
Biosensor-Enhanced Organ-on-a-Chip Models for Investigating Glioblastoma Tumor Microenvironment Dynamics




Abstract
:1. Introduction
2. Glioblastoma Tumor Microenvironment
2.1. Extracellular Matrix
2.2. The Immune Composition within the Glioma Microenvironment
2.2.1. Microglia and Glioma-Associated Macrophages (GAMs)
2.2.2. Neutrophils
2.2.3. Dendritic Cells
2.2.4. Natural Killer Cells
2.2.5. Lymphocytes
2.3. Neural Components of the Glioblastoma Microenvironment
2.4. Chemical Constituents in the GBM Microenvironment
2.4.1. Hypoxia
2.4.2. Acidosis
2.5. Glioblatoma Stem Cells
2.6. Deciphering Glioblastoma Complexity: Insights from Organ-on-Chip Modeling
3. GBM-on-Chip Models
3.1. Microfluidic GBM-on-Chip Models
3.2. Bioprinting GBM-on-a-Chip
4. Biosensor Technology in Glioblastoma Research
4.1. Advancing Organ-on-Chip Technology with Integrated Biosensors
4.1.1. Electrochemical Biosensors
4.1.2. Optical Biosensors
4.1.3. Electrical Biosensors
4.1.4. Multiple Biosensors
4.1.5. Other Types of Biosensors
4.2. Glioblastoma with Biosensor-Integrated Organ-on-Chip Models
4.3. Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GBM | glioblastoma multiforme |
| WHO | World Health Organization |
| IDH | isocitrate dehydrogenase |
| CNS | central nervous system |
| TMZ | temozolomide |
| TME | tumor microenvironment |
| ECM | extracellular matrix |
| OOC | organ-on-a-chip |
| HA | hyaluronic acid |
| CSPG | chondroitin sulfate proteoglycan |
| TN-C | tenascin-C |
| FAK | focal adhesion kinase |
| GSCs | glioblastoma stem cells |
| FN | fibronectin |
| IL-33 | interleukin-33 |
| NFκB | nuclear factor kappa B |
| GBP2 | interferon-inducible large GTPase |
| MMPs | matrix metalloproteinases |
| uPA/uPAR | urokinase plasminogen activator/receptor |
| ERK | extracellular signal-regulated kinase |
| PI3K/AKT | phosphoinositide 3-kinase/protein kinase B |
| HMGB1 | high-mobility group box 1 |
| NETs | neutrophil extracellular traps |
| MPO | myeloperoxidase |
| CitH3 | citrullinated histone H3 |
| Nrf | nuclear factor erythroid 2-related factor |
| HLA-I | human leukocyte antigen class I |
| NK | natural killer |
| CTLs | cytotoxic T lymphocytes |
| PD-L1 | programmed cell death ligand 1 |
| PDGF | platelet-derived growth factor |
| EGFR | epidermal growth factor receptor |
| PD1 | programmed cell death protein 1 |
| DCVs | dendritic cell vaccines |
| STING | stimulator of interferon genes |
| SHH | sonic hedgehog |
| CTGF | connective tissue growth factor |
| BDNF | brain-derived neurotrophic factor |
| NLGN3 | neuroligin-3 |
| mTOR | mammalian target of rapamycin |
| EMT | epithelial–mesenchymal transition |
| PKR | protein kinase R |
| UPR | unfolded protein response |
| CA9 | carbonic anhydrase 9 |
| HIF | hypoxia-inducible factor |
| TAMs | tumor-associated macrophages |
| MDSCs | myeloid-derived suppressor cells |
| PLOD2 | procollagen-lysine 2-oxoglutarate 5-dioxygenase 2 |
| ORR | objective response rate |
| PFS | progression-free survival |
| OS | overall survival |
| ACZ | acetazolamide |
| CD133 (PROM-1) | cluster of differentiation 133 (prominin-1) |
| LGR5 | leucine-rich repeat-containing G protein-coupled receptor 5 |
| NPM1 | nucleophosmin 1 |
| GPD1 | glycerol-3-phosphate dehydrogenase 1 |
| Notch | Notch signaling pathway |
| Wnt | Wnt signaling pathway |
| RTK | receptor tyrosine kinase |
| GSIs | γ-secretase inhibitors |
| PDGFR | platelet-derived growth factor receptor |
| VEGFR | vascular endothelial growth factor receptor |
| BMPs | bone morphogenic proteins |
| miRNAs | microRNAs |
| OV | oncolytic virotherapy |
| PDMS | polydimethylsiloxane |
| MIMIC | micromolding in capillaries |
| HPLC-UV | high-performance liquid chromatography combined with ultraviolet detection |
| VC | vessel co-option |
| PAA | polyacrylamide |
| TG | transglutaminase |
| DOX | doxorubicin |
| AKRs | aldo-keto reductases |
| ROS | reactive oxygen species |
| BBB | blood–brain barrier |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| PEGDA | poly(ethylene) glycol diacrylate |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| µG | microgravity |
| HUVECs | human umbilical vein endothelial cells |
| MST | microscale thermophoresis |
| SCPL | solid contact polymer layer |
| pEDOT | poly(3,4-ethylenedioxythiophene) |
| GBM | glioblastoma multiforme |
| DPV | differential pulse voltammetry |
| LOD | limit of detection |
| POC | point-of-care |
| FRET | fluorescence resonance energy transfer |
| ROC | receiver operating characteristic |
| PhCs | photonic crystals |
| FOM | figure of merit |
| RIU | refractive index unit |
| THz | terahertz |
| EIT | electromagnetic-induced transparency |
| GHz | gigahertz |
| PDMS | polydimethylsiloxane |
| PTFE | polytetrafluoroethylene |
| hiPSC | human-induced pluripotent stem cell |
| GST-α | glutathione S-transferase alpha |
| SP | surface plasmon |
| HSP-70 | heat shock protein 70 |
| CPNA-LAMP | colorimetric peptide nucleic acid loop-mediated isothermal amplification |
| MAS | multivalent aptamer nanoscaffold |
| HRP | horseradish peroxidase |
| CSF | cerebrospinal fluid |
| ADMA | asymmetric dimethylarginine |
| MRB | mitochondrial ribosomes |
| ECM | extracellular matrix |
| CICs | cancer-initiating cells |
| CAFs | cancer-associated fibroblasts |
| GEM | glioma extracellular matrix |
| MMPs | matrix metalloproteinases |
| PDLSCs | periodontal ligament stem cells |
| CAR | chimeric antigen receptor |
| TCR | T-cell receptor |
| LAMP | loop-mediated isothermal amplification |
| MGB | minor groove binder |
| EMA | European Medicines Agency |
| FDG | fluorodeoxyglucose |
| PET | positron emission tomography |
| HSCs | hematopoietic stem cells |
| CICs | cancer-initiating cells |
| 5mC | 5-methylcytosine |
| Fe3O4 | Iron(II,III) oxide |
| β-CD | beta-cyclodextrin |
| Ru(NH3)63+ | tris(2,2′-bipyridine)ruthenium(II) |
| H2O2 | hydrogen peroxide |
| EGFRvIII | epidermal growth factor receptor variant III |
| Zr-MOFs | zirconium-based metal–organic frameworks |
References
- Lin, S.; Xu, H.; Zhang, A.; Ni, Y.; Xu, Y.; Meng, T.; Wang, M.; Lou, M. Prognosis Analysis and Validation of m6A Signature and Tumor Immune Microenvironment in Glioma. Front. Oncol. 2020, 10, 541401. [Google Scholar] [CrossRef] [PubMed]
- Thenuwara, G.; Curtin, J.; Tian, F. Advances in diagnostic tools and therapeutic approaches for gliomas: A comprehensive review. Sensors 2023, 23, 9842. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Packer, R.J. The 2021 WHO Classification of Tumors of the Central Nervous System: Clinical Implications. Neuro-Oncology 2021, 23, 1215–1217. [Google Scholar] [CrossRef]
- Fisher, J.P.; Adamson, D.C. Current FDA-Approved Therapies for High-Grade Malignant Gliomas. Biomedicines 2021, 9, 324. [Google Scholar] [CrossRef]
- Lapointe, S.; Perry, A.; Butowski, N.A. Primary brain tumours in adults. Lancet 2018, 392, 432–446. [Google Scholar] [CrossRef]
- Bi, J.; Chowdhry, S.; Wu, S.; Zhang, W.; Masui, K.; Mischel, P.S. Altered cellular metabolism in gliomas—An emerging landscape of actionable co-dependency targets. Nat. Rev. Cancer 2020, 20, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Mariappan, A.; Goranci-Buzhala, G.; Ricci-Vitiani, L.; Pallini, R.; Gopalakrishnan, J. Trends and challenges in modeling glioma using 3D human brain organoids. Cell Death Differ. 2021, 28, 15–23. [Google Scholar] [CrossRef]
- Prager, B.C.; Bhargava, S.; Mahadev, V.; Hubert, C.G.; Rich, J.N. Glioblastoma stem cells: Driving resilience through chaos. Trends Cancer 2020, 6, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lage, M.; Lynch, T.M.; Bi, Y.; Cocito, C.; Way, G.P.; Pal, S.; Haller, J.; Yan, R.E.; Ziober, A.; Nguyen, A.; et al. Immune landscapes associated with different glioblastoma molecular sub-types. Acta Neuropathol. Commun. 2019, 7, 203. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Frisch, J.; Angenendt, A.; Hoth, M.; Prates Roma, L.; Lis, A. STIM-Orai channels and reactive oxygen species in the tumor microenvironment. Cancers 2019, 11, 457. [Google Scholar] [CrossRef]
- Taiarol, L.; Formicola, B.; Fagioli, S.; Sierri, G.; D’aloia, A.; Kravicz, M.; Renda, A.; Viale, F.; Magro, R.D.; Ceriani, M.; et al. The 3.0 cell communication: New insights in the usefulness of tunneling nanotubes for glioblastoma treatment. Cancers 2021, 13, 4001. [Google Scholar] [CrossRef]
- Seidi, K.; Neubauer, H.A.; Moriggl, R.; Jahanban-Esfahlan, R.; Javaheri, T. Tumor target amplification: Implications for nano drug delivery systems. J. Control. Release 2018, 275, 142–161. [Google Scholar] [CrossRef]
- Baghban, R.; Roshangar, L.; Jahanban-Esfahlan, R.; Seidi, K.; Ebrahimi-Kalan, A.; Jaymand, M.; Kolahian, S.; Javaheri, T.; Zare, P. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun. Signal. 2020, 18, 59. [Google Scholar] [CrossRef]
- Xiao, Y.; Yu, D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol. Ther. 2021, 221, 107753. [Google Scholar] [CrossRef]
- Liu, P.; Griffiths, S.; Veljanoski, D.; Vaughn-Beaucaire, P.; Speirs, V.; Brüning-Richardson, A. Preclinical models of glioblastoma: Limitations of current models and the promise of new developments. Expert Rev. Mol. Med. 2021, 23, e20. [Google Scholar] [CrossRef]
- Giles, B.; Nakhjavani, M.; Wiesa, A.; Knight, T.; Shigdar, S.; Samarasinghe, R.M. Unravelling the glioblastoma tumour microenvironment: Can aptamer targeted delivery become successful in treating brain cancers? Cancers 2023, 15, 4376. [Google Scholar] [CrossRef]
- Bergmann, N.; Delbridge, C.; Gempt, J.; Feuchtinger, A.; Walch, A.; Schirmer, L.; Bunk, W.; Aschenbrenner, T.; LiescheStarnecker, F.; Schlegel, J. The Intratumoral Heterogeneity Reflects the Intertumoral Subtypes of Glioblastoma Multiforme: A Regional Immunohistochemistry Analysis. Front. Oncol. 2020, 10, 494. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.; Ruzevick, J.; Phallen, J.; Belcaid, Z.; Lim, M. Challenges in immunotherapy presented by the glioblastoma multiforme microenvironment. J. Immunol. Res. 2011, 1, 2011. [Google Scholar] [CrossRef]
- Sun, W.; Luo, Z.; Lee, J.; Kim, H.J.; Lee, K.; Tebon, P.; Feng, Y.; Dokmeci, M.R.; Sengupta, S.; Khademhosseini, A. Organ-on-a-chip for cancer and immune organs modeling. Adv. Healthc. Mater. 2019, 8, 1801363. [Google Scholar] [CrossRef]
- Imparato, G.; Urciuolo, F.; Netti, P.A. Organ on chip technology to model cancer growth and metastasis. Bioengineering 2022, 9, 28. [Google Scholar] [CrossRef]
- Ma, C.; Peng, Y.; Li, H.; Chen, W. Organ-on-a-chip: A new paradigm for drug development. Trends Pharmacol. Sci. 2021, 42, 119–133. [Google Scholar] [CrossRef]
- Gonçalves, I.M.; Rodrigues, R.O.; Moita, A.S.; Hori, T.; Kaji, H.; Lima, R.A.; Minas, G. Recent trends of biomaterials and biosensors for organ-on-chip platforms. Bioprinting 2022, 26, e00202. [Google Scholar] [CrossRef]
