sequence typing scheme for Enterococcus faecium. J. Clin. Microbiol. 2002, 40, 1963–1971. [Google Scholar] [CrossRef]
- de Been, M.; Pinholt, M.; Top, J.; Bletz, S.; Mellmann, A.; Van Schaik, W.; Brouwer, E.; Rogers, M.; Kraat, Y.; Bonten, M.; et al. Core Genome Multilocus Sequence Typing Scheme for High-Resolution Typing of Enterococcus faecium. J. Clin. Microbiol. 2015, 53, 3788–3797. [Google Scholar] [CrossRef]
- Garibyan, L.; Avashia, N. Polymerase chain reaction. J. Investig. Dermatol. 2013, 133, 1–4. [Google Scholar] [CrossRef]
- Bezdicek, M.; Hanslikova, J.; Nykrynova, M.; Dufkova, K.; Kocmanova, I.; Kubackova, P.; Mayer, J.; Lengerova, M. New Multilocus Sequence Typing Scheme for Enterococcus faecium Based on Whole Genome Sequencing Data. Microbiol. Spectr. 2023, 11, e0510722. [Google Scholar] [CrossRef]
- Raven, K.E.; Reuter, S.; Reynolds, R.; Brodrick, H.J.; Russell, J.E.; Török, M.E.; Parkhill, J.; Peacock, S.J. A decade of genomic history for healthcare-associated Enterococcus faecium in the United Kingdom and Ireland. Genome Res. 2016, 26, 1388–1396. [Google Scholar] [CrossRef]
- van Hal, S.J.; Ip, C.L.; Ansari, M.A.; Wilson, D.J.; Espedido, B.A.; Jensen, S.O.; Bowden, R. Evolutionary dynamics of Enterococcus faecium reveals complex genomic relationships between isolates with independent emergence of vancomycin resistance. Microb. Genom. 2016, 2, e000048. [Google Scholar] [CrossRef]
- Rios, R.; Reyes, J.; Carvajal, L.P.; Rincon, S.; Panesso, D.; Echeverri, A.M.; Dinh, A.; Kolokotronis, S.O.; Narechania, A.; Tran, T.T.; et al. Genomic Epidemiology of Vancomycin-Resistant Enterococcus faecium (VREfm) in Latin America: Revisiting The Global VRE Population Structure. Sci. Rep. 2020, 10, 5636. [Google Scholar] [CrossRef]
- Menghwar, H.; Guo, A.; Chen, Y.; Lysnyansky, I.; Parker, A.M.; Prysliak, T.; Perez-Casal, J. A core genome multilocus sequence typing (cgMLST) analysis of Mycoplasma bovis isolates. Vet. Microbiol. 2022, 273, 09532. [Google Scholar] [CrossRef]
- Jolley, K.A.; Maiden, M.C. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinform. 2010, 11, 595. [Google Scholar] [CrossRef]
- Maiden, M.C.; Van Rensburg, M.J.J.; Bray, J.E.; Earle, S.G.; Ford, S.A.; Jolley, K.A.; McCarthy, N.D. MLST revisited: The gene-by-gene approach to bacterial genomics. Nat. Rev. Microbiol. 2013, 11, 728–736. [Google Scholar] [CrossRef]
