1. Introduction
Surface plasmon resonance (SPR) is a powerful label-free technique for real-time measurements of binding affinity and kinetics [
1,
2,
3,
4]. The method leverages the phenomenon that occurs when light at a given angle couples with electrons on the layer between a conducting metal and a dielectric insulator, typically gold on glass. This charged electron wave through the metal is defined as surface plasmons, and the oscillation of plasmons on the sensor surface generates an electric field [
4,
5,
6,
7]. The formation of plasmons is sensitive to the angle of incident light, which, in turn, is affected by the refractive index at the surface of the chip. When the refractive index of the sensor changes (such as upon the immobilization of a ligand), the ability of the light to cause surface plasmons is altered, allowing the researcher to quantify the binding on the surface by monitoring the intensity or shift in the angle of the reflected light [
4,
8]. The instrument we used in this study utilizes nanoparticles rather than planar metal to create the sensor surface. This version of the technique is called localized surface plasmon resonance (LSPR) [
9,
10]. When the surface comprises nanoparticles that are the same size as or smaller than the wavelength of the light, the electrons collectively oscillate and generate localized surface plasmons [
9,
10]. The localized plasmons have a maximum absorbance at a given resonance wavelength, and by monitoring the shift in wavelength where this maximum absorbance is observed, the binding on the surface can be measured [
10]. The hybrid immobilization technique we present here would work equally well for traditional SPR; the method used to measure binding on the surface is independent of the mechanism used to immobilize the ligand.
Although SPR does not require analyte labeling, it does necessitate the immobilization of one binding partner to the surface of the sensor chip. The immobilized binding partner is typically referred to as the ligand; the binding partner free in solution is referred to as the analyte. A variety of strategies are commonly used to achieve ligand immobilization, ranging from direct approaches like thiol or carboxyl coupling to potentially reversible methods that use affinity capture tags [
8]. Streptavidin–biotin is one of the most common of these capture systems for a variety of reasons (
Figure 1A). Biotinylated tags are small compared to other tags such as glutathione-S-transferase (GST) and maltose-binding protein (MBP), minimizing possible interference with the structure or binding ability of the ligand [
4]. Biotin can be conjugated to a variety of ligands, and the strong affinity of streptavidin–biotin binding results in an essentially permanent attachment of the biotinylated ligand to streptavidin-functionalized surfaces [
4,
8,
11,
12]. The position of the tag on the ligand offers the ability to control the orientation of the ligand relative to the sensor surface [
4]. A review of 200 studies utilizing the Nicoya OpenSPR between 2016 and 2022 included 22 that opted for streptavidin–biotin immobilization [
13].
Another popular strategy for immobilization is capturing ligands tagged with a poly histidine tag (6xHis) onto a nitrilotriacetic acid (NTA)-functionalized surface primed with Ni
2+ ions [
4,
14] (
Figure 1B). 6xHis-Ni-NTA capture has some of the benefits of streptavidin–biotin immobilization, such as small tag size and directionality of attachment, but offers the ability to regenerate the ligand surface by disrupting the complex between 6xHis tags and NTA groups by using ethylenediaminetetraacetic acid (EDTA) to remove the Ni
2+ ions necessary for binding [
4,
8].
Aptamers are small, single-stranded nucleic acids that can be used as affinity reagents against a variety of targets, including proteins and small molecules [
15,
16]. They are smaller than antibodies, easily synthesized, and can be conjugated with a variety of modifications. Because of their fixed sequence, aptamers can be made to the exact same composition and specifications every time, and they can be shared across research laboratories by sharing a simple text string (the aptamer sequence). No such universal transfer is possible for antibodies [
17,
18]. Aptamers are primarily developed using a process called systematic evolution of ligands by exponential enrichment (SELEX), in which a large pool of random-sequence nucleic acids of a given length are allowed to bind to the target, separated from non-binders in the bulk library, and amplified. By performing this process multiple times, the aptamer sequences with the highest affinity for the target are separated from the pool of all potential aptamers and amplified [
16,
17]. Typically, the input DNA or RNA library, along with material archived during several rounds of selection, are sequenced and analyzed to determine the sequences that have become the most abundant. Often, a set of aptamers from the final round is chosen to experimentally characterize binding affinity to the target of interest. One of the most widely used model systems for protein–aptamer affinity comprises thrombin and two ssDNA aptamers referred to as the 15mer and the 29mer [
18,
19,
20]. While both aptamers exhibit cooperative binding to thrombin with high affinity, the 29mer has been reported to have a higher affinity [
17,
20], though apparent affinity has been shown to depend on the method of measurement and the ionic environment [
18].