- Yi, H.G.; Jeong, Y.H.; Kim, Y.; Choi, Y.J.; Moon, H.E.; Park, S.H.; Kang, K.S.; Bae, M.; Jang, J.; Youn, H.; et al. A bioprinted human-glioblastoma-on-a-chip for the identification of patient-specific responses to chemoradiotherapy. Nat. Biomed. Eng. 2019, 3, 509–519. [Google Scholar] [CrossRef]
- Xiao, Y.; Kim, D.; Dura, B.; Zhang, K.; Yan, R.; Li, H.; Han, E.; Ip, J.; Zou, P.; Liu, J.; et al. Ex vivo dynamics of human glioblastoma cells in a microvasculature-on-a-chip system correlates with tumor heterogeneity and subtypes. Adv. Sci. 2019, 6, 1801531. Available online: https://onlinelibrary.wiley.com/authored-by/Vortmeyer/Alexander+O (accessed on 26 April 2024). [CrossRef]
- Lin, Z.; Luo, G.; Du, W.; Kong, T.; Liu, C.; Liu, Z. Recent advances in microfluidic platforms applied in cancer metastasis: Circulating tumor cells’ (CTCs) isolation and tumor-on-a-chip. Small 2020, 16, 1903899. [Google Scholar] [CrossRef]
- Takebe, T.; Zhang, B.; Radisic, M. Synergistic engineering: Organoids meet organs-on-a-chip. Cell Stem Cell 2017, 21, 297–300. [Google Scholar] [CrossRef]
- Orcheston-Findlay, L.; Hashemi, A.; Garrill, A.; Nock, V. A microfluidic gradient generator to simulate the oxygen microenvironment in cancer cell culture. Microelectron. Eng. 2018, 195, 107–113. [Google Scholar] [CrossRef]
- DePalma, T.J.; Sivakumar, H.; Skardal, A. Strategies for developing complex multi-component in vitro tumor models: Highlights in glioblastoma. Adv. Drug Deliv. Rev. 2022, 180, 114067. [Google Scholar] [CrossRef] [PubMed]
- Logun, M.; Zhao, W.; Mao, L.; Karumbaiah, L. Microfluidics in malignant glioma research and precision medicine. Adv. Biosyst. 2018, 2, 1700221. [Google Scholar] [CrossRef]
- Fang, L.; Liu, Y.; Qiu, J.; Wan, W. Bioprinting and its Use in Tumor-On-A-Chip Technology for Cancer Drug Screening: A Review. Int. J. Bioprint. 2022, 8, 46–64. [Google Scholar] [CrossRef]
- Xie, Z.; Chen, M.; Lian, J.; Wang, H.; Ma, J. Glioblastoma-on-a-chip construction and therapeutic applications. Front. Oncol. 2023, 13, 1183059. [Google Scholar] [CrossRef]
- Ustun, M.; Rahmani Dabbagh, S.; Ilci, I.S.; Bagci-Onder, T.; Tasoglu, S. Glioma-on-a-chip models. Micromachines 2021, 12, 490. [Google Scholar] [CrossRef]
- Fedi, A.; Vitale, C.; Giannoni, P.; Caluori, G.; Marrella, A. Biosensors to monitor cell activity in 3D hydrogel-based tissue models. Sensors 2022, 22, 1517. [Google Scholar] [CrossRef]
- Shinde, A.; Illath, K.; Kasiviswanathan, U.; Nagabooshanam, S.; Gupta, P.; Dey, K.; Chakrabarty, P.; Nagai, M.; Rao, S.; Kar, S.; et al. Recent Advances of Biosensor-Integrated Organ-on-a-Chip Technologies for Diagnostics and Therapeutics. Anal. Chem. 2023, 95, 3121–3146. [Google Scholar] [CrossRef]
- Hosseini, A.; Ashraf, H.; Rahimi, F.; Alipourfard, I.; Alivirdiloo, V.; Hashemi, B.; Yazdani, Y.; Ghazi, F.; Eslami, M.; Ameri Shah Reza, M.; et al. Recent advances in the detection of glioblastoma, from imaging-based methods to proteomics and biosensors: A narrative review. Cancer Cell Int. 2023, 23, 98. [Google Scholar] [CrossRef]
- Deng, S.; Li, C.; Cao, J.; Cui, Z.; Du, J.; Fu, Z.; Yang, H.; Chen, P. Organ-on-a-chip meets artificial intelligence in drug evaluation. Theranostics 2023, 13, 4526. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef]
- Fanelli, G.N.; Grassini, D.; Ortenzi, V.; Pasqualetti, F.; Montemurro, N.; Perrini, P.; Naccarato, A.G.; Scatena, C. Decipher the glioblastoma microenvironment: The first milestone for new groundbreaking therapeutic strategies. Genes 2021, 12, 445. [Google Scholar] [CrossRef]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining tumor niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef]
- Lau, L.W.; Cua, R.; Keough, M.B.; Haylock-Jacobs, S.; Yong, V.W. Pathophysiology of the brain extracellular matrix: A new target for remyelination. Nat. Rev. Neurosci. 2013, 14, 722–729. [Google Scholar] [CrossRef]
- Yamaguchi, Y. Lecticans: Organizers of the brain extracellular matrix. Cell Mol. Life Sci. 2000, 57, 276–289. [Google Scholar] [CrossRef]
- Sood, D.; Tang-Schomer, M.; Pouli, D.; Mizzoni, C.; Raia, N.; Tai, A.; Arkun, K.; Wu, J.; Black, L.D., III; Scheffler, B.; et al. 3D extracellular matrix microenvironment in bioengineered tissue models of primary pediatric and adult brain tumors. Nat. Commun. 2019, 10, 4529. [Google Scholar] [CrossRef]
- Yan, T.; Chen, X.; Zhan, H.; Yao, P.; Wang, N.; Yang, H.; Zhang, C.; Wang, K.; Hu, H.; Li, J.; et al. Interfering with hyaluronic acid metabolism suppresses glioma cell proliferation by regulating autophagy. Cell Death Dis. 2021, 12, 486. [Google Scholar] [CrossRef]
- Schittenhelm, J.; Schwab, E.I.; Sperveslage, J.; Tatagiba, M.; Meyermann, R.; Fend, F.; Goodman, S.L.; Sipos, B. Longitudinal expression analysis of αv integrins in human gliomas reveals upregulation of integrin αvβ3 as a negative prognostic factor. J. Neuropathol. Exp. Neurol. 2013, 72, 194–210. [Google Scholar] [CrossRef]
- Ellert-Miklaszewska, A.; Poleszak, K.; Pasierbinska, M.; Kaminska, B. Integrin signaling in glioma pathogenesis: From biology to therapy. Int. J. Mol. Sci. 2020, 21, 888. [Google Scholar] [CrossRef]
- Brosicke, N.; Faissner, A. Role of tenascins in the ECM of gliomas. Cell Adh. Migr. 2015, 9, 131–140. [Google Scholar] [CrossRef]
- Xia, S.; Lal, B.; Tung, B.; Wang, S.; Goodwin, C.R.; Laterra, J. Tumor microenvironment tenascin-C promotes glioblastoma invasion and negatively regulates tumor proliferation. Neuro Oncol. 2016, 18, 507–517. [Google Scholar] [CrossRef]
- Hirata, E.; Arakawa, Y.; Shirahata, M.; Yamaguchi, M.; Kishi, Y.; Okada, T.; Takahashi, J.A.; Matsuda, M.; Hashimoto, N. Endogenous tenascin-C enhances glioblastoma invasion with reactive change of surrounding brain tissue. Cancer Sci. 2009, 100, 1451–1459. [Google Scholar] [CrossRef]
- Zhang, J.-F.; Tao, T.; Wang, K.; Zhang, G.-X.; Yan, Y.; Lin, H.-R.; Li, Y.; Guan, M.-W.; Yu, J.-J.; Wang, X.-D. IL-33/ST2 axis promotes glioblastoma cell invasion by accumulating tenascin-C. Sci. Rep. 2019, 9, 20276. [Google Scholar] [CrossRef]
- Sameni, M.; Mikkelsen, T.; Sloane, B. Degradation of extracellular matrix protein tenascin-C by cathepsin B: An interaction involved in the progression of gliomas. Biol. Chem. 2002, 383, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Nuttall, R.K.; Liu, S.; Edwards, D.R.; Yong, V.W. Tenascin-C stimulates glioma cell invasion through matrix metalloproteinase-12. Cancer Res. 2006, 66, 11771–11780. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Xue, Y.; Liu, J.; Xi, Z.; Li, Z.; Liu, Y. Fibronectin promotes the malignancy of glioma stem-like cells via modulation of cell adhesion, differentiation, proliferation and chemoresistance. Front. Mol. Neurosci. 2018, 11, 130. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, T.; Hiraga, S.; Izumoto, S.; Matsumura, H.; Kanemura, Y.; Arita, N.; Hayakawa, T. Role of fibronectin-stimulated tumor cell migration in glioma invasion in vivo: Clinical significance of fibronectin and fibronectin receptor expressed in human glioma tissues. Clin. Exp. Metastasis 1998, 16, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Caffo, M.; Caruso, G.; Meli, F.; Galatioto, S.; Sciacca, M.P.; Tomasello, F.; Germano, A. An immunohistochemical study of extracellular matrix proteins laminin, fibronectin and type IV collagen in paediatric glioblastoma multiforme. Acta Neurochir. 2004, 146, 113–1118. [Google Scholar] [CrossRef] [PubMed]
- Chintala, S.K.; Sawaya, R.; Gokaslan, Z.L.; Fuller, G.; Rao, J.S. Immunohistochemical localization of extracellular matrix proteins in human glioma, both in vivo and in vitro. Cancer Lett. 1996, 101, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Serres, E.; Debarbieux, F.; Stanchi, F.; Maggiorella, L.; Grall, D.; Turchi, L.; Burel-Vandenbos, F.; Figarella-Branger, D.; Virolle, T.; Rougon, G.; et al. Fibronectin expression in glioblastomas promotes cell cohesion, collective invasion of basement membrane in vitro and orthotopic tumor growth in mice. Oncogene 2013, 33, 3451–3462. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Nandi, S.; Hindi, E.S.; Wainwright, D.A.; Han, Y.; Lesniak, M.S. Short hairpin RNA-mediated fibronectin knockdown delays tumor growth in a mouse glioma model. Neoplasia 2010, 12, 837–847. [Google Scholar] [CrossRef]
- Yu, S.; Yu, X.; Sun, L.; Zheng, Y.; Chen, L.; Xu, H.; Jin, J.; Lan, Q.; Chen, C.C.; Li, M. GBP2 enhances glioblastoma invasion through Stat3/fibronectin pathway. Oncogene 2020, 39, 5042–5055. [Google Scholar] [CrossRef]
- Kabir, F.; Apu, M.N.H. Multi-omics analysis predicts fibronectin 1 as a prognostic biomarker in glioblastoma multiforme. Genomics 2022, 114, 110378. [Google Scholar] [CrossRef]
- Hu, B.; Nandhu, M.S.; Sim, H.; Agudelo-Garcia, P.A.; Saldivar, J.C.; Dolan, C.E.; Mora, M.E.; Nuovo, G.J.; Cole, S.E.; Viapiano, M.S. Fibulin-3 promotes glioma growth and resistance through a novel paracrine regulation of Notch signaling. Cancer Res. 2012, 72, 3873–3885. [Google Scholar] [CrossRef]
- Nandhu, M.S.; Behera, P.; Bhaskaran, V.; Longo, S.L.; Barrera-Arenas, L.M.; Sengupta, S.; Rodriguez-Gil, D.J.; Chiocca, E.A.; Viapiano, M.S. Development of a function-blocking antibody against fibulin-3 as a targeted reagent for glioblastoma. Clin. Cancer Res. 2018, 24, 821–833. [Google Scholar] [CrossRef]
- Tysnes, B.B.; Mahesparan, R.; Thorsen, F.; Haugland, H.K.; Porwol, T.; Enger, P.Ø.; Lund-Johansen, M.; Bjerkvig, R. Laminin expression by glial fibrillary acidic protein positive cells in human gliomas. Int. J. Dev. Neurosci. 1999, 17, 531–539. [Google Scholar] [CrossRef]
- Ljubimova, J.Y.; Fujita, M.; Khazenzon, N.M.; Ljubimov, A.V.; Black, K.L. Changes in laminin isoforms associated with brain tumor invasion and angiogenesis. Front. Biosci. 2006, 11, 81. [Google Scholar] [CrossRef]
- Sun, T.; Patil, R.; Galstyan, A.; Klymyshyn, D.; Ding, H.; Chesnokova, A.; Cavenee, W.K.; Furnari, F.B.; Ljubimov, V.A.; Shatalova, E.S.; et al. Blockade of a laminin-411–notch axis with CRISPR/Cas9 or a nanobioconjugate inhibits glioblastoma growth through tumor-microenvironment cross-talk. Cancer Res. 2019, 79, 1239–1251. [Google Scholar] [CrossRef]
- Khazenzon, N.M.; Ljubimov, A.V.; Lakhter, A.J.; Fujita, M.; Fujiwara, H.; Sekiguchi, K.; Sorokin, L.M.; Petajaniemi, N.; Virtanen, I.; Black, K.L.; et al. Antisense inhibition of laminin-8 expression reduces invasion of human gliomas in vitro. Mol. Cancer Ther. 2003, 2, 985–994. [Google Scholar]
- Calori, I.R.; Alves, S.R.; Bi, H.; Tedesco, A.C. Type-I collagen/collagenase modulates the 3D structure and behavior of glioblastoma spheroid models. ACS Appl. Bio Mater. 2022, 5, 723–733. [Google Scholar] [CrossRef]
- Wang, Y.; Sakaguchi, M.; Sabit, H.; Tamai, S.; Ichinose, T.; Tanaka, S.; Kinoshita, M.; Uchida, Y.; Ohtsuki, S.; Nakada, M. COL1A2 inhibition suppresses glioblastoma cell proliferation and invasion. J. Neurosurg. 2022, 138, 639–648. [Google Scholar] [CrossRef]