- Pinholt, M.; Gumpert, H.; Bayliss, S.; Nielsen, J.B.; Vorobieva, V.; Pedersen, M.; Feil, E.; Worning, P.; Westh, H. Genomic analysis of 495 vancomycin-resistant Enterococcus faecium reveals broad dissemination of a vanA plasmid in more than 19 clones from Copenhagen, Denmark. J. Antimicrob. Chemother. 2017, 72, 40–47. [Google Scholar] [CrossRef]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Werner, A.; Mölling, P.; Fagerström, A.; Dyrkell, F.; Arnellos, D.; Johansson, K.; Sundqvist, M.; Norén, T. Whole genome sequencing of Clostridioides difficile PCR ribotype 046 suggests transmission between pigs and humans. PLoS ONE 2020, 15, e0244227. [Google Scholar] [CrossRef]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef]
- Aparna, G.M.; Tetala, K.K.R. Recent Progress in Development and Application of DNA, Protein, Peptide, Glycan, Antibody, and Aptamer Microarrays. Biomolecules 2023, 13, 602. [Google Scholar] [CrossRef]
- Howbrook, D.N.; van der Valk, A.M.; O’Shaughnessy, M.C.; Sarker, D.K.; Baker, S.C.; Lloyd, A.W. Developments in microarray technologies. Drug Discov. Today 2003, 8, 642–651. [Google Scholar] [CrossRef]
- Shiu, S.H.; Borevitz, J.O. The next generation of microarray research: Applications in evolutionary and ecological genomics. Heredity 2008, 100, 141–149. [Google Scholar] [CrossRef]
- Trevino, V.; Falciani, F.; Barrera-Saldaña, H.A. DNA Microarrays: A Powerful Genomic Tool for Biomedical and Clinical Research. Mol. Med. 2007, 13, 527–541. [Google Scholar] [CrossRef]
- Monecke, S.; Slickers, P.; Ehricht, R. Assignment of Staphylococcus aureus isolates to clonal complexes based on microarray analysis and pattern recognition. FEMS Immunol. Med. Microbiol. 2008, 53, 237–251. [Google Scholar] [CrossRef]
- Monecke, S.; Coombs, G.; Shore, A.C.; Coleman, D.C.; Akpaka, P.; Borg, M.; Chow, H.; Ip, M.; Jatzwauk, L.; Jonas, D.; et al. A field guide to pandemic, epidemic and sporadic clones of methicillin-resistant Staphylococcus aureus. PLoS ONE 2011, 6, e17936. [Google Scholar] [CrossRef]
- Monecke, S.; Jatzwauk, L.; Weber, S.; Slickers, P.; Ehricht, R.D.N.A. DNA microarray-based genotyping of methicillin-resistant Staphylococcus aureus strains from Eastern Saxony. Clin. Microbiol. Infect. 2008, 14, 534–545. [Google Scholar] [CrossRef]
- Monecke, S.; Ehricht, R.; Slickers, P.; Wernery, R.; Johnson, B.; Jose, S.; Wernery, U. Microarray-based genotyping of Staphylococcus aureus isolates from camels. Vet. Microbiol. 2011, 150, 309–314. [Google Scholar] [CrossRef]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef]
- Collatz, M.; Braun, S.D.; Monecke, S.; Ehricht, R. ConsensusPrime—A Bioinformatic Pipeline for Ideal Consensus Primer Design. BioMedInformatics 2022, 2, 637–642. [Google Scholar] [CrossRef]
- Collatz, M.; Reinicke, M.; Diezel, C.; Braun, S.D.; Monecke, S.; Reissig, A.; Ehricht, R. ConsensusPrime—A Bioinformatic Pipeline for Efficient Consensus Primer Design—Detection of Various Resistance and Virulence Factors in MRSA—A Case Study. BioMedInformatics 2024, 4, 1249–1261. [Google Scholar] [CrossRef]