While the strength of streptavidin–biotin binding is, in many ways, beneficial when immobilizing ligands of interest, it reduces the efficiency of optimizing experimental conditions. Because biotin–streptavidin binding is essentially permanent, every immobilization condition tested requires the sacrifice of a sensor chip. Even after optimizing an experimental protocol, using biotin immobilization to attach aptamers to sensor chips vastly limits the throughput of aptamer candidates an investigator can test by limiting each sensor chip to a single aptamer ligand, which is expensive and time-consuming to assess.
In this study, we report a simple strategy for immobilizing biotinylated aptamers to SPR sensing surfaces that combines the benefits of stable and reproducible biotin immobilization with the flexibility of 6xHis-Ni-NTA immobilization. The method is demonstrated using thrombin and the thrombin-binding 29mer aptamer as a model system (
Figure 1C). A similar method for immobilizing proteins to measure the affinity of small molecule analytes was reported in 2021 [
21], but our independent development of this technique for the assessment of aptamers adds several benefits. Our method is used to immobilize a different type of ligand target, attaching oligonucleotides to the sensor rather than proteins. By immobilizing the smaller binding partner of interest (the aptamer), the possible binding signal from the larger protein analyte is maximized. In addition, the two-reagent strategy for ligand regeneration we report is simple and quickly removes the ligand, enabling surface preparation and further ligand immobilization with fewer steps and reagents required than the protocol they report for regenerating immobilized proteins off the surface. While Gunnarsson et al. note that ligand regeneration will depend on the ligand of interest [
21], it is likely that different aptamer candidates will behave similarly when regenerating aptamer-6xHis–streptavidin complexes off the surface. They report the immobilization of proteins that contained both 6xHis and biotin tags, which does not allow for clarity whether the mechanism of immobilization occurs via the biotin tag binding to streptavidin protein on the sensor or due to the 6xHis tag on the protein. Our method verifies that immobilization results from the specific interaction of the biotin tag and the streptavidin protein by demonstrating binding with a complementary binding affinity technique and by using ligands that contain only a singular capture tag. This demonstrates that ligands do not require a 6xHis tag in order to use this hybrid immobilization method.