- Chintala, S.K.; Sawaya, R.; Gokaslan, Z.L.; Rao, J.S. The effect of type III collagen on migration and invasion of human glioblastoma cell lines in vitro. Cancer Lett. 1996, 102, 57–63. [Google Scholar] [CrossRef]
- Senner, V.; Ratzinger, S.; Mertsch, S.; Grässel, S.; Paulus, W. Collagen XVI expression is upregulated in glioblastomas and promotes tumor cell adhesion. FEBS Lett. 2008, 582, 3293–3300. [Google Scholar] [CrossRef]
- Mammoto, T.; Jiang, A.; Jiang, E.; Panigrahy, D.; Kieran, M.W.; Mammoto, A. Role of collagen matrix in tumor angiogenesis and glioblastoma multiforme progression. Am. J. Pathol. 2013, 183, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Jiang, L.; Wang, X.; Wei, W.; Song, C.; Cui, Y.; Wu, X.; Qiu, G. P4HA2 promotes epithelial-to-mesenchymal transition and glioma malignancy through the collagen-dependent PI3K/AKT pathway. J. Oncol. 2021, 2021, 1406853. [Google Scholar] [CrossRef] [PubMed]
- Huijbers, I.J.; Iravani, M.; Popov, S.; Robertson, D.; Al-Sarraj, S.; Jones, C.; Isacke, C.M. A role for fibrillar collagen deposition and the collagen internalization receptor Endo180 in glioma invasion. PLoS ONE 2010, 5, e9808. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhou, T.; Wang, Z.; Qi, B.; Xia, H. HSP47 promotes glioblastoma stemlike cell survival by modulating tumor microenvironment extracellular matrix through TGF-β pathway. ACS Chem. Neurosci. 2017, 8, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De la Rosa, C.; Ramirez-Acuna, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The roles of matrix metalloproteinases and their inhibitors in human diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, C.; Anacker, J.; Ernestus, R.I.; Vince, G.H. A complete compilation of matrix metalloproteinase expression in human malignant gliomas. World J. Clin. Oncol. 2012, 3, 67. [Google Scholar] [CrossRef] [PubMed]
- Kargiotis, O.; Chetty, C.; Gondi, C.S.; Tsung, A.J.; Dinh, D.H.; Gujrati, M.; Lakka, S.S.; Kyritsis, A.P.; Rao, J.S. Adenovirus-mediated transfer of siRNA against MMP-2 mRNA results in impaired invasion and tumor-induced angiogenesis, induces apoptosis in vitro and inhibits tumor growth in vivo in glioblastoma. Oncogene 2008, 27, 4830–4840. [Google Scholar] [CrossRef]
- Li, Q.; Chen, B.; Cai, J.; Sun, Y.; Wang, G.; Li, Y.; Li, R.; Feng, Y.; Han, B.; Li, J.; et al. Comparative analysis of matrix metalloproteinase family members reveals that MMP9 predicts survival and response to temozolomide in patients with primary glioblastoma. PLoS ONE 2016, 11, e0151815. [Google Scholar] [CrossRef]
- Schuler, P.J.; Bendszus, M.; Kuehnel, S.; Wagner, S.; Hoffmann, T.K.; Goldbrunner, R.; Vince, G.H. Urokinase plasminogen activator, uPAR, MMP-2, and MMP-9 in the C6-glioblastoma rat model. In Vivo 2012, 26, 571–576. [Google Scholar]
- Yue, J.; Zhang, K.; Chen, J. Role of integrins in regulating proteases to mediate extracellular matrix remodeling. Cancer Microenviron. 2012, 5, 275–283. [Google Scholar] [CrossRef]
- Zhao, Y.; Lyons, C.E., Jr.; Xiao, A.; Templeton, D.J.; Sang, Q.A.; Brew, K.; Hussaini, I.M. Urokinase directly activates matrix metalloproteinases-9: A potential role in glioblastoma invasion. Biochem. Biophys. Res. Commun. 2008, 369, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Liu, H.B.; Wang, Q.S.; Zhang, P.; Li, C.L.; Du, W.Z.; Wang, H.J.; Liu, X.; Zhang, Z.R.; Jiang, C.L. Activation of sonic hedgehog signaling enhances cell migration and invasion by induction of matrix metalloproteinase-2 and-9 via the phosphoinositide-3 kinase/AKT signaling pathway in glioblastoma. Mol. Med. Rep. 2015, 12, 6702–6710. [Google Scholar] [CrossRef]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the immune system in glioblastoma. Br. J. Cancer 2018, 119, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Klemm, F.; Maas, R.R.; Bowman, R.L.; Kornete, M.; Soukup, K.; Nassiri, S.; Brouland, J.-P.; Iacobuzio-Donahue, C.A.; Brennan, C.; Tabar, V.; et al. Interrogation of the microenvironmental landscape in brain tumors reveals disease-specific alterations of immune cells. Cell 2020, 181, 1643–1660.e17. [Google Scholar] [CrossRef] [PubMed]
- Szulzewsky, F.; Pelz, A.; Feng, X.; Synowitz, M.; Markovic, D.; Langmann, T.; Holtman, I.R.; Wang, X.; Eggen, B.J.; Boddeke, H.W.; et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS ONE 2015, 10, e0116644. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. The microenvironmental landscape of brain tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.F.; Yang, D.; Suki, D.; Aldape, K.; Grimm, E.; Heimberger, A.B. The role of human glioma-infiltrating microglia/macrophages in mediating anti-tumor immune responses. Neuro-Oncology 2006, 8, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Quiceno, D.G.; Zabaleta, J.; Ortiz, B.; Zea, A.H.; Piazuelo, M.B.; Delgado, A.; Correa, P.; Brayer, J.; Sotomayor, E.M.; et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. 2004, 64, 5839–5849. [Google Scholar] [CrossRef]
- Schartner, J.M.; Hagar, A.R.; Van Handel, M.; Zhang, L.; Nadkarni, N.; Badie, B. Impaired capacity for upregulation of MHC class II in tumor-associated microglia. Glia 2005, 51, 279–285. [Google Scholar] [CrossRef]
- Buonfiglioli, A.; Hambardzumyan, D. Macrophages and microglia: The cerberus of glioblastoma. Acta Neuropathol. Commun. 2021, 9, 54. [Google Scholar] [CrossRef] [PubMed]
- Poon, C.C.; Gordon, P.M.; Liu, K.; Yang, R.; Sarkar, S.; Mirzaei, R.; Ahmad, S.T.; Hughes, M.L.; Yong, V.W.; Kelly, J.J. Differential microglia and macrophage profiles in human IDH-mutant and-wild type glioblastoma. Oncotarget 2019, 10, 3129. [Google Scholar] [CrossRef] [PubMed]
- Bettinger, I.; Thanos, S.; Paulus, W. Microglia promote glioma migration. Acta Neuropathol. 2002, 103, 351–355. [Google Scholar] [CrossRef] [PubMed]
- Wick, W.; Platten, M.; Weller, M. Glioma cell invasion: Regulation of metalloproteinase activity by TGF-beta. J. Neurooncol. 2001, 53, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Wesolowska, A.; Kwiatkowska, A.; Slomnicki, L.; Dembinski, M.; Master, A.; Sliwa, M.; Franciszkiewicz, K.; Chouaib, S.; Kaminska, B. Microglia-derived TGF-beta as an important regulator of glioblastoma invasion–an inhibition of TGF-beta-dependent effects by shRNA against human TGF-beta type II receptor. Oncogene 2008, 27, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Vinnakota, K.; Hu, F.; Ku, M.C.; Georgieva, P.B.; Szulzewsky, F.; Pohlmann, A.; Waiczies, S.; Waiczies, H.; Niendorf, T.; Lehnardt, S.; et al. Toll-like receptor 2 mediates microglia/brain macrophage MT1-MMP expression and glioma expansion. Neuro-Oncology 2013, 15, 1457–1468. [Google Scholar] [CrossRef] [PubMed]
- Schi, Y.; Ping, Y.F.; Zhou, W.; He, Z.-C.; Chen, C.; Bian, B.-S.J.; Zhang, L.; Chen, L.; Lan, X.; Zhang, X.-C.; et al. Tumour-associated macrophages secrete pleiotrophin to promote PTPRZ1 signalling in glioblastoma stem cells for tumour growth. Nat. Commun. 2017, 8, 15080. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sarkar, S.; Cua, R.; Zhou, Y.; Hader, W.; Yong, V.W. A dialog between glioma and microglia that promotes tumor invasiveness through the CCL2/CCR2/interleukin-6 axis. Carcinogenesis 2012, 33, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef]
- De, I.; Steffen, M.D.; Clark, P.A.; Patros, C.J.; Sokn, E.; Bishop, S.M.; Litscher, S.; Maklakova, V.I.; Kuo, J.S.; Rodriguez, F.J.; et al. CSF1 overexpression promotes high-grade glioma formation without impacting the polarization status of glioma-associated microglia and macrophages. Cancer Res. 2016, 76, 2552–2560. [Google Scholar] [CrossRef]
- Brandenburg, S.; Müller, A.; Turkowski, K.; Radev, Y.T.; Rot, S.; Schmidt, C.; Bungert, A.D.; Acker, G.; Schorr, A.; Hippe, A.; et al. Resident microglia rather than peripheral macrophages promote vascularization in brain tumors and are source of alternative pro-angiogenic factors. Acta Neuropathol. 2016, 131, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, L.; Zhang, I.Y.; Liang, J.; Wang, H.; Ouyang, M.; Wu, S.; da Fonseca, A.C.C.; Weng, L.; Yamamoto, Y.; et al. RAGE expression in tumor-associated macrophages promotes angiogenesis in glioma. Cancer Res. 2014, 74, 7285–7297. [Google Scholar] [CrossRef]
- Nijaguna, M.B.; Patil, V.; Urbach, S.; Shwetha, S.D.; Sravani, K.; Hegde, A.S.; Chandramouli, B.A.; Arivazhagan, A.; Marin, P.; Santosh, V.; et al. Glioblastoma-derived macrophage colony-stimulating factor (MCSF) induces microglial release of insulin-like growth factor-binding protein 1 (IGFBP1) to promote angiogenesis. J. Biol. Chem. 2015, 290, 23401–23415. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, L.; Dang, W.Q.; Cao, M.-F.; Xiao, J.-F.; Lv, S.-Q.; Jiang, W.-J.; Yao, X.-H.; Lu, H.-M.; Miao, J.-Y.; et al. CCL8 secreted by tumor-associated macrophages promotes invasion and stemness of glioblastoma cells via ERK1/2 signaling. Lab. Investig. 2020, 100, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Markovic, D.S.; Vinnakota, K.; Chirasani, S.; Synowitz, M.; Raguet, H.; Stock, K.; Sliwa, M.; Lehmann, S.; Kälin, R.; van Rooijen, N.; et al. Gliomas induce and exploit microglial MT1-MMP expression for tumor expansion. Proc. Natl. Acad. Sci. USA 2009, 106, 12530–12535. [Google Scholar] [CrossRef] [PubMed]
- Galarneau, H.; Villeneuve, J.; Gowing, G.; Julien, J.P., Jr.; Vallières, L. Increased glioma growth in mice depleted of macrophages. Cancer Res. 2007, 67, 8874–8881. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Lu, Y.; Chen, W.; Fu, J.; Fan, R. In silico experimentation of glioma microenvironment development and anti-tumor therapy. PLoS Comput. Biol. 2012, 8, e1002355. [Google Scholar] [CrossRef] [PubMed]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.-M.; Ries, C.H.; Rüttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef]
- Hutter, G.; Theruvath, J.; Graef, C.M.; Zhang, M.; Schoen, M.K.; Manz, E.M.; Bennett, M.L.; Olson, A.; Azad, T.D.; Sinha, R.; et al. Microglia are effector cells of CD47-SIRPα antiphagocytic axis disruption against glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 997–1006. [Google Scholar] [CrossRef]
- Gholamin, S.; Mitra, S.S.; Feroze, A.H.; Liu, J.; Kahn, S.A.; Zhang, M.; Esparza, R.; Richard, C.; Ramaswamy, V.; Remke, M.; et al. Disrupting the CD47-SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 2017, 9, eaaf2968. [Google Scholar] [CrossRef] [PubMed]
- Held-Feindt, J.; Hattermann, K.; Müerköster, S.S.; Wedderkopp, H.; Knerlich-Lukoschus, F.; Ungefroren, H.; Mehdorn, H.M.; Mentlein, R. CX3CR1 promotes recruitment of human glioma-infiltrating microglia/macrophages (GIMs). Exp. Cell Res. 2010, 316, 1553–1566. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Mittal, S.; McGee, K.; Alfaro-Munoz, K.D.; Majd, N.; Balasubramaniyan, V.; de Groot, J.F. Role of neutrophils and myeloid-derived suppressor cells in glioma progression and treatment resistance. Int. J. Mol. Sci. 2020, 21, 1954. [Google Scholar] [CrossRef]