- Caplunik-Pratsch, A.; Kieninger, B.; Donauer, V.A.; Brauer, J.M.; Meier, V.M.; Seisenberger, C.; Rath, A.; Loibl, D.; Eichner, A.; Fritsch, J.; et al. Introduction and spread of vancomycin-resistant Enterococcus faecium (VREfm) at a German tertiary care medical center from 2004 until 2010: A retrospective whole-genome sequencing (WGS) study of the molecular epidemiology of VREfm. Antimicrob. Resist. Infect. Control 2024, 13, 20. [Google Scholar] [CrossRef]
- Rath, A.; Kieninger, B.; Caplunik-Pratsch, A.; Fritsch, J.; Mirzaliyeva, N.; Holzmann, T.; Bender, J.K.; Werner, G.; Schneider-Brachert, W. Concerning emergence of a new vancomycin-resistant Enterococcus faecium strain ST1299/CT1903/vanA at a tertiary university centre in South Germany. J. Hosp. Infect. 2024, 143, 25–32. [Google Scholar] [CrossRef]
- Hajian-Tilaki, K. Receiver Operating Characteristic (ROC) Curve Analysis for Medical Diagnostic Test Evaluation. Casp. J. Intern. Med. 2013, 4, 627–635. [Google Scholar]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Monecke, S.; Schaumburg, F.; Shittu, A.O.; Schwarz, S.; Mühldorfer, K.; Brandt, C.; Braun, S.D.; Collatz, M.; Diezel, C.; Gawlik, D.; et al. Description of Staphylococcal Strains from Straw-Coloured Fruit Bat (Eidolon helvum) and Diamond Firetail (Stagonopleura guttata) and a Review of their Phylogenetic Relationships to Other Staphylococci. Front. Cell. Infect. Microbiol. 2022, 12, 878137. [Google Scholar] [CrossRef]
- Monecke, S.; Müller, E.; Braun, S.D.; Armengol-Porta, M.; Bes, M.; Boswihi, S.; El-Ashker, M.; Engelmann, I.; Gawlik, D.; Gwida, M.; et al. Characterisation of S. aureus/MRSA CC1153 and review of mobile genetic elements carrying the fusidic acid resistance gene fusC. Sci. Rep. 2021, 11, 8128. [Google Scholar] [CrossRef]
- Nappi, F. Current Knowledge of Enterococcal Endocarditis: A Disease Lurking in Plain Sight of Health Providers. Pathogens 2024, 13, 235. [Google Scholar] [CrossRef]
- Madsen, K.T.; Skov, M.N.; Gill, S.; Kemp, M. Virulence Factors Associated with Enterococcus faecalis Infective Endocarditis: A Mini Review. Open Microbiol. J. 2017, 11, 1–11. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef]
- The Lancet Global Health. Fighting antimicrobial resistance on all fronts. Lancet Glob. Health 2017, 5, e1161. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Microarray | Target Probes on the Array | Number of Target Probes on the Microarray |
|---|---|---|
| 1st generation | Biotin, resistance, and virulence markers | 111 |
| 2nd generation | Biotin, resistance, virulence, and species-specific markers | 139 |