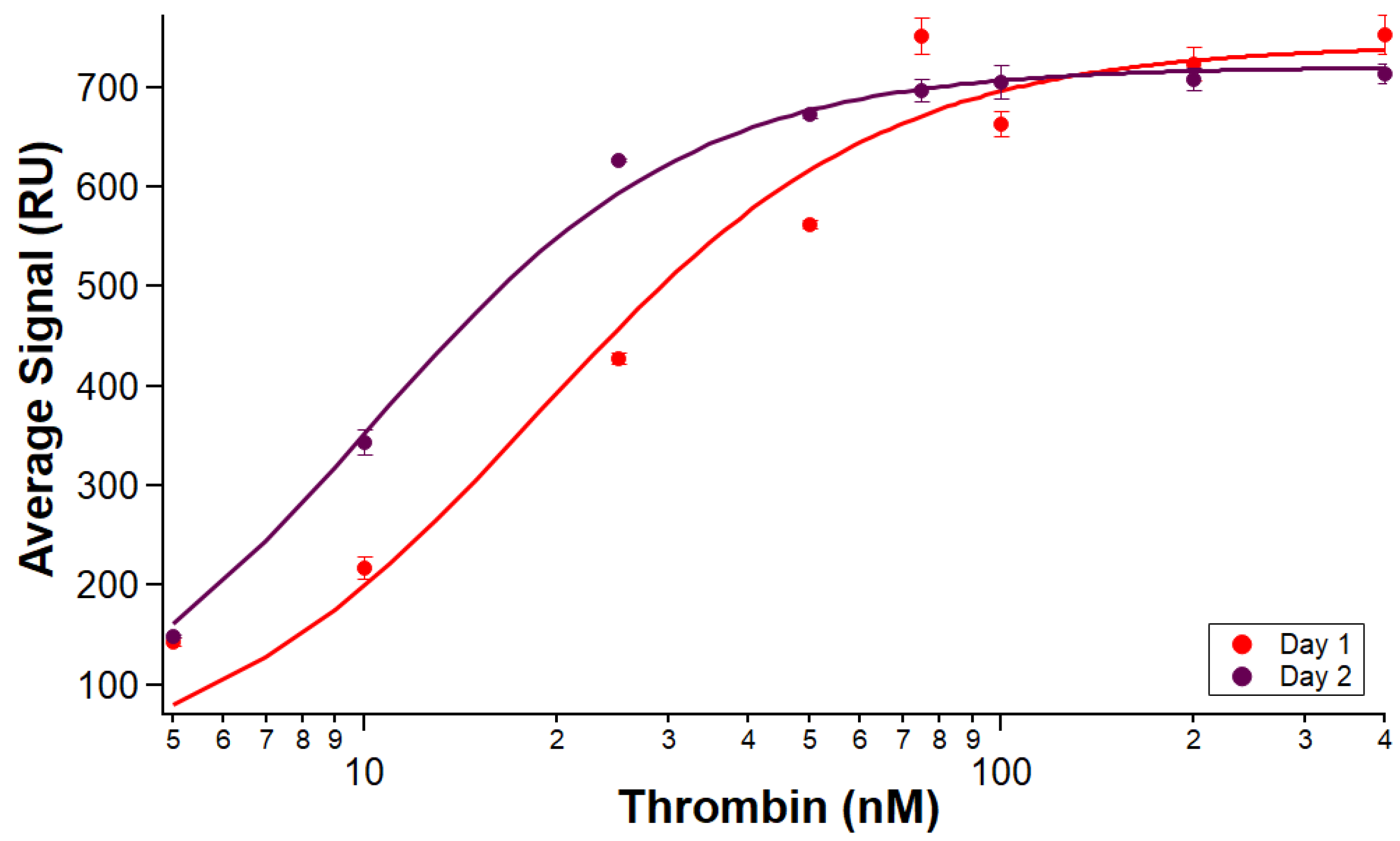
In addition to reproducible immobilization and binding affinity measurements, the stability of the aptamer-immobilized surface over time was tested. Binding affinity characterizations between an immobilized aptamer and five sets of thrombin samples over the course of two days were reproducible, giving researchers the opportunity to confidently use the same surface for multiple experiments to measure several protein targets or collect sets of replicate measurements. While the present study only tested the stability of an immobilized aptamer surface through two full days of experiments, it is probable that the surface could have been used for additional experiments. Finding the limit of exactly how long the same aptamer-immobilized surface can be used for binding affinity assays will require further experimentation. In terms of immobilization reproducibility rather than immobilization stability, Nicoya has demonstrated the ability to reproducibly immobilize 6xHis–streptavidin up to 27 cycles and measure 6xHis protein A–IgG binding for 18 cycles in their NTA sensor ligand recapture on OpenSPR
TM tech note [
22]. The ability to perform many different reproducible immobilizations, along with the ability to use individual aptamer-immobilized surfaces for multiple assays, provides investigators with a great deal of flexibility while designing experiments.
2. Materials and Methods
2.1. Materials
Experiments were performed on an OpenSPR-XT 2-channel instrument from Nicoya Lifesciences (Kitchener, ON, Canada) with NTA-functionalized sensors and well plate foils from the same vendor. In the two-channel set-up, the first channel (Ch1) serves as a reference and the second (Ch2) is where ligand is immobilized. The calculated corrected signal corresponds to the response from Ch1 subtracted from Ch2 to measure only the interaction of the bound ligand and the free analyte, rather than any non-specific binding on the surface.