- Lin, Y.J.; Wei, K.C.; Chen, P.Y.; Lim, M.; Hwang, T.-L. Roles of neutrophils in glioma and brain metastases. Front. Immunol. 2021, 12, 701383. [Google Scholar] [CrossRef]
- Kasahara, T.; Mukaida, N.; Yamashita, K.; Yagisawa, H.; Akahoshi, T.; Matsushima, K. IL-1 and TNF-Alpha Induction of IL-8 and Monocyte Chemotactic and Activating Factor (MCAF) mRNA Expression in a Human Astrocytoma Cell Line. Immunology 1991, 74, 60–67. [Google Scholar] [PubMed]
- Zha, C.; Meng, X.; Li, L.; Mi, S.; Qian, D.; Li, Z.; Wu, P.; Hu, S.; Zhao, S.; Cai, J.; et al. Neutrophil Extracellular Traps Mediate the Crosstalk between Glioma Progression and the Tumor Microenvironment via the HMGB1/RAGE/IL-8 Axis. Cancer Biol. Med. 2020, 17, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Immunological and Inflammatory Functions of the Interleukin-1 Family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Chow, K.H.; Park, H.J.; George, J.; Yamamoto, K.; Gallup, A.D.; Graber, J.H.; Chen, Y.; Jiang, W.; Steindler, D.A.; Neilson, E.G.; et al. S100A4 Is a Biomarker and Regulator of Glioma Stem Cells That Is Critical for Mesenchymal Transition in Glioblastoma. Cancer Res. 2017, 77, 5360–5373. [Google Scholar] [CrossRef]
- Liang, J.; Piao, Y.; Holmes, L.; Fuller, G.N.; Henry, V.; Tiao, N.; de Groot, J.F. Neutrophils Promote the Malignant Glioma Phenotype through S100A4. Clin. Cancer Res. 2014, 20, 187–198. [Google Scholar] [CrossRef]
- Chang, Y.; Cai, X.; Syahirah, R.; Yao, Y.; Xu, Y.; Jin, G.; Bhute, V.J.; Torregrosa-Allen, S.; Elzey, B.D.; Won, Y.-Y.; et al. CAR-neutrophil mediated delivery of tumor-microenvironment responsive nanodrugs for glioblastoma chemo-immunotherapy. Nat. Commun. 2023, 14, 2266. [Google Scholar] [CrossRef]
- Serot, J.-M.; Béné, M.-C.; Foliguet, B.; Faure, G.C. Monocyte-derived IL-10-secreting dendritic cells in choroid plexus epithelium. J. Neuroimmunol. 2000, 105, 115–119. [Google Scholar] [CrossRef]
- Matyszak, M.K.; Perry, V.H. The potential role of dendritic cells in immune-mediated inflammatory diseases in the central nervous system. Neuroscience 1996, 74, 599–608. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, P.M.; Gottfried-Blackmore, A.; Anandasabapathy, N.; Bulloch, K. Brain dendritic cells: Biology and pathology. Acta Neuropathol. 2012, 124, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Zong, J.; Keskinov, A.A.; Shurin, G.V.; Shurin, M.R. Tumor-derived factors modulating dendritic cell function. Cancer Immunol. Immunother. 2016, 65, 821–833. [Google Scholar] [CrossRef] [PubMed]
- De Smedt, T.; Van Mechelen, M.; De Becker, G.; Urbain, J.; Leo, O.; Moser, M. Effect of interleukin-10 on dendritic cell maturation and function. Eur. J. Immunol. 1997, 27, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Tsumura, H.; Miwa, M.; Inaba, K. Contrasting effects of TGF-β1 and TNF-α on the development of dendritic cells from progenitors in mouse bone marrow. Stem Cells 1997, 15, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Nitta, T.; Hishii, M.; Sato, K.; Okumura, K. Selective expression of interleukin-10 gene within glioblastoma multiforme. Brain Res. 1994, 649, 122–128. [Google Scholar] [CrossRef]
- Ludewig, P.; Gallizioli, M.; Urra, X.; Behr, S.; Brait, V.H.; Gelderblom, M.; Magnus, T.; Planas, A.M. Dendritic cells in brain diseases. Biochim. Biophys. Acta 2016, 1862, 352–367. [Google Scholar] [CrossRef]
- Wang, J.; Liu, P.; Xin, S.; Wang, Z.; Li, J. Nrf2 suppresses the function of dendritic cells to facilitate the immune escape of glioma cells. Exp. Cell Res. 2017, 360, 66–73. [Google Scholar] [CrossRef]
- Han, D.; Liu, J.; Chen, C.; Dong, L.; Liu, Y.; Chang, R.; Huang, X.; Liu, Y.; Wang, J.; Dougherty, U.; et al. Anti-tumour immunity controlled through mRNA m6A methylation and YTHDF1 in dendritic cells. Nature 2019, 566, 270–274. [Google Scholar] [CrossRef]
- Siesjö, P.; Visse, E.; Sjögren, H.O. Cure of established, intracerebral rat gliomas induced by therapeutic immunizations with tumor cells and purified APC or adjuvant IFN-γ treatment. J. Immunother. 1996, 19, 334–345. [Google Scholar] [CrossRef]
- Kmiecik, J.; Poli, A.; Brons, N.H.; Waha, A.; Eide, G.E.; Enger, P.Ø.; Zimmer, J.; Chekenya, M. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol. 2013, 264, 71–83. [Google Scholar] [CrossRef]
- Friebel, E.; Kapolou, K.; Unger, S.; Núñez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-cell mapping of human brain cancer reveals tumor-specific instruction of tissue-invading leukocytes. Cell 2020, 181, 1626–1642.e20. [Google Scholar] [CrossRef]
- Crane, C.A.; Han, S.J.; Barry, J.J.; Ahn, B.J.; Lanier, L.L.; Parsa, A.T. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro-Oncology 2010, 12, 7–13. [Google Scholar] [CrossRef]
- Castriconi, R.; Daga, A.; Dondero, A.; Zona, G.; Poliani, P.L.; Melotti, A.; Griffero, F.; Marubbi, D.; Spaziante, R.; Bellora, F.; et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J. Immunol. 2009, 182, 3530–3539. [Google Scholar] [CrossRef]
- Shaim, H.; Shanley, M.; Basar, R.; Daher, M.; Gumin, J.; Zamler, D.B.; Uprety, N.; Wang, F.; Huang, Y.; Gabrusiewicz, K.; et al. Targeting the alphav integrin/TGF-beta axis improves natural killer cell function against glioblastoma stem cells. J. Clin. Investig. 2021, 131, 7195–7208. [Google Scholar] [CrossRef] [PubMed]
- Boussiotis, V.A.; Charest, A. Immunotherapies for malignant glioma. Oncogene 2018, 37, 1121–1141. [Google Scholar] [CrossRef]
- Woroniecka, K.I.; Rhodin, K.E.; Chongsathidkiet, P.; Keith, K.A.; Fecci, P.E. T-cell dysfunction in glioblastoma: Applying a new framework. Clin. Cancer Res. 2018, 24, 3792–3802. [Google Scholar] [CrossRef] [PubMed]
- Sandru, A.; Voinea, S.; Panaitescu, E.; Blidaru, A. Survival rates of patients with metastatic malignant melanoma. J. Med. Life 2014, 7, 572–576. [Google Scholar] [PubMed]
- Goldberg, S.B.; Schalper, K.A.; Gettinger, S.N.; Mahajan, A.; Herbst, R.S.; Chiang, A.C.; Lilenbaum, R.; Wilson, F.H.; Omay, S.B.; Yu, J.B.; et al. Pembrolizumab for management of patients with NSCLC and brain metastases: Long-term results and biomarker analysis from a non-randomised, open-label, phase 2 trial. Lancet Oncol. 2020, 21, 655–663. [Google Scholar] [CrossRef]
- Bunse, L.; Bunse, T.; Kramer, C.; Chih, Y.C.; Platten, M. Clinical and translational advances in glioma immunotherapy. Neurotherapeutics 2022, 19, 1799–1817. [Google Scholar] [CrossRef] [PubMed]
- Andersen, B.M.; Reardon, D.A. Immunotherapy approaches for adult glioma: Knowledge gained from recent clinical trials. Curr. Opin. Neurol. 2022, 35, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Dalrymple, S.J.; Parisi, J.E.; Roche, P.C.; Ziesmer, S.C.; Scheithauer, B.W.; Kelly, P.J. Changes in proliferating cell nuclear antigen expression in glioblastoma multiforme cells along a stereotactic biopsy trajectory. Neurosurgery 1994, 35, 1036–1044. [Google Scholar] [CrossRef]
- Stoecklein, V.M.; Stoecklein, S.; Galiè, F.; Ren, J.; Schmutzer, M.; Unterrainer, M.; Albert, N.L.; Kreth, F.-W.; Thon, N.; Liebig, T.; et al. Resting-state fMRI detects alterations in whole brain connectivity related to tumor biology in glioma patients. Neuro-Oncology 2020, 22, 1388–1398. [Google Scholar] [CrossRef]
- Krishna, S.; Hervey-Jumper, S.L. Neural regulation of cancer: Cancer-induced remodeling of the central nervous system. Adv. Biol. 2022, 6, e2200047. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, S.; Spray, D.C. Glioblastoma–Astrocyte Connexin 43 Gap Junctions Promote Tumor Invasion. Mol. Cancer Res. 2022, 20, 319–331. [Google Scholar] [CrossRef]
- Pei, Z.; Lee, K.C.; Khan, A.; Erisnor, G.; Wang, H.Y. Pathway analysis of glutamate-mediated, calcium-related signaling in glioma progression. Biochem. Pharmacol. 2020, 176, 113814. [Google Scholar] [CrossRef] [PubMed]
- MacVicar, B.A.; Newman, E.A. Astrocyte regulation of blood flow in the brain. Cold Spring Harb. Perspect. Biol. 2015, 7, a020388. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Jin, X.; Sohn, Y.W.; Jin, X.; Jeon, H.Y.; Kim, E.J.; Ham, S.W.; Jeon, H.M.; Chang, S.Y.; Oh, S.Y.; et al. Tumoral RANKL activates astrocytes that promote glioma cell invasion through cytokine signaling. Cancer Lett. 2014, 353, 194–200. [Google Scholar] [CrossRef]
- Ugbode, C.I.; Smith, I.; Whalley, B.J.; Hirst, W.D.; Rattray, M. Sonic hedgehog signalling mediates astrocyte crosstalk with neurons to confer neuroprotection. J. Neurochem. 2017, 142, 429–443. [Google Scholar] [CrossRef]
- Song, Z.B.; Yang, H.P.; Xu, A.Q.; Zhan, Z.M.; Song, Y.; Li, Z.Y. Connective tissue growth factor as an unfavorable prognostic marker promotes the proliferation, migration, and invasion of gliomas. Chin. Med. J. 2020, 133, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Edwards, L.A.; Woolard, K.; Son, M.J.; Li, A.; Lee, J.; Ene, C.; Mantey, S.A.; Maric, D.; Song, H.; Belova, G.; et al. Effect of brain- and tumor-derived connective tissue growth factor on glioma invasion. J. Natl. Cancer Inst. 2011, 103, 1162–1178. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Shin, S.H.; Chun, Y.S.; Shin, H.W.; Shin, Y.J.; Lee, Y.; Kim, D.; Nam, D.H.; Park, J.W. Astrocyte-derived CCL20 reinforces HIF-1-mediated hypoxic responses in glioblastoma by stimulating the CCR6-NF-kappaB signaling pathway. Oncogene 2018, 37, 3070–3087. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.I.; Anjo, S.I.; Vieira de Castro, J.; Serra, S.C.; Salgado, A.J.; Manadas, B.; Costa, B.M. Crosstalk between glial and glioblastoma cells triggers the “go-or-grow” phenotype of tumor cells. Cell Commun. Signal. 2017, 15, 37. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Liu, J.; Xu, P.; Deng, G.; Liu, B.; Yuan, F.; Tan, Y.; Sun, Q.; Xu, Y.; Zhang, H.; et al. AKT Inhibitor SC66 Inhibits Proliferation and Induces Apoptosis in Human Glioblastoma through Down-Regulating AKT/beta-Catenin Pathway. Front. Pharmacol. 2020, 11, 1102. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Choy, P.M.; Bubici, C. The ERK and JNK pathways in the regulation of metabolic reprogramming. Oncogene 2019, 38, 2223–2240. [Google Scholar] [CrossRef]
- Qiu, X.Y.; Hu, D.X.; Chen, W.Q.; Chen, R.Q.; Qian, S.R.; Li, C.Y.; Li, Y.J.; Xiong, X.X.; Liu, D.; Pan, F.; et al. PD-L1 confers glioblastoma multiforme malignancy via Ras binding and Ras/Erk/EMT activation. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1754–1769. [Google Scholar] [CrossRef]
- Litak, J.; Mazurek, M.; Grochowski, C.; Kamieniak, P.; Rolinski, J. PD-L1/PD-1 Axis in Glioblastoma Multiforme. Int. J. Mol. Sci. 2019, 20, 5347. [Google Scholar] [CrossRef] [PubMed]