| GenBank Number | Target Genes | Target Gene Description | No. of Positives | No. of Negatives | Step Threshold | * ROC AUC |
|---|---|---|---|---|---|---|
| Resistance markers | ||||||
| ACAX01000144.1 [280:1017] | ermB | Macrolide resistance | 144 | 12 | 0.5 | 1 |
| ACBB01000372.1 [37:1476] | aacA-aphD | Aminoglycoside resistance | 47 | 93 | 0.5 | 1 |
| CP003583.1 [1443763:1445799] | pbp5_3 | Penicillin-binding protein 5 (associated with ampicillin resistance) [8] | 179 | 4 | 0.6 | 0.98 |
| CP071931.1 [853137:854165] | vanB | Vancomycin resistance (van operon B) | 90 | 66 | 0.64 | 1 |
| GQ484956.1 [28460:29491] | vanA | D-alanine-D-lactate ligase/vancomycin resistance | 41 | 97 | 0.5 | 1 |
| CP068244.1 [894269:894844] | vanZ | Transmembrane teicoplanin-resistance protein (van operon Z) | 140 | 1 | 0.49 | 1 |
| CP014452.1 [34706:35674] | vanH_1 | D-alanine-D-serine ligase/vancomycin resistance | 107 | 1 | 0.49 | 1 |
| AF162694.1 [3008:5104] | vanC | Membrane-bound serine racemase (glycopeptide resistance gene) | 0 | 183 | - | - |
| CP019989.1 [102852:103547] | vanR | Two component sensor/regulator, transcriptional regulator | 55 | 101 | 0.5 | 1 |
| CP003583.1 [1174985:1175809] | vanYB | D-Ala-D-Ala carboxypeptidase (glycopeptide resistance gene) | 132 | 1 | 0.49 | 1 |
| CP012454.1 [2804893:2806020] | tetM | Tetracycline resistance through ribosomal protection | 125 | 29 | 0.5 | 1 |
| CP006030.1 [2020974:2022182] | qnr | Quinolone resistance | 1 | 159 | 0.5 | 1 |
| KT892063.1 [105:2072] | poxtA | Linezolid resistance | 2 | 160 | 0.55 | 1 |
| CP066673.1 [1667698:1668903] | tetK | Tetracycline resistance | 0 | 183 | - | - |
| CP068244.1 [1940965:1942392] | fexA | Florfenicol resistance | 0 | 183 | - | - |
| AEBZ01000030.1 [377201:377944] | lsa(A) | Quinupristin + dalfopristin resistance intrinsic in E. faecalis | 1 | 150 | 0.51 | 1 |
| Virulence markers | ||||||
| CP003583.1 [2228117:2230282] | acm | Collagen-binding microbial surface components recognizing adhesive matrix molecules (MSCRAMM) in E. faecium | 113 | 1 | 0.40 | 1 |
| CP018070.1 [73480:75456] | pilA | Major pilin subunit | 62 | 98 | 0.61 | 1 |
| AE016830.1 [537810:538529] | efaA | Adhesion protein plays role in endocarditis | 109 | 6 | 0.49 | 1 |
| CP012465.1 [759897:761699] | bepA | Permease | 182 | 1 | - | 1 |
| CP018071.1 [530776:534003] | ecbA | Collagen-binding MSCRAMM | 118 | 19 | 0.5 | 1 |
| CP012465.1 [1664985:1665647] | eep | Protease said to play a role in endocarditis [54,55] | 138 | 1 | 0.48 | 1 |
| CP012522.1 [252130:253836] | efbA | Adhesion protein plays role in endocarditis [54,55] | 135 | 1 | 0.64 | 1 |
| CP018065.1 [2272402:2273688] | efmM | Ribosomal RNA (rRNA) methyltransferase | 123 | 1 | 0.48 | 1 |