Lyophilized bovine serum albumin (BSA) was purchased from MilliporeSigma (Burlington, MS, USA) and reconstituted in water to a concentration of 1 mg/mL. Lyophilized thrombin derived from human plasma was purchased from MilliporeSigma and reconstituted to 1000 U/mL in 1 mg/mL BSA. 6xHis tagged streptavidin (His-SA) at a stock concentration of 1 mg/mL in 20 mM Tris-HCl pH 7.5 was purchased from Fitzgerald Industries, (Acton, MA, USA), My Biosource (San Diego, CA, USA), and Abcam (Waltham, MA, USA). Aptamers were purchased from Integrated DNA Technologies (Coralville, IA, USA) both with a biotin tag (B-29mer) and without (unlabeled 29mer) on the 5′ end (sequence: /5Biosg/AG TCC GTG GTA GGG CAG GTT GGG GTG ACT). Lyophilized aptamers were reconstituted in 1X Tris EDTA (TE) buffer (VWR, Radnor, PA, USA) to a concentration of 100 µM and stored at −20 °C. NiCl2 was purchased from BeanTown Chemical (Hudson, NH, USA) and reconstituted to 40 mM in water. EDTA was purchased from TCI America (Portland, OR, USA). Glycine, D-(+)-Biotin, NaCl, NaOH, and HCl were all purchased from VWR. A solution of 10 mM glycine–HCl was prepared from solid glycine and pH adjusted to 1.5 with HCl. The 10X phosphate-buffered saline (PBS) and Tween 20 were purchased from Thermo Fisher (Waltham, MS, USA). We then prepared two modified buffers in house for our experiments. PBS-T was 1X PBS and 0.05% Tween 20 (v/v) and used as the sample buffer. Some experiments also utilized an immobilization buffer (PBS-I) that was 1X PBS with NaCl added to a total concentration of 300 mM, with pH adjusted with NaOH to a final pH of 8.0. Data were analyzed using TraceDrawer Analysis 1.9.2 (Ridgeview Instruments, Uppsala, Sweden) and IGOR Pro (WaveMetrics, Portland, OR, USA).
2.2. General Experimental Overview
In brief, each experiment entailed cleaning the surface of the sensor chip with HCl and EDTA, followed by priming the surface with NiCl
2 and immobilizing His-SA into both channels. B-29mer was then immobilized into only channel 2, and a range of concentrations of thrombin protein were injected to measure the protein–aptamer binding signal. Average corrected curve signals were calculated from a given interval towards the end of the injection and plotted versus concentration in order to fit the data with the Hill equation:
Then, we calculated the binding constants, as described by Mears et al. [
18], where the fraction bound is represented as f
b, B
max is a fit parameter for the maximum observed signal, the n coefficient accounts for cooperative binding, protein concentration is given as P, and K
A is a dissociation constant that corresponds to the concentration of protein at half the maximum binding signal. Specific experimental details that sometimes differed between experiments can be found in
Table 1.