- Felcht, M.; Luck, R.; Schering, A.; Seidel, P.; Srivastava, K.; Hu, J.; Bartol, A.; Kienast, Y.; Vettel, C.; Loos, E.K.; et al. Angiopoietin-2 differentially regulates angiogenesis through TIE2 and integrin signaling. J. Clin. Investig. 2012, 122, 1991–2005. [Google Scholar] [CrossRef]
- Kawashima, T.; Yashiro, M.; Kasashima, H.; Terakawa, Y.; Uda, T.; Nakajo, K.; Umaba, R.; Tanoue, Y.; Tamrakar, S.; Ohata, K. Oligodendrocytes Up-regulate the Invasive Activity of Glioblastoma Cells via the Angiopoietin-2 Signaling Pathway. Anticancer Res. 2019, 39, 577–584. [Google Scholar] [CrossRef]
- Xiong, J.; Zhou, L.I.; Lim, Y.; Yang, M.; Zhu, Y.H.; Li, Z.W.; Fu, D.L.; Zhou, X.F. Mature brain-derived neurotrophic factor and its receptor TrkB are upregulated in human glioma tissues. Oncol. Lett. 2015, 10, 223–227. [Google Scholar] [CrossRef]
- Zheng, B.; Chen, T. MiR-489-3p inhibits cell proliferation, migration, and invasion, and induces apoptosis, by targeting the BDNF-mediated PI3K/AKT pathway in glioblastoma. Open Life Sci. 2020, 15, 274–283. [Google Scholar] [CrossRef]
- Liu, S.; Jiang, T.; Zhong, Y.; Yu, Y. miR-210 inhibits cell migration and invasion by targeting the brain-derived neurotrophic factor in glioblastoma. J. Cell Biochem. 2019, 120, 11375–11382. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, H.S.; Tam, L.T.; Woo, P.J.; Lennon, J.; Nagaraja, S.; Gillespie, S.M.; Ni, J.; Duveau, D.Y.; Morris, P.J.; Zhao, J.J.; et al. Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 2017, 549, 533–537. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef]
- Ge, T.; Gu, X.; Jia, R.; Ge, S.; Chai, P.; Zhuang, A.; Fan, X. Crosstalk between metabolic reprogramming and epigenetics in cancer: Updates on mechanisms and therapeutic opportunities. Cancer Commun. 2022, 42, 1049–1082. [Google Scholar] [CrossRef]
- Masui, K.; Onizuka, H.; Cavenee, W.K.; Mischel, P.S.; Shibata, N. Metabolic reprogramming in the pathogenesis of glioma: Update. Neuropathology 2019, 39, 3–13. [Google Scholar] [CrossRef]
- Strickland, M.; Stoll, E.A. Metabolic reprogramming in glioma. Front. Cell Dev. Biol. 2017, 5, 43. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Erecinska, M.; Silver, I.A. Tissue oxygen tension and brain sensitivity to hypoxia. Respir. Physiol. 2001, 128, 263–276. [Google Scholar] [CrossRef]
- Beppu, T.; Kamada, K.; Yoshida, Y.; Arai, H.; Ogasawara, K.; Ogawa, A. Change of oxygen pressure in glioblastoma tissue under various conditions. J. Neurooncol. 2002, 58, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Brat, D.J.; Van Meir, E.G. Glomeruloid microvascular proliferation orchestrated by VPF/VEGF: A new world of angiogenesis research. Am. J. Pathol. 2001, 158, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.O. Vascular endothelial growth factors and vascular permeability. Cardiovasc. Res. 2010, 87, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kim, I.K.; Han, S.; Park, I.; Kim, C.; Bae, J.; Oh, S.J.; Lee, S.; Kim, J.H.; Woo, D.C.; et al. Normalization of Tumor Vessels by Tie2 Activation and Ang2 Inhibition Enhances Drug Delivery and Produces a Favorable Tumor Microenvironment. Cancer Cell 2016, 30, 953–967. [Google Scholar] [CrossRef] [PubMed]
- Marotta, D.; Karar, J.; Jenkins, W.T.; Kumanova, M.; Jenkins, K.W.; Tobias, J.W.; Baldwin, D.; Hatzigeorgiou, A.; Alexiou, P.; Evans, S.M.; et al. In vivo profiling of hypoxic gene expression in gliomas using the hypoxia marker EF5 and laser-capture microdissection. Cancer Res. 2011, 71, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Maciaczyk, D.; Picard, D.; Zhao, L.; Koch, K.; Herrera-Rios, D.; Li, G.; Marquardt, V.; Pauck, D.; Hoerbelt, T.; Zhang, W.; et al. CBF1 is clinically prognostic and serves as a target to block cellular invasion and chemoresistance of EMT-like glioblastoma cells. Br. J. Cancer 2017, 117, 102–112. [Google Scholar] [CrossRef]
- Joseph, J.V.; Conroy, S.; Pavlov, K.; Sontakke, P.; Tomar, T.; Eggens-Meijer, E.; Balasubramaniyan, V.; Wagemakers, M.; den Dunnen, W.F.; Kruyt, F.A. Hypoxia enhances migration and invasion in glioblastoma by promoting a mesenchymal shift mediated by the HIF1alpha-ZEB1 axis. Cancer Lett. 2015, 359, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Skuli, N.; Monferran, S.; Delmas, C.; Favre, G.; Bonnet, J.; Toulas, C.; Cohen-Jonathan Moyal, E. Alphavbeta3/alphavbeta5 integrins-FAK-RhoB: A novel pathway for hypoxia regulation in glioblastoma. Cancer Res. 2009, 69, 3308–3316. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, L.; Wei, Y.; Zhang, X.; Xu, R.; Han, M.; Huang, B.; Chen, A.; Li, W.; Zhang, Q.; et al. Procollagen-lysine 2-oxoglutarate 5-dioxygenase 2 promotes hypoxia-induced glioma migration and invasion. Oncotarget 2017, 8, 23401–23413. [Google Scholar] [CrossRef]
- Liu, Z.; Han, L.; Dong, Y.; Tan, Y.; Li, Y.; Zhao, M.; Xie, H.; Ju, H.; Wang, H.; Zhao, Y.; et al. EGFRvIII/integrin beta3 interaction in hypoxic and vitronectin-enriching microenvironment promote GBM progression and metastasis. Oncotarget 2016, 7, 4680–4694. [Google Scholar] [CrossRef]
- Braunstein, S.; Karpisheva, K.; Pola, C.; Goldberg, J.; Hochman, T.; Yee, H.; Cangiarella, J.; Arju, R.; Formenti, S.C.; Schneider, R.J. A hypoxia-controlled cap-dependent to cap-independent translation switch in breast cancer. Mol. Cell 2007, 28, 501–512. [Google Scholar] [CrossRef]
- Van den Beucken, T.; Magagnin, M.G.; Jutten, B.; Seigneuric, R.; Lambin, P.; Koritzinsky, M.; Wouters, B.G. Translational control is a major contributor to hypoxia induced gene expression. Radiother. Oncol. 2011, 99, 379–384. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Ohta, A.; Diwanji, R.; Kini, R.; Subramanian, M.; Ohta, A.; Sitkovsky, M. In vivo T cell activation in lymphoid tissues is inhibited in the oxygen-poor microenvironment. Front. Immunol. 2011, 2, 27. [Google Scholar] [CrossRef]
- Nakamura, H.; Makino, Y.; Okamoto, K.; Poellinger, L.; Ohnuma, K.; Morimoto, C.; Tanaka, H. TCR engagement increases hypoxia-inducible factor-1 alpha protein synthesis via rapamycin-sensitive pathway under hypoxic conditions in human peripheral T cells. J. Immunol. 2005, 174, 7592–7599. [Google Scholar] [CrossRef]
- Doedens, A.L.; Phan, A.T.; Stradner, M.H.; Fujimoto, J.K.; Nguyen, J.V.; Yang, E.; Johnson, R.S.; Goldrath, A.W. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat. Immunol. 2013, 14, 1173–1182. [Google Scholar] [CrossRef]
- Peng, W.; Wang, H.Y.; Miyahara, Y.; Peng, G.; Wang, R.-F. Tumor-associated galectin-3 modulates the function of tumor-reactive T cells. Cancer Res. 2008, 68, 7228–7236. [Google Scholar] [CrossRef]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef]
- Nizet, V.; Johnson, R.S. Interdependence of hypoxic and innate immune responses. Nat. Rev. Immunol. 2009, 9, 609–617. [Google Scholar] [CrossRef]
- Gerweck, L.E.; Seetharaman, K. Cellular pH gradient in tumor versus normal tissue: Potential exploitation for the treatment of cancer. Cancer Res. 1996, 56, 1194–1198. [Google Scholar]
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer 2017, 17, 577–593. [Google Scholar] [CrossRef]
- Vaupel, P.; Mayer, A. Hypoxia in tumors: Pathogenesis-related classification, characterization of hypoxia subtypes, and associated biological and clinical implications. Adv. Exp. Med. Biol. 2014, 812, 19–24. [Google Scholar] [CrossRef]
- Wiedmann, R.M.; von Schwarzenberg, K.; Palamidessi, A.; Schreiner, L.; Kubisch, R.; Liebl, J.; Schempp, C.; Trauner, D.; Vereb, G.; Zahler, S.; et al. The V-ATPase-inhibitor archazolid abrogates tumor metastasis via inhibition of endocytic activation of the Rho-GTPase Rac1. Cancer Res. 2012, 72, 5976–5987. [Google Scholar] [CrossRef]
- Boyd, N.H.; Tran, A.N.; Bernstock, J.D.; Etminan, T.; Jones, A.B.; Gillespie, G.Y.; Friedman, G.K.; Hjelmeland, A.B. Glioma stem cells and their roles within the hypoxic tumor microenvironment. Theranostics 2021, 11, 665. [Google Scholar] [CrossRef]
- Hulikova, A.; Swietach, P. Rapid CO2 permeation across biological membranes: Implications for CO2 venting from tissue. FASEB J. 2014, 28, 2762–2774. [Google Scholar] [CrossRef]
- Huber, V.; Camisaschi, C.; Berzi, A.; Ferro, S.; Lugini, L.; Triulzi, T.; Tuccitto, A.; Tagliabue, E.; Castelli, C.; Rivoltini, L. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. Cancer Biol. 2017, 43, 74–89. [Google Scholar] [CrossRef]
- Duraj, T.; Garcia-Romero, N.; Carrion-Navarro, J.; Madurga, R.; de Mendivil, A.O.; Prat-Acin, R.; Garcia-Cañamaque, L.; Ayuso-Sacido, A. Beyond the Warburg effect: Oxidative and glycolytic phenotypes coexist within the metabolic heterogeneity of glioblastoma. Cells 2021, 10, 202. [Google Scholar] [CrossRef]
- Yamini, B.; Lyne, S.; Driscoll, R.; Bernal, G.; Wu, L.; Nicholas, M.; Chmura, S.; Collins, J.; Park, D.; Pytel, P.; et al. Interim results of NCT03011671: A multi-institutional phase I study of acetazolamide with temozolomide in adults with newly diagnosed mgmt-methylated malignant glioma. Neuro-Oncology 2021, 23 (Suppl. S6), vi70. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yuan, X.; Zeng, Z.; Tunici, P.; Ng, H.; Abdulkadir, I.R.; Lu, L.; Irvin, D.; Black, K.L.; Yu, J.S. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol. Cancer 2006, 5, 67. [Google Scholar] [CrossRef]
- Tang, X.; Zuo, C.; Fang, P.; Liu, G.; Qiu, Y.; Huang, Y.; Tang, R. Targeting glioblastoma stem cells: A review on biomarkers, signal pathways and targeted therapy. Front. Oncol. 2021, 11, 701291. [Google Scholar] [CrossRef] [PubMed]
- Saito, N.; Aoki, K.; Hirai, N.; Fujita, S.; Iwabuchi, S. Abstract 774: Notch Pathway Activation Predicts Resistance to Bevacizumab Therapy in Glioblastoma. Cancer Res. 2017, 77 (Suppl. S13), 774. [Google Scholar] [CrossRef]
- Dantas-Barbosa, C.; Bergthold, G.; Daudigeos-Dubus, E.; Blockus, H.; Boylan, J.F.; Ferreira, C.; Puget, S.; Abely, M.; Vassal, G.; Grill, J.; et al. Inhibition of the NOTCH Pathway Using Gamma-Secretase Inhibitor RO4929097 has Limited Antitumor Activity in Established Glial Tumors. Anti-Cancer Drugs 2015, 26, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Liebelt, B.D.; Shingu, T.; Zhou, X.; Ren, J.; Shin, S.A.; Hu, J. Glioma Stem Cells: Signaling, Microenvironment, and Therap. Stem Cells Int. 2016, 2016, 7849890. [Google Scholar] [CrossRef]
- Wee, B.; Pietras, A.; Ozawa, T.; Bazzoli, E.; Podlaha, O.; Antczak, C.; Westermark, B.; Nelander, S.; Uhrbom, L.; Forsberg-Nilsson, K.; et al. ABCG2 Regulates Self-Renewal and Stem Cell Marker Expression but not Tumorigenicity or Radiation Resistance of Glioma Cells. Sci. Rep. 2016, 6, 25956. [Google Scholar] [CrossRef]
- Hombach-Klonisch, S.; Mehrpour, M.; Shojaei, S.; Harlos, C.; Ghavami, S. Glioblastoma and Chemoresistance to Alkylating Agents: Involvement of Apoptosis, Autophagy, and Unfolded Protein Respons. Pharmacol. Ther. 2017, 184, 13–41. [Google Scholar] [CrossRef] [PubMed]