| CP006620.1 [2865948:2866424] | esp | Enterococcal surface protein | 124 | 25 | 0.5 | 1 |
| CP003351.1 [950876:951718] | srtA2C | Biofilm and pilus-associated sortase | 94 | 34 | 0.5 | 1 |
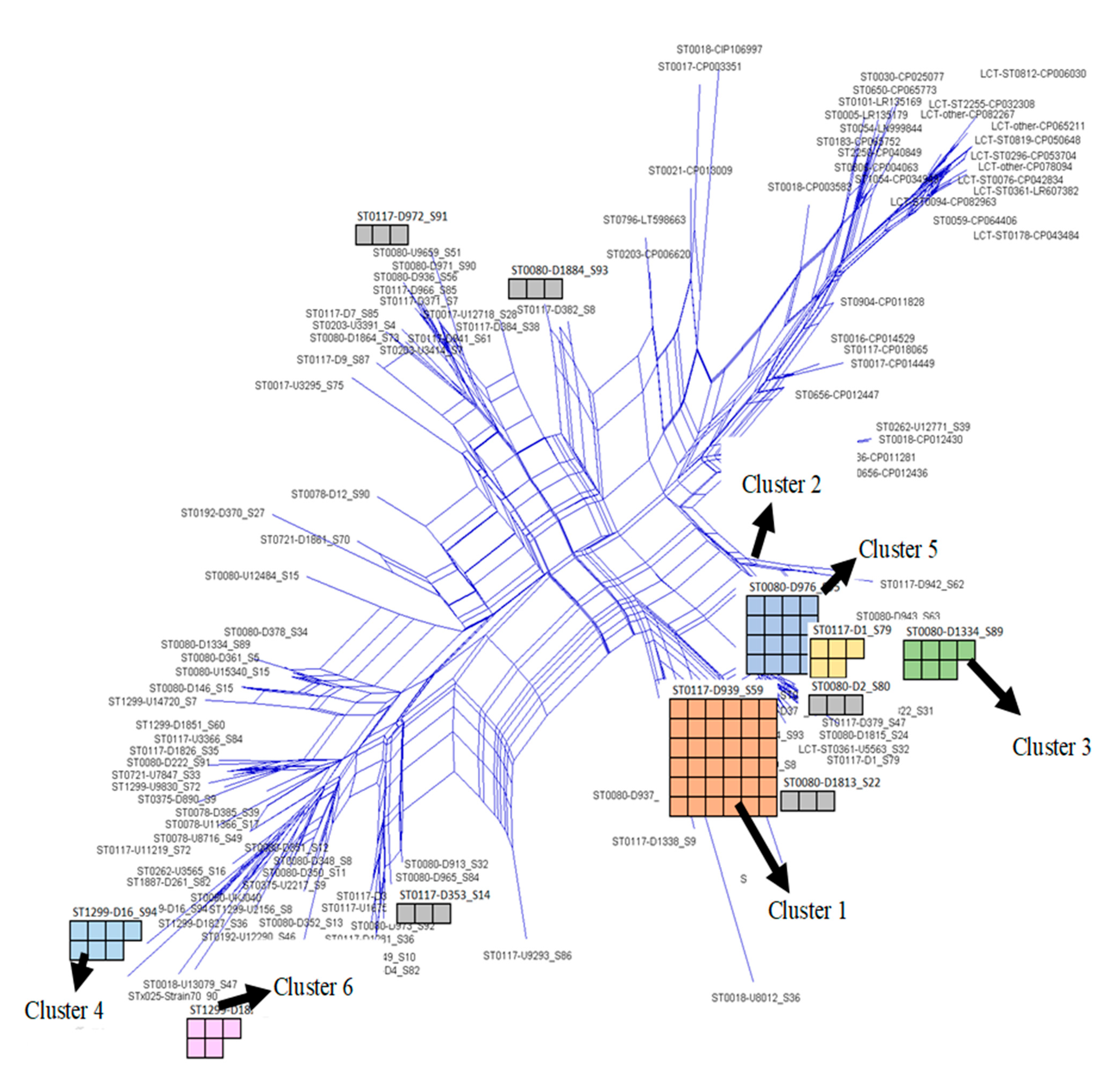
| Clusters | * MLST | Bezdíček MLST | CT | Array Patterns | No. of VRE Strains with the Same Array Pattern | No. of VRE Strains within the Cluster with Traceable Epidemiological Link |
|---|---|---|---|---|---|---|
| 1 | 117/262 | 17/57 | 71/7077/1686/1917/1775 | RGB-[D939_S59] | 36 | 4 |
| 2 | 80/117 | 17/21 | 71/1065/3243 | RGB-[D379_S47] | 16 | 0 |
| 3 | 80/117 | 143/327/318 | 1473/7078/7059/2406/2403 | RGB-[D1_S79] | 7 | 0 |
| 4 | 80 | 504 | 1470 | RGB-[D1334_S89] | 7 | 0 |
| 5 | 1299 | N/A | 3109/1903 | RGB-[D16_S94] | 5 | 0 |
| 6 | 1299 | N/A | 1903 | RGB-[D1855_S64] | 3 | 0 |
| 7 | 117 | 18 | 2505/7074 | RGB-[D353_S14] | 3 | 0 |
| 8 | 0721 | N/A | 1573 | RGB-[U7847_S33] | 3 | 0 |
| 9 | 80 | N/A | 7050 | RGB-[D1813_S22] | 3 | 2 |
| 10 | 80 | 21 | 1065 | RGB-[D1884_S93] | 3 | 2 |
| 11 | 80 | 21 | 1065/7063 | RGB-[D1860_S69] | 3 | 0 |
| 12 | - | 18 | 929/2967/3292 | RGB-[U3453_S12] | 3 | 2 |
| 13 | 117 | 17/18 | 469/7081 | RGB-[D941_S61] | 2 | 0 |
| 14 | 80/117 | 143 | 7056/7060 | RGB-[D1815_S24] | 2 | 0 |
| 15 | 80 | 102 | 1565 | RGB-[D0965_S84] | 2 | 0 |
| 16 | 80 | 102 | 1565 | RGB-[D973_S92] | 2 | 0 |
| 17 | 117 | 166 | 6002 | RGB-[D9_S87] | 2 | 0 |
| 18 | - | 123/18 | 2035/7163 | RGB-[D363_S21] | 2 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osadare, I.E.; Monecke, S.; Abdilahi, A.; Müller, E.; Collatz, M.; Braun, S.; Reissig, A.; Schneider-Brachert, W.; Kieninger, B.; Eichner, A.; et al. Fast and Economic Microarray-Based Detection of Species-, Resistance-, and Virulence-Associated Genes in Clinical Strains of Vancomycin-Resistant Enterococci (VRE). Sensors 2024, 24, 6476. https://doi.org/10.3390/s24196476
Osadare IE, Monecke S, Abdilahi A, Müller E, Collatz M, Braun S, Reissig A, Schneider-Brachert W, Kieninger B, Eichner A, et al. Fast and Economic Microarray-Based Detection of Species-, Resistance-, and Virulence-Associated Genes in Clinical Strains of Vancomycin-Resistant Enterococci (VRE). Sensors. 2024; 24(19):6476. https://doi.org/10.3390/s24196476
Chicago/Turabian StyleOsadare, Ibukun Elizabeth, Stefan Monecke, Abdinasir Abdilahi, Elke Müller, Maximilian Collatz, Sascha Braun, Annett Reissig, Wulf Schneider-Brachert, Bärbel Kieninger, Anja Eichner, and et al. 2024. "Fast and Economic Microarray-Based Detection of Species-, Resistance-, and Virulence-Associated Genes in Clinical Strains of Vancomycin-Resistant Enterococci (VRE)" Sensors 24, no. 19: 6476. https://doi.org/10.3390/s24196476