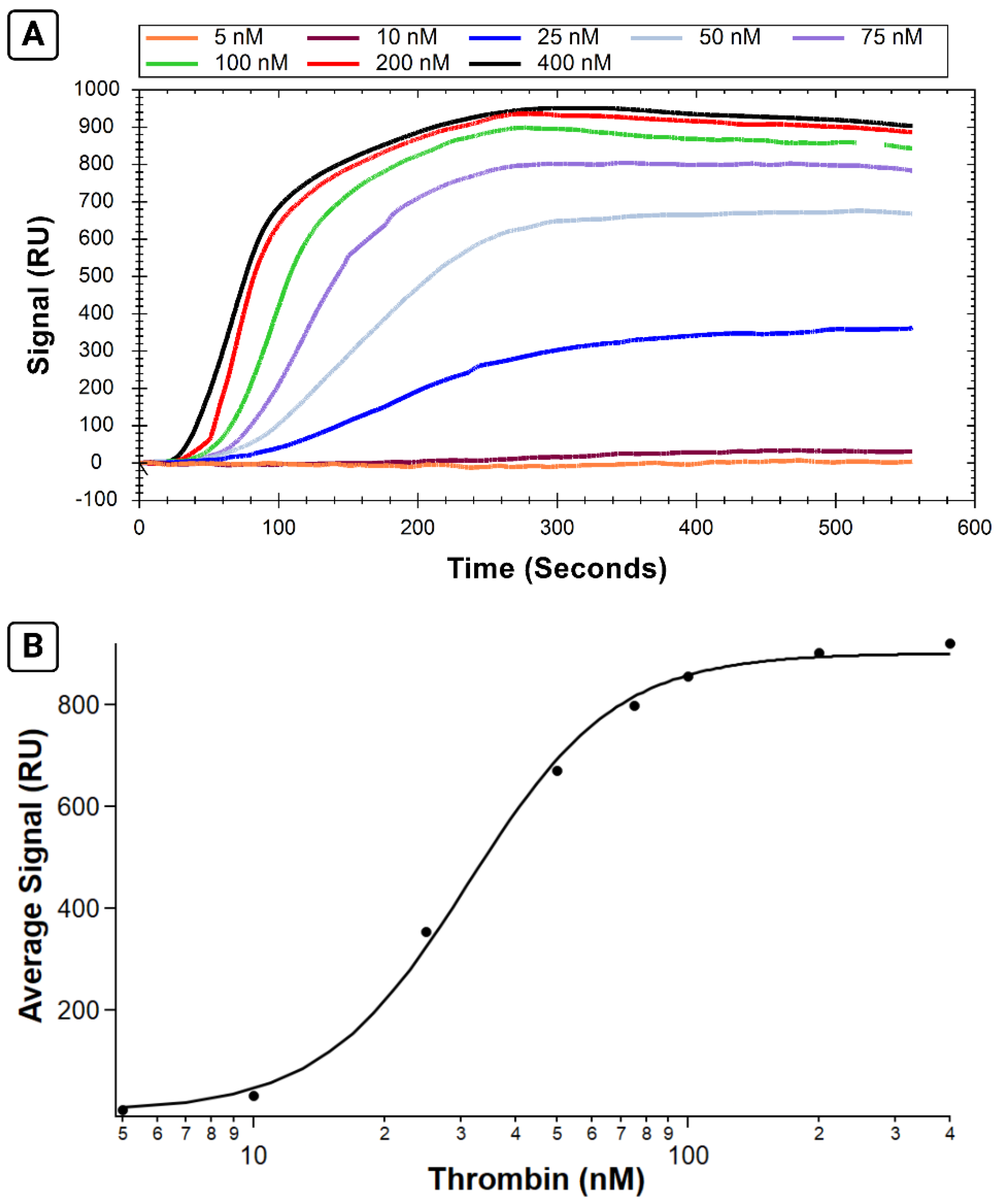
2.3. Demonstration of a Typical Binding Affinity Assay (Experiment 1)
B-29mer, His-SA, and thrombin samples were diluted in PBS-T sample buffer. B-29mer was diluted into PBS (to avoid heat degradation of Tween20) at a concentration of 10 µM and heated to 95 °C for 3 min then cooled to 4 °C and held at that temperature until further sample preparation. B-29mer was then diluted to a final concentration of 1 µM in PBS-T. His-SA was prepared to a concentration of 1 µM. Thrombin samples were prepared from stocks reconstituted in 1 mg/mL BSA, and an appropriate volume of 1 mg/mL BSA was added to each sample so that the volume contributed between thrombin and stock BSA in each was 10 µL. Samples of thrombin were then diluted in 200 µL PBS-T to their final concentrations, which ranged from 5 to 400 nM.
Unless otherwise noted, all injections used a flow rate of 20 µL/min, a dissociation time of 270 s, and were injected into both channels. The sensor surface was cleaned with two injections of 10 mM glycine–HCl, pH 1.5 (at 150 µL/min), and one injection of 350 mM EDTA (at 100 µL/min). The NTA surface was then primed with 40 mM NiCl2, and His-SA was immobilized into both channels. B-29mer was then injected into channel 2 only. Both His-SA and B-29mer had extended dissociation times of 600 s each. Following immobilization, injections alternated between regeneration using 2 M NaCl (at 150 µL/min) and thrombin analyte injections (concentration order was scrambled across the test). At the end of the test, an additional NaCl regeneration step along with two glycine–HCl injections and one EDTA injection were performed to fully remove the ligand from the surface and clean the sensor for future experiments.