- Shahi, M.H.; Farheen, S.; Mariyath, M.; Castresana, J.S. Potential Role of Shh-Gli1-BMI1 Signaling Pathway Nexus in Glioma Chemoresistance. Tumour Biol. 2016, 37, 15107–15114. [Google Scholar] [CrossRef]
- Ciechomska, I.A.; Gielniewski, B.; Wojtas, B.; Kaminska, B. EGFR/FOXO3a/BIM Signaling Pathway Determines Chemosensitivity of BMP4-Differentiated Glioma Stem Cells to Temozolomide. Exp. Mol. Med. 2020, 52, 1326–1340. [Google Scholar] [CrossRef]
- Cardoso, A.M.; Morais, C.M.; Pena, F.; Marante, T.; Cunha, P.P.; Jurado, A.S.; de Lima, M.C.P. Differentiation of Glioblastoma Stem Cells Promoted by miR-128 or miR-302a Overexpression Enhances Senescence-Associated Cytotoxicity of Axitinib. Hum. Mol. Genet. 2021, 30, 160–171. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, W.; Li, X.; Ren, J.; Ji, G.; Du, J.; de Lima, M.C.P. Graphene Oxide Suppresses the Growth and Malignancy of Glioblastoma Stem Cell-Like Spheroids via Epigenetic Mechanisms. J. Res. Transl. Med. 2020, 18, 200. [Google Scholar] [CrossRef] [PubMed]
- Dey, M.; Ulasov, I.V.; Tyler, M.A.; Sonabend, A.M.; Lesniak, M.S. Cancer Stem Cells: The Final Frontier for Glioma Virotherapy. Stem Cell Rev. Rep. 2011, 7, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Weller, M.; Lee, E.Q.; Alexander, B.M.; Barnholtz-Sloan, J.S.; Barthel, F.P.; Batchelor, T.T.; Bindra, R.S.; Chang, S.M.; Chiocca, E.A.; et al. Glioblastoma in Adults: A Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) Consensus Review on Current Management and Future Directions. Neuro Oncol. 2020, 22, 1073–1133. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lee, H.; Schmitt, P.; Choy, C.J.; Miller, D.M.; Williams, B.J.; Bearer, E.L.; Frieboes, H.B. Bioengineered models to study microenvironmental regulation of glioblastoma metabolism. J. Neuropathol. Exp. Neurol. 2021, 80, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Rape, A.; Ananthanarayanan, B.; Kumar, S. Engineering strategies to mimic the glioblastoma microenvironment. Adv. Drug Deliv. Rev. 2014, 79, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Ingber, D.E. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat. Rev. Genet. 2022, 23, 467–491. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Korolj, A.; Lai, B.F.L.; Radisic, M. Advances in organ-on-a-chip engineering. Nat. Rev. Mater. 2018, 3, 257–278. [Google Scholar] [CrossRef]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef]
- Park, S.E.; Georgescu, A.; Huh, D. Organoids-on-a-chip. Science 2019, 364, 960–965. [Google Scholar] [CrossRef] [PubMed]
- Aziz, A.U.R.; Geng, C.; Fu, M.; Yu, X.; Qin, K.; Liu, B. The role of microfluidics for organ on chip simulations. Bioengineering 2017, 4, 39. [Google Scholar] [CrossRef]
- Puryear Iii, J.R.; Yoon, J.K.; Kim, Y. Advanced fabrication techniques of microengineered physiological systems. Micromachines 2020, 11, 730. [Google Scholar] [CrossRef] [PubMed]
- Nie, J.; Fu, J.; He, Y. Hydrogels: The next generation body materials for microfluidic chips? Small 2020, 16, 2003797. [Google Scholar] [CrossRef] [PubMed]
- Nazari, H.; Heirani-Tabasi, A.; Ghorbani, S.; Eyni, H.; Razavi Bazaz, S.; Khayati, M.; Gheidari, F.; Moradpour, K.; Kehtari, M.; Tafti, S.M.A.; et al. Microfluidic-based droplets for advanced regenerative medicine: Current challenges and future trends. Biosensors 2021, 12, 20. [Google Scholar] [CrossRef]
- Zhao, Z.; Chen, X.; Dowbaj, A.M.; Sljukic, A.; Bratlie, K.; Lin, L.; Fong, E.L.S.; Balachander, G.M.; Chen, Z.; Soragni, A.; et al. Organoids. Nat. Rev. Methods Primers 2022, 2, 94. [Google Scholar] [CrossRef] [PubMed]
- Park, T.E.; Mustafaoglu, N.; Herland, A.; Hasselkus, R.; Mannix, R.; FitzGerald, E.A.; Prantil-Baun, R.; Watters, A.; Henry, O.; Benz, M.; et al. Hypoxia-enhanced Blood-Brain Barrier Chip recapitulates human barrier function and shuttling of drugs and antibodies. Nat. Commun. 2019, 10, 2621. [Google Scholar] [CrossRef] [PubMed]
- Samadian, H.; Jafari, S.; Sepand, M.R.; Alaei, L.; Malvajerd, S.S.; Jaymand, M.; Ghobadinezhad, F.; Jahanshahi, F.; Hamblin, M.; Derakhshankhah, H.; et al. 3D bioprinting technology to mimic the tumor microenvironment: Tumor-on-a-chip concept. Mater. Today Adv. 2021, 12, 100160. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Song, S.R.; Lv, S.Q.; Qin, J.H.; Yu, S.C. Models for evaluating glioblastoma invasion along white matter tracts. Trends Biotechnol. 2023, 42, 293–309. [Google Scholar] [CrossRef]
- Gupta, N.; Liu, J.R.; Patel, B.; Solomon, D.E.; Vaidya, B.; Gupta, V. Microfluidics-based 3D cell culture models: Utility in novel drug discovery and delivery research. Bioeng. Transl. Med. 2016, 1, 63–81. [Google Scholar] [CrossRef]
- Shi, Y.; He, X.; Wang, H.; Dai, J.; Fang, J.; He, Y.; Chen, X.; Hong, Z.; Chai, Y. Construction of a novel blood brain barrier-glioma microfluidic chip model: Applications in the evaluation of permeability and anti-glioma activity of traditional Chinese medicine components. Talanta 2023, 253, 123971. [Google Scholar] [CrossRef]
- Bae, J.; Kim, M.H.; Han, S.; Park, S. Development of tumor-vasculature interaction on chip mimicking vessel co-option of glioblastoma. BioChip J. 2023, 17, 77–84. [Google Scholar] [CrossRef]
- Hosni, I.; Iles, A.; Greenman, J.; Wade, M.A. A Robust, Flow-Based, Microfluidic Device for siRNA-Mediated Gene Knockdown in Glioblastoma Spheroids. Innov. Emerg. Technol. 2023, 10, 2340005. [Google Scholar] [CrossRef]
- Dou, J.; Mao, S.; Li, H.; Lin, J.-M. Combination Stiffness Gradient with Chemical Stimulation Directs Glioma Cell Migration on a Microfluidic Chip. Anal. Chem. 2020, 92, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Olubajo, F.; Achawal, S.; Greenman, J. Development of a Microfluidic Culture Paradigm for Ex Vivo Maintenance of Human Glioblastoma Tissue: A New Glioblastoma Model? Transl. Oncol. 2020, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Jie, M.; He, Z.; Li, H.F.; Lin, J.M. Study of Antioxidant Effects on Malignant Glioma Cells by Constructing a Tumor-Microvascular Structure on Microchip. Anal. Chim. Acta 2017, 978, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Nguyen, D.T.; Akay, Y.; Xu, F.; Akay, M. Engineering a Brain Cancer Chip for High-Throughput Drug Screening. Sci. Rep. 2016, 6, 25062. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Jun, Y.; Kim, S.H.; Hoang, H.H.; Jung, Y.; Kim, S.; Kim, J.; Austin, R.H.; Lee, S.; Park, S. Rapid Emergence and Mechanisms of Resistance by U87 Glioblastoma Cells to Doxorubicin in an In Vitro Tumor Microfluidic Ecology. Proc. Natl. Acad. Sci. USA 2016, 113, 14283–14288. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Chung, J.; Lee, K.; Balaj, L.; Min, C.; Carter, B.S.; Hochberg, F.H.; Breakefield, X.O.; Lee, H.; Weissleder, R. Chip-Based Analysis of Exosomal mRNA Mediating Drug Resistance in Glioblastoma. Nat. Commun. 2015, 6, 6999. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Xie, Q.; Gimple, R.C.; Zhong, Z.; Tam, T.; Tian, J.; Kidwell, R.L.; Wu, Q.; Prager, B.C.; Qiu, Z.; et al. Three-Dimensional Bioprinted Glioblastoma Microenvironments Model Cellular Dependencies and Immune Interactions. Cell Res. 2020, 30, 833–853. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Tiwari, S.K.; Agrawal, K.; Tan, M.; Dang, J.; Tam, T.; Tian, J.; Wan, X.; Schimelman, J.; You, S.; et al. Rapid 3D Bioprinting of Glioblastoma Model Mimicking Native Biophysical Heterogeneity. Small 2021, 17, 2006050. [Google Scholar] [CrossRef]
- Silvani, G.; Basirun, C.; Wu, H.; Mehner, C.; Poole, K.; Bradbury, P.; Chou, J. A 3D-Bioprinted Vascularized Glioblastoma-on-a-Chip for Studying the Impact of Simulated Microgravity as a Novel Pre-Clinical Approach in Brain Tumor Therapy. Adv. Ther. 2021, 4, 2100106. [Google Scholar] [CrossRef]
- Heinrich, M.A.; Bansal, R.; Lammers, T.; Zhang, Y.S.; Michel Schiffelers, R.; Prakash, J. 3D-Bioprinted Mini-Brain: A Glioblastoma Model to Study Cellular Interactions and Therapeutics. Adv. Mater. 2019, 31, 1806590. [Google Scholar] [CrossRef]
- Neufeld, L.; Yeini, E.; Reisman, N.; Shtilerman, Y.; Ben-Shushan, D.; Pozzi, S.; Madi, A.; Tiram, G.; Eldar-Boock, A.; Ferber, S.; et al. Microengineered Perfusable 3D-Bioprinted Glioblastoma Model for In Vivo Mimicry of Tumor Microenvironment. Sci. Adv. 2021, 7, eabi9119. [Google Scholar] [CrossRef]
- Amereh, M.; Seyfoori, A.; Dallinger, B.; Azimzadeh, M.; Stefanek, E.; Akbari, M. 3D-Printed Tumor-on-a-Chip Model for Investigating the Effect of Matrix Stiffness on Glioblastoma Tumor Invasion. Biomimetics 2023, 8, 421. [Google Scholar] [CrossRef]
- Keusgen, M. Biosensors: New Approaches in Drug Discovery. Naturwissenschaften 2002, 89, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Birkó, Z.; Nagy, B.; Klekner, Á.; Virga, J. Novel Molecular Markers in Glioblastoma-Benefits of Liquid Biopsy. Int. J. Mol. Sci. 2020, 21, 7522. [Google Scholar] [CrossRef]
- Zhou, E.; Li, Y.; Wu, F.; Guo, M.; Xu, J.; Wang, S.; Tan, Q.; Ma, P.; Song, S.; Jin, Y. Circulating Extracellular Vesicles Are Effective Biomarkers for Predicting Response to Cancer Therapy. EBioMedicine 2021, 67, 103365. [Google Scholar] [CrossRef]
- Amirsaadat, S.; Jafari-Gharabaghlou, D.; Dadashpour, M.; Zarghami, N. Potential Anti-Proliferative Effect of Nano-Formulated Curcumin through Modulating Micro RNA-132, Cyclin D1, and hTERT Genes Expression in Breast Cancer Cell Lines. J. Clust. Sci. 2023, 34, 2537–2546. [Google Scholar] [CrossRef]
- Figueroa, J.M.; Carter, B.S. Detection of Glioblastoma in Biofluids. J. Neurosurg. 2017, 129, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Castillo, M. History and Evolution of Brain Tumor Imaging: Insights through Radiology. Radiology 2014, 273, S111–S125. [Google Scholar] [CrossRef]
- Liguori, C.; Frauenfelder, G.; Massaroni, C.; Saccomandi, P.; Giurazza, F.; Pitocco, F.; Marano, R.; Schena, E. Emerging Clinical Applications of Computed Tomography. Med. Devices Evid. Res. 2015, 8, 265–278. [Google Scholar] [CrossRef]
- Bernstock, J.D.; Gary, S.E.; Klinger, N.; Valdes, P.A.; Ibn Essayed, W.; Olsen, H.E.; Chagoya, G.; Elsayed, G.; Yamashita, D.; Schuss, P.; et al. Standard Clinical Approaches and Emerging Modalities for Glioblastoma Imaging. Neuro-Oncol. Adv. 2022, 4, vdac080. [Google Scholar] [CrossRef] [PubMed]
- Drake, L.R.; Hillmer, A.T.; Cai, Z. Approaches to PET Imaging of Glioblastoma. Molecules 2020, 25, 568. [Google Scholar] [CrossRef] [PubMed]
- Verger, A.; Langen, K.J. PET Imaging in Glioblastoma: Use in Clinical Practice. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; pp. 155–174. [Google Scholar] [CrossRef]