The data were exported into TraceDrawer and the average signal for each curve over the interval between 475–500 s was calculated. The average curve interval for each concentration was plotted in IGOR Pro and fit with the Hill equation using values of n calculated from a Hill plot of the data.
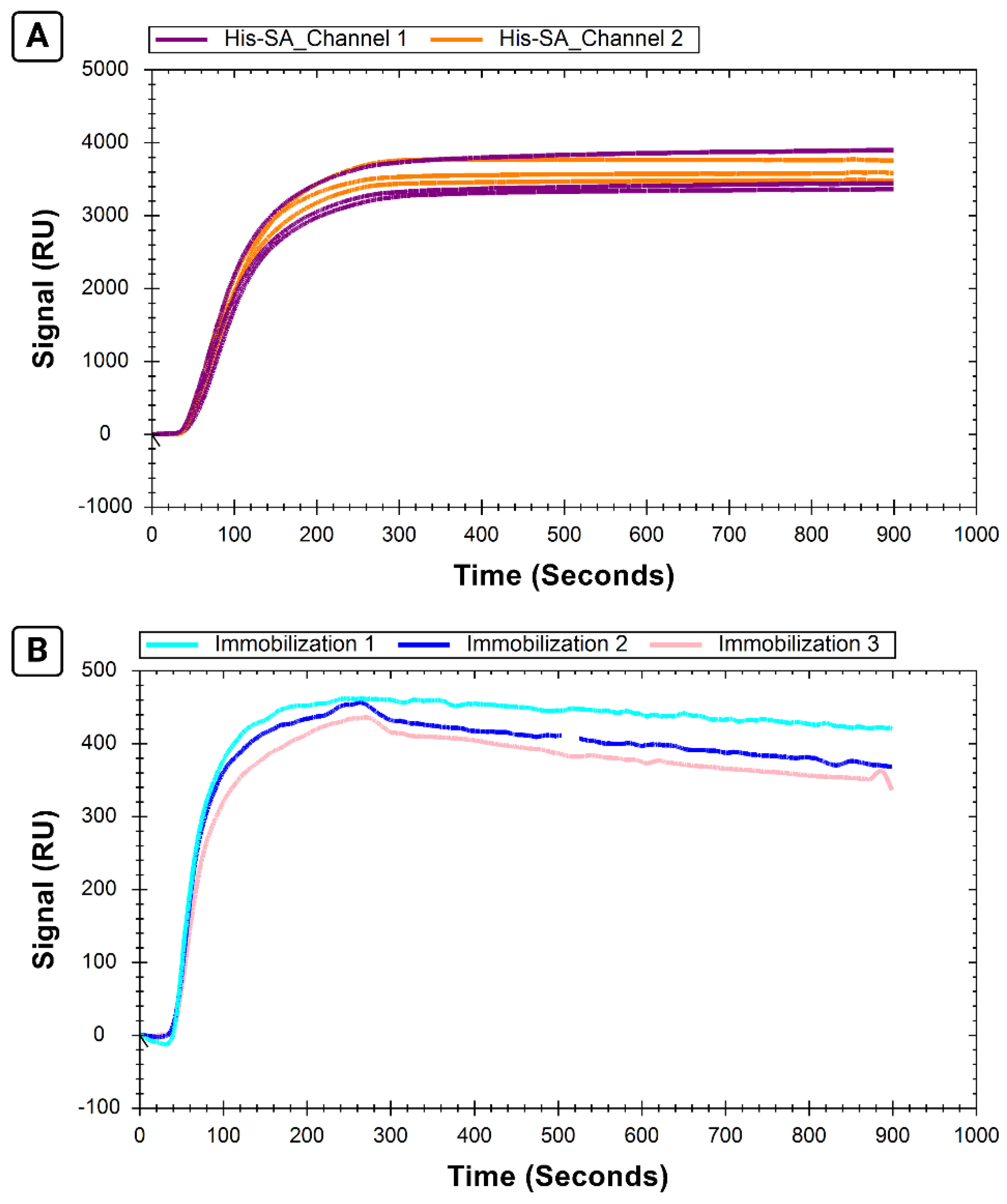
2.4. Immobilization Reproducibility (Experiment 2)
His-SA and B-29mer were prepared as for Experiment 1 but at triple the volume to account for 3 separate immobilizations onto the same chip. Representative thrombin concentrations of 10, 100, and 400 nM were prepared as in Experiment 1, performing the initial dilution in the appropriate volume of 1 mg/mL BSA so that the background contribution to each sample was the same (with tripled volume to allow for three replicates). Immobilization of His-SA and B-29mer used the same parameters as Experiment 1. Following immobilization, injections alternated between 2 M NaCl to regenerate the surface and thrombin samples. After the last thrombin sample, the surface was regenerated with NaCl and EDTA. This protocol was repeated in triplicate, starting with two glycine–HCl injections and one EDTA injection to ensure the surface was fully regenerated before reimmobilizing His-SA and B-29mer.
The data were exported into TraceDrawer and the average signal for each curve over the interval of 800–825 s was calculated for the corrected B-29mer signals and both His-SA channels 1 and 2.
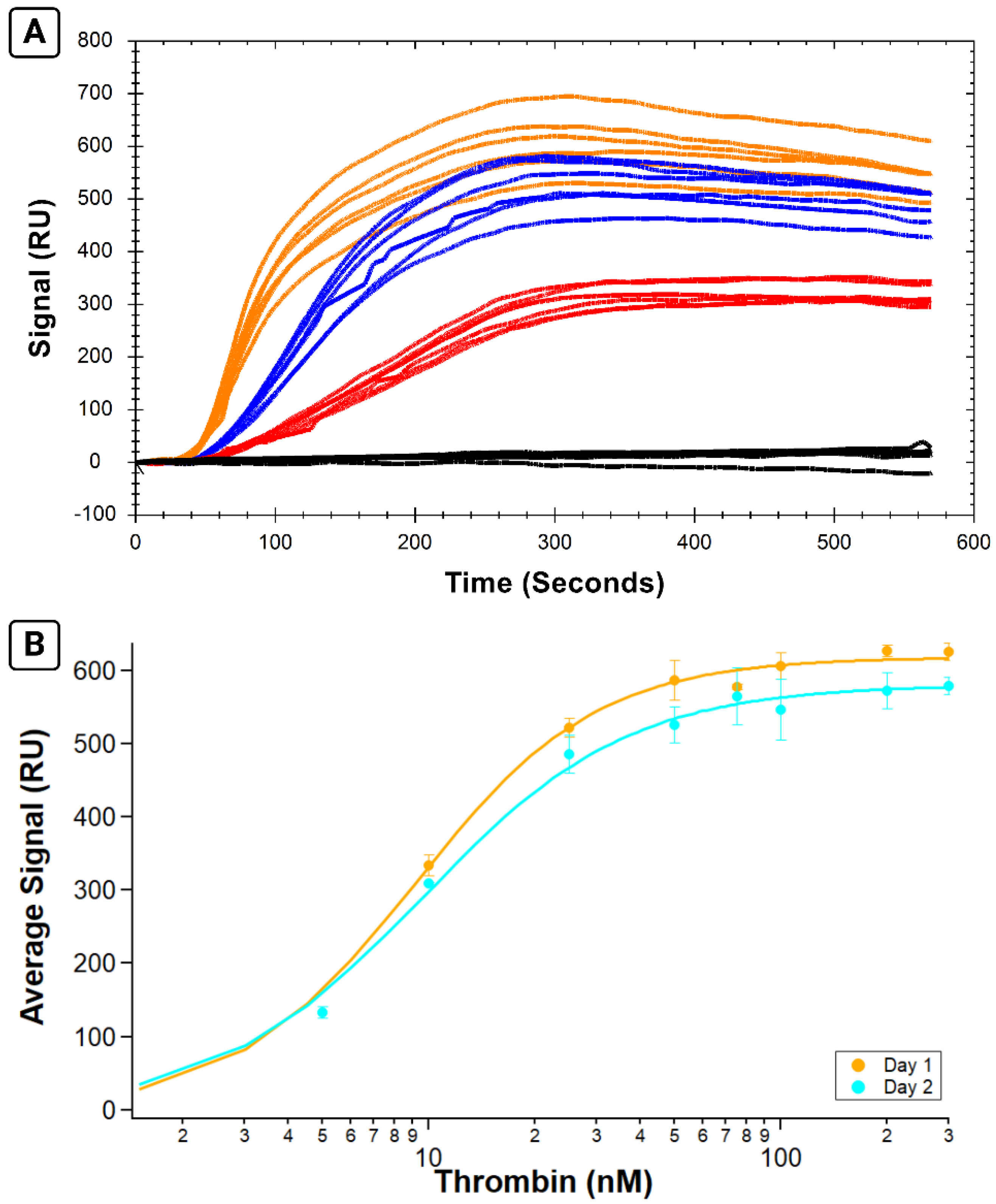
2.5. Experimental Reproducibility (Experiments 3 and 4)
The same parameters as above were used to prepare and immobilize His-SA and B-29mer onto a fresh NTA chip. Thrombin concentrations ranging from 10 to 300 nM were prepared (using the same strategy as earlier experiments with regards to BSA dilutions), and each was prepared with enough volume for 3 analyte injections. In addition, a sample of 0 nM protein was prepared (with the same volume of BSA as samples) to assess the non-specific binding of samples due to the presence of BSA. The instrumental protocol used the same strategy for surface cleaning and B-29mer immobilization, but a single B-29mer-immobilized surface was used for all three sets of thrombin analytes. Following immobilization, each set of thrombin samples was injected in a scrambled order with NaCl injected between thrombin samples to remove analyte from the surface. Once a full set of thrombin concentrations had been run, an additional injection of NaCl was used to ensure analyte was fully removed from the surface, and the next set of analytes was injected. At the end of the test, His-SA and B-29mer were regenerated off the surface using two glycine–HCl injections and one EDTA injection, and the sensor chip was put into standby overnight. Experiment 4 used an almost identical protocol as above but included a 5 nM thrombin sample in each set of analytes.
The average analyte signal for each curve was calculated over the interval of 475–500 s, and the average signal of each day’s three replicates was plotted versus the concentration and fit with the Hill equation on IGOR Pro.
2.6. Immobilization Stability (Experiments 5–8)