- Law, I.; Albert, N.L.; Arbizu, J.; Boellaard, R.; Drzezga, A.; Galldiks, N.; La Fougère, C.; Langen, K.J.; Lopci, E.; Lowe, V.; et al. Joint EANM/EANO/RANO Practice Guidelines/SNMMI Procedure Standards for Imaging of Gliomas Using PET with Radio-labelled Amino Acids and [18F] FDG: Version 1.0. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 540–557. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.A.; Batara, J.F. Current Management of Glioblastoma Multiforme. Semin Oncol. 2004, 31, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Krol, I.; Castro-Giner, F.; Maurer, M.; Gkountela, S.; Szczerba, B.M.; Scherrer, R.; Coleman, N.; Carreira, S.; Bachmann, F.; Anderson, S.; et al. Detection of Circulating Tumour Cell Clusters in Human Glioblastoma. Br. J. Cancer 2018, 119, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, Z.; Zhang, X.; Wang, H.; Friedman, G.K.; Ding, Q.; Zhao, X.; Li, H.; Kim, K.; Yu, X.; et al. Generation of Chromosome 1p/19q Co-Deletion by CRISPR/Cas9-Guided Genomic Editing. Neuro-Oncol. Adv. 2022, 4, vdac131. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; McNamara, C.; Mankad, K.; Thust, S.; Dixon, L.; Limback-Stanic, C.; D’Arco, F.; Jacques, T.S.; Löbel, U. ATRX Loss in Glioma Results in Dysregulation of Cell-Cycle Phase Transition and ATM Inhibitor Radiosensitization. Cell Rep. 2022, 38, 110216. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Patel, C.B.; Xu, G.; Iagaru, A.; Zhu, Z.; Zhang, L.; Cheng, Z. Visualization of Diagnostic and Therapeutic Targets in Glioma with Molecular Imaging. Front. Immunol. 2020, 11, 592389. [Google Scholar] [CrossRef] [PubMed]
- Arita, H.; Narita, Y.; Fukushima, S.; Tateishi, K.; Matsushita, Y.; Yoshida, A.; Miyakita, Y.; Ohno, M.; Collins, V.P.; Kawahara, N.; et al. Upregulating Mutations in the TERT Promoter Commonly Occur in Adult Malignant Gliomas and Are Strongly Associated with Total 1p19q Loss. Acta Neuropathol. 2013, 126, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Richards, R.; Jensen, R. Role of Chromosomal 1p/19q Co-Deletion on the Prognosis of Oligodendrogliomas: A Systematic Review and Meta-Analysis. Interdiscip. Neurosurg. 2016, 5, 58–63. [Google Scholar] [CrossRef]
- Kang, Y.; Lin, X.; Kang, D. Diagnostic Value of Circulating Tumor DNA in Molecular Characterization of Glioma: A Meta-Analysis. Medicine 2020, 99, e21196. [Google Scholar] [CrossRef] [PubMed]
- Marei, H.E.; Althani, A.; Afifi, N.; Hasan, A.; Caceci, T.; Cifola, I.; Caratelli, S.; Sconocchia, G.; D’Agnano, I.; Cenciarelli, C. Glioma Extracellular Vesicles for Precision Medicine: Prognostic and Theragnostic Application. Discov. Oncol. 2022, 13, 49. [Google Scholar] [CrossRef] [PubMed]
- Śledzińska, P.; Bebyn, M.G.; Furtak, J.; Kowalewski, J.; Lewandowska, M.A. Prognostic and Predictive Biomarkers in Gliomas. Int. J. Mol. Sci. 2021, 22, 10373. [Google Scholar] [CrossRef]
- Bagley, S.J.; Nabavizadeh, S.A.; Mays, J.J.; Till, J.E.; Ware, J.B.; Levy, S.; Sarchiapone, W.; Hussain, J.; Prior, T.; Guiry, S.; et al. Clinical Utility of Plasma Cell-Free DNA in Adult Patients with Newly Diagnosed Glioblastoma: A Pilot Prospective Study. Clin. Cancer Res. 2020, 26, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Choate, K.A.; Raack, E.J.; Line, V.F.; Jennings, M.J.; Belton, R.J., Jr.; Winn, R.J.; Mann, P.B. Rapid extraction-free detection of the R132H isocitrate dehydrogenase mutation in glioma using colorimetric peptide nucleic acid-loop mediated isothermal amplification (CPNA-LAMP). PLoS ONE 2023, 18, e0291666. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.Y.; Huang, C.H.; Hsu, P.W.; Lu, Y.J.; Chou, P.C.; Chen, H.C.; Liu, V.Y. Protein Typing and mRNA Analysis of Circulating Exosomes for Glioblastoma Therapy Using Plasmonic-Enhanced Integrated Magneto-Electrochemical Sensor. Neuro-Oncology 2021, 23 (Suppl. S6), vi108. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, Y.; Tang, S.; Chen, S.; Zou, F.; Yuan, H.; Jiao, J. Multivalent aptamer nanoscaffold cytosensor for glioma circulating tumor cells during Epithelial–Mesenchymal transition. Biosens. Bioelectron. 2023, 226, 115140. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ren, J.; Zhao, M.; Chen, C.; Wang, F.; Chen, Z. Novel electrochemical immunosensor for O6-methylguanine-DNA methyltransferase gene methylation based on graphene oxide-magnetic nanoparticles-β-cyclodextrin nanocomposite. Bioelectrochemistry 2022, 146, 108111. [Google Scholar] [CrossRef] [PubMed]
- Scoggin, J.L.; Tan, C.; Nguyen, N.H.; Kansakar, U.; Madadi, M.; Siddiqui, S.; Arumugam, P.U.; DeCoster, M.A.; Murray, T.A. An enzyme-based electrochemical biosensor probe with sensitivity to detect astrocytic versus glioma uptake of glutamate in real time in vitro. Biosens. Bioelectron. 2019, 126, 751–757. [Google Scholar] [CrossRef]
- Yao, C.Y.; Qin, Y.; Fan, W.T.; Yan, L.P.; Chen, M.; Liu, Y.L.; Huang, W.H. A three-dimensional electrochemical biosensor integrated with hydrogel for cells culture and lactate release monitoring. J. Electroanal. Chem. 2022, 915, 116338. [Google Scholar] [CrossRef]
- Chen, L.; Zhu, S.; Wang, X. Detection of Glioma Cells based on Electrochemical Sensor Based on an Aptamer Method Recognition. Int. J. Electrochem. Sci. 2022, 17, 221258. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, L.; Wu, S.; Pan, Y.; Dong, Y.; Zhu, S.; Yang, J.; Yin, Y.; Li, G. An electrochemical biosensor designed by using Zr-based metal–organic frameworks for the detection of glioblastoma-derived exosomes with practical application. Anal. Chem. 2020, 92, 3819–3826. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Zhang, X.; Zhao, S.; Wang, X.; Wang, J.; Liu, R. Construction of a label-free electrochemical biosensing system utilizing Fe3O4/α-Fe2O3@ Au with magnetic-induced self-assembly for the detection of EGFR glycoprotein. Vacuum 2024, 222, 112975. [Google Scholar] [CrossRef]
- Calabretta, M.M.; Lopreside, A.; Montali, L.; Zangheri, M.; Evangelisti, L.; D‘Elia, M.; Michelini, E. Portable light detectors for bioluminescence biosensing applications: A comprehensive review from the analytical chemist’s perspective. Anal. Chim. Acta 2022, 1200, 339583. [Google Scholar] [CrossRef] [PubMed]
- Dey, D.; Goswami, T. Optical biosensors: A revolution towards quantum nanoscale electronics device fabrication. J. Biomed. Biotechnol. 2011, 2011, 348218. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Kaur, P.; Ahuja, S. Förster resonance energy transfer (FRET) and applications thereof. Anal. Methods 2020, 12, 5532–5550. [Google Scholar] [CrossRef] [PubMed]
- Hall, W.P.; Ngatia, S.N.; Van Duyne, R.P. LSPR biosensor signal enhancement using nanoparticle−antibody conjugates. J. Phys. Chem. C 2011, 115, 1410–1414. [Google Scholar] [CrossRef]
- Wu, L.; Huang, C.; Emery, B.P.; Sedgwick, A.C.; Bull, S.D.; He, X.-P.; Tian, H.; Yoon, J.; Sessler, J.L.; James, T.D. Förster resonance energy transfer (FRET)-based small-molecule sensors and imaging agents. Chem. Soc. Rev. 2020, 49, 5110–5139. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.H.; Park, J.; Kang, S.; Kim, M. Surface plasmon resonance: A versatile technique for biosensor applications. Sensors 2015, 15, 10481–10510. [Google Scholar] [CrossRef]
- Singh, P. SPR biosensors: Historical perspectives and current challenges. Sens. Actuators B Chem. 2016, 229, 110–130. [Google Scholar] [CrossRef]
- García-Milán, V.; Franco, A.; Zvezdanova, M.E.; Marcos, S.; Martin-Laez, R.; Moreno, F.; Velasquez, C.; Fernandez-Luna, J.L. Discriminating Glioblastoma from Peritumoral Tissue by a Nanohole Array-Based Optical and Label-Free Biosensor. Biosensors 2023, 13, 591. [Google Scholar] [CrossRef] [PubMed]
- Nouman, W.M.; Abd El-Ghany, S.E.S.; Sallam, S.M.; Dawood, A.F.B.; Aly, A.H. Biophotonic sensor for rapid detection of brain lesions using 1D photonic crystal. Opt. Quantum Electron. 2020, 52, 287. [Google Scholar] [CrossRef]
- Asuvaran, A.; Elatharasan, G. Design of two-dimensional photonic crystal-based biosensor for abnormal tissue analysis. Silicon 2022, 14, 7203–7210. [Google Scholar] [CrossRef]
- Mohammed, N.A.; Khedr, O.E.; El-Rabaie, E.S.M.; Khalaf, A.A. Brain tumors biomedical sensor with high-quality factor and ultra-compact size based on nanocavity 2D photonic crystal. Alex. Eng. J. 2023, 64, 527–540. [Google Scholar] [CrossRef]
- Zhang, J.; Mu, N.; Liu, L.; Xie, J.; Feng, H.; Yao, J.; Chen, T.; Zhu, W. Highly sensitive detection of malignant glioma cells using metamaterial-inspired THz biosensor based on electromagnetically induced transparency. Biosens. Bioelectron. 2021, 185, 113241. [Google Scholar] [CrossRef] [PubMed]
- Kilic, T.; Navaee, F.; Stradolini, F.; Renaud, P.; Carrara, S. Organs-on-chip monitoring: Sensors and other strategies. Microphysiol. Syst. 2018, 1, 1. [Google Scholar] [CrossRef]
- Modena, M.M.; Chawla, K.; Misun, P.M.; Hierlemann, A. Smart Cell Culture Systems: Integration of Sensors and Actuators into Microphysiological Systems. ACS Chem. Biol. 2018, 13, 1767–1784. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, J.; Jin, Y.; Cho, S.W. In situ biosensing technologies for an organ-on-a-chip. Biofabrication 2023, 15, 042002. [Google Scholar] [CrossRef] [PubMed]
- Kratz, S.R.A.; Höll, G.; Schuller, P.; Ertl, P.; Rothbauer, M. Latest Trends in Biosensing for Microphysiological Organs-on-a-Chip and Body-on-a-Chip Systems. Biosensors 2019, 9, 110. [Google Scholar] [CrossRef]
- Feng, S.; Zhang, Q.; Xie, T.; Hou, Y.; Lin, J.M. In-situ monitoring calcium signaling through tumor microtubes for single cell-cell communication via an open microfluidic probe. Biosens. Bioelectron. 2022, 206, 114137. [Google Scholar] [CrossRef]
- Arlett, J.; Myers, E.; Roukes, M. Comparative advantages of mechanical biosensors. Nat. Nanotechnol. 2011, 6, 203–215. [Google Scholar] [CrossRef]
- Damborsky, P.; Svitel, J.; Katrlik, J. Optical biosensors. Essays Biochem. 2016, 60, 91–100. [Google Scholar] [CrossRef]
- Karimi-Maleh, H.; Karimi, F.; Alizadeh, M.; Sanati, A.L. Electrochemical sensors, a bright future in the fabrication of portable kits in analytical systems. Chem. Rec. 2020, 20, 682–692. [Google Scholar] [CrossRef]
- Pemberton, R.M.; Cox, T.; Tuffin, R.; Drago, G.A.; Griffiths, J.; Pittson, R.; Johnson, G.; Xu, J.; Sage, I.C.; Davies, R.; et al. Fabrication and evaluation of a micro(bio)sensor array chip for multiple parallel measurements of important cell biomarkers. Sensors 2014, 14, 20519–20532. [Google Scholar] [CrossRef]
- Cho, I.H.; Kim, D.H.; Park, S. Electrochemical biosensors: Perspective on functional nanomaterials for on-site analysis. Biomater. Res. 2020, 24, 6. [Google Scholar] [CrossRef]
- Grieshaber, D.; MacKenzie, R.; Vörös, J.; Reimhult, E. Electrochemical Biosensors—Sensor Principles and Architectures. Sensors 2008, 8, 1400–1458. [Google Scholar] [CrossRef]
- Misun, P.M.; Rothe, J.; Schmid, Y.R.F.; Hierlemann, A.; Frey, O. Multi-analyte biosensor interface for real-time monitoring of 3D microtissue spheroids in hanging-drop networks. Microsyst. Nanoeng. 2016, 2, 16022. [Google Scholar] [CrossRef]
- Dornhof, J.; Kieninger, J.; Muralidharan, H.; Maurer, J.; Urban, G.A.; Weltin, A. Microfluidic organ-on-chip system for multi-analyte monitoring of metabolites in 3D cell cultures. Lab Chip 2022, 22, 225–239. [Google Scholar] [CrossRef]