Experiments 5 and 6 were an initial scouting run to assess the feasibility of using the same immobilized surface on separate days. Experiments 7 and 8 were similar protocols run with triplicate and duplicate sets of samples, respectively.
Experiments 5 and 7 followed similar sample preparation and instrumental protocols as Experiments 1–4, with a few differences. PBS-I was used to prepare the samples for immobilization (B-29mer, His-SA, and an added injection of 255 µM biotin as a blocker), and they were each injected at 20 µL/min with a dissociation time of 270 s. B-29mer and His-SA concentrations were prepared in excess, i.e., to 10 µM and 2.5 µM, respectively. Following biotin injection, instrumental protocol incorporated a buffer swap from PBS-I to PBS-T for the rest of the run. Thrombin samples were prepared in PBS-T, with the same strategy to standardize BSA volume across samples. Thrombin samples were prepared fresh each day; Experiments 5 and 6 each had one replicate of each analyte concentration and Experiments 7 and 8 had enough sample prepared for triplicate injections of each analyte concentration. Instrumental protocols used the same parameters as earlier experiments but did not include the regeneration injections to remove His-SA and B-29mer from the surface of the chip at the end of Experiments 5 and 7. Following the experiment on the first day, the instrument was put into standby overnight with the sensor temperature set to 12 °C before preparing and running the next set of analytes the following day on the same immobilized surface. Data were analyzed as in Experiments 3 and 4.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}