- McCauley, T.G.; Hamaguchi, N.; Stanton, M. Aptamer-based biosensor arrays for detection and quantification of biological macromolecules. Anal. Biochem. 2003, 319, 244–250. [Google Scholar] [CrossRef]
- Liu, Y.; Tuleouva, N.; Ramanculov, E.; Revzin, A. Aptamer-based electrochemical biosensor for interferon gamma detection. Anal. Chem. 2010, 82, 8131–8136. [Google Scholar] [CrossRef]
- Shin, S.R.; Zhang, Y.S.; Kim, D.J.; Manbohi, A.; Avci, H.; Silvestri, A.; Aleman, J.; Hu, N.; Kilic, T.; Keung, W.; et al. Aptamer-based microfluidic electrochemical biosensor for monitoring cell-secreted trace cardiac biomarkers. Anal. Chem. 2016, 88, 10019–10027. [Google Scholar] [CrossRef]
- Chen, Y.A.; Shie, M.Y.; Ho, C.C.; Ye, S.W.; Chen, I.P.; Shih, Y.Y.; Shen, Y.F.; Chen, Y.W. A novel label-free electrochemical immunosensor for the detection of heat shock protein 70 of lung adenocarcinoma cell line following paclitaxel treatment using l-cysteine-functionalized Au@MnO2/MoO3 nanocomposites. RSC Adv. 2023, 13, 29847–29861. [Google Scholar] [CrossRef]
- Ortega, M.A.; Fernandez-Garibay, X.; Castano, A.G.; De Chiara, F.; HernandezAlbors, A.; Balaguer-Trias, J.; Ramon-Azcon, J. Muscle-on-a-chip with an on-site multiplexed biosensing system for in situ monitoring of secreted IL-6 and TNFalpha. Lab Chip 2019, 19, 2568–2580. [Google Scholar] [CrossRef]
- Jin, Z.H.; Liu, Y.L.; Fan, W.T.; Huang, W.H. Integrating flexible electrochemical sensor into microfluidic chip for simulating and monitoring vascular mechanotransduction. Small 2020, 16, e1903204. [Google Scholar] [CrossRef]
- Shang, W.; Chen, C.Y.; Lo, K.; Payne, G.F.; Bentley, W.E. Chip modularity enables molecular information access from organ-on-chip devices with quality control. Sens. Actuators B 2019, 295, 30–39. [Google Scholar] [CrossRef]
- Chen, C.; Wang, J. Optical biosensors: An exhaustive and comprehensive review. Analyst 2020, 145, 1605–1628. [Google Scholar] [CrossRef]
- Sang, S.; Wang, Y.; Feng, Q.; Wei, Y.; Ji, J.; Zhang, W. Progress of new label-free techniques for biosensors: A review. Crit. Rev. Biotechnol. 2016, 36, 465–481. [Google Scholar] [CrossRef]
- Sun, D.; Fan, T.; Liu, F.; Wang, F.; Gao, D.; Lin, J.M. A microfluidic chemiluminescence biosensor based on multiple signal amplification for rapid and sensitive detection of E. coli O157:H7. Biosens. Bioelectron. 2022, 212, 114390. [Google Scholar] [CrossRef]
- Rothbauer, M.; Holl, G.; Eilenberger, C.; Kratz, S.R.A.; Farooq, B.; Schuller, P.; Olmos Calvo, I.; Byrne, R.A.; Meyer, B.; Niederreiter, B.; et al. Monitoring tissue-level remodeling during inflammatory arthritis using a three-dimensional synoviumon-a-chip with non-invasive light scattering biosensing. Lab Chip 2020, 20, 1461–1471. [Google Scholar] [CrossRef]
- Ortega, M.A.; Rodriguez-Comas, J.; Yavas, O.; Velasco-Mallorqui, F.; BalaguerTrias, J.; Parra, V.; Novials, A.; Servitja, J.M.; Quidant, R.; Ramon-Azcon, J. In situ LSPR sensing of secreted insulin in organ-on-chip. Biosensors 2021, 11, 138. [Google Scholar] [CrossRef]
- Schneider, O.; Moruzzi, A.; Fuchs, S.; Grobel, A.; Schulze, H.S.; Mayr, T.; Loskill, P. Fusing spheroids to aligned mu-tissues in a heart-on-chip featuring oxygen sensing and electrical pacing capabilities. Mater. Today Bio. 2022, 15, 100280. [Google Scholar] [CrossRef]
- Azimzadeh, M.; Khashayar, P.; Amereh, M.; Tasnim, N.; Hoorfar, M.; Akbari, M. Microfluidic-based oxygen (O2) sensors for on-chip monitoring of cell, tissue and organ metabolism. Biosensors 2021, 12, 6. [Google Scholar] [CrossRef]
- Kobuszewska, A.; Kolodziejek, D.; Wojasinski, M.; Ciach, T.; Brzozka, Z.; Jastrzebska, E. Study of stem cells influence on cardiac cells cultured with a cyanide-p-trifluoromethoxyphenylhydrazone in organ-on-a-chip system. Biosensors 2021, 11, 131. [Google Scholar] [CrossRef]
- Srinivasan, B.; Kolli, A.R.; Esch, M.B.; Abaci, H.E.; Shuler, M.L.; Hickman, J.J. Teer measurement techniques for in vitro barrier model systems. J. Lab. Autom. 2015, 20, 107–126. [Google Scholar] [CrossRef]
- Henry, O.Y.F.; Villenave, R.; Cronce, M.J.; Leineweber, W.D.; Benz, M.A.; Ingber, D.E. Organs-on-chips with integrated electrodes for trans-epithelial electrical resistance (teer) measurements of human epithelial barrier function. Lab Chip 2017, 17, 2264–2271. [Google Scholar] [CrossRef]
- Vatine, G.D.; Barrile, R.; Workman, M.J.; Sances, S.; Barriga, B.K.; Rahnama, M.; Barthakur, S.; Kasendra, M.; Lucchesi, C.; Kerns, J.; et al. Human iPSC-derived blood-brain barrier chips enable disease modeling and personalized medicine applications. Cell Stem Cell 2019, 24, 995–1005. [Google Scholar] [CrossRef]
- Giampetruzzi, L.; Blasi, L.; Barca, A.; Sciurti, E.; Verri, T.; Casino, F.; Siciliano, P.; Francioso, L. Advances in trans-epithelial electrical resistance (TEER) monitoring integration in an intestinal barrier-on-chip (iBoC) platform with microbubbles-tolerant analytical method. Sens. Bio-Sens. Res. 2022, 37, 100512. [Google Scholar] [CrossRef]
- Mermoud, Y.; Felder, M.; Stucki, J.D.; Stucki, A.O.; Guenat, O.T. Microimpedance tomography system to monitor cell activity and membrane movements in a breathing lung-on-chip. Sensor. Actuator B Chem. 2018, 255, 3647–3653. [Google Scholar] [CrossRef]
- Shaik, F.A.; Ihida, S.; Ikeuchi, Y.; Tixier-Mita, A.; Toshiyoshi, H. Tft sensor array for real-time cellular characterization, stimulation, impedance measurement and optical imaging of in-vitro neural cells. Biosens. Bioelectron. 2020, 169, 112546. [Google Scholar] [CrossRef]
- Oleaga, C.; Riu, A.; Rothemund, S.; Lavado, A.; McAleer, C.W.; Long, C.J.; Persaud, K.; Narasimhan, N.S.; Tran, M.; Roles, J.; et al. Investigation of the effect of hepatic metabolism on off-target cardiotoxicity in a multi-organ human-on-a-chip system. Biomaterials 2018, 182, 176–190. [Google Scholar] [CrossRef]
- Shah, P.; Fritz, J.V.; Glaab, E.; Desai, M.S.; Greenhalgh, K.; Frachet, A.; Niegowska, M.; Estes, M.; Jäger, C.; Seguin-Devaux, C.; et al. A microfluidics-based in vitro model of the gastrointestinal human-microbe interface. Nat. Commun. 2016, 7, 11535. [Google Scholar] [CrossRef] [PubMed]
- Khalid, M.A.U.; Kim, K.H.; Chethikkattuveli Salih, A.R.; Hyun, K.; Park, S.H.; Kang, B.; Soomro, A.M.; Ali, M.; Jun, Y.; Huh, D.; et al. High performance inkjet printed embedded electrochemical sensors for monitoring hypoxia in a gut bilayer microfluidic chip. Lab Chip 2022, 22, 1764–1778. [Google Scholar] [CrossRef] [PubMed]
- Curto, V.F.; Marchiori, B.; Hama, A.; Pappa, A.M.; Ferro, M.P.; Braendlein, M.; Rivnay, J.; Fiocchi, M.; Malliaras, G.G.; Ramuz, M.; et al. Organic transistor platform with integrated microfluidics for in-line multi-parametric in vitro cell monitoring. Microsyst. Nanoeng. 2017, 3, 17028. [Google Scholar] [CrossRef]
- Asif, A.; Kim, K.H.; Jabbar, F.; Kim, S.; Choi, K.H. Real-time sensors for live monitoring of disease and drug analysis in microfluidic model of proximal tubule. Microfluid. Nanofluidics 2020, 24, 43. [Google Scholar] [CrossRef]
- Aleman, J.; Kilic, T.; Mille, L.S.; Shin, S.R.; Zhang, Y.S. Microfluidic integration of regeneratable electrochemical affinity-based biosensors for continual monitoring of organ-on-a-chip devices. Nat. Protoc. 2021, 16, 2564–2593. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Green, R.; Howell, M.; Martinez, T.; Dutta, R.; Mohapatra, S.S. The design and characterization of a gravitational microfluidic platform for drug sensitivity assay in colorectal perfused tumoroid cultures. Nanomedicine 2020, 30, 102294. [Google Scholar] [CrossRef]
- Khalid, M.A.U.; Kim, Y.S.; Ali, M.; Lee, B.G.; Cho, Y.-J.; Choi, K.H. A lung cancer-on-chip platform with integrated biosensors for physiological monitoring and toxicity assessment. Biochem. Eng. J. 2020, 155, 107469. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Aleman, J.; Shin, S.R.; Kilic, T.; Kim, D.; Shaegh, S.A.M.; Massa, S.; Riahi, R.; Chae, S.; Hu, N.; et al. Multisensor-integrated organs-on-chips platform for automated and continual in situ monitoring of organoid behaviors. Proc. Natl. Acad. Sci. USA 2017, 114, E2293–E2302. [Google Scholar] [CrossRef]
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the decline in pharmaceutical r&d efficiency. Nat. Rev. Drug Discov. 2012, 11, 191–200. [Google Scholar] [CrossRef]
- Farooqi, H.M.U.; Khalid, M.A.U.; Kim, K.H.; Lee, S.R.; Choi, K.H. Real-time physiological sensor-based liver-on-chip device for monitoring drug toxicity. J. Micromech. Microeng. 2020, 30, 115013. [Google Scholar] [CrossRef]
- Sciurti, E.; Blasi, L.; Prontera, C.T.; Barca, A.; Giampetruzzi, L.; Verri, T.; Siciliano, P.A.; Francioso, L. TEER and ion selective transwell-integrated sensors system for Caco-2 cell model. Micromachines 2023, 14, 469. [Google Scholar] [CrossRef]
- Azizgolshani, H.; Coppeta, J.R.; Vedula, E.M.; Marr, E.E.; Cain, B.P.; Luu, R.J.; Lech, M.P.; Tandon, V.; Tan, K.; Tan, N.J.; et al. High-throughput organ-on-chip platform with integrated programmable fluid flow and real-time sensing for complex tissue models in drug development work. Lab Chip 2021, 21, 1454–1474. [Google Scholar] [CrossRef]
- Bavli, D.; Prill, S.; Ezra, E.; Levy, G.; Cohen, M.; Vinken, M.; Vanfleteren, J.; Jaeger, M.; Nahmias, Y. Real-time monitoring of metabolic function in liver-on-chip microdevices tracks the dynamics of mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 2016, 113, E2231–E2240. [Google Scholar] [CrossRef]
- Liebisch, F.; Weltin, A.; Marzioch, J.; Urban, G.A.; Kieninger, J. Zero-consumption clark-type microsensor for oxygen monitoring in cell culture and organ-on-chip systems. Sens. Actuators B Chem. 2020, 322, 128652. [Google Scholar] [CrossRef]
- Moya, A.; Ortega-Ribera, M.; Guimera, X.; Sowade, E.; Zea, M.; Illa, X.; Ramon, E.; Villa, R.; Gracia-Sancho, J.; Gabriel, G. Online oxygen monitoring using integrated inkjet-printed sensors in a liver-on-a-chip system. Lab Chip 2018, 18, 2023–2035. [Google Scholar] [CrossRef]
- Tajeddin, A.; Mustafaoglu, N.; Yapici, M.K. On-chip measurement of pH using a microcantilever: A biomimetic design approach. In 2021 Symposium on Design, Test, Integration & Packaging of MEMS and MOEMS; IEEE: Piscataway, NJ, USA, 2021; pp. 1–5. [Google Scholar] [CrossRef]
- Liang, T.; Gu, C.L.; Gan, Y.; Wu, Q.; He, C.J.; Tu, J.W.; Pan, Y.X.; Qiu, Y.; Kong, L.B.; Wan, H.; et al. Flexible organic electrochemical transistors for real-time monitoring of pH, lactate, and glucose. J. Mater. Chem. B 2021, 9, 10618–10627. [Google Scholar]
- da Ponte, R.M.; Gaio, N.; van Zeijl, H.; Vollebregt, S.; Dijkstra, P.; Dekker, R.; Serdijn, W.A.; Giagka, V. Monolithic integration of a smart temperature sensor on a modular silicon-based organ-on-a-chip device. Sens. Actuator A Phys. 2021, 317, 112439. [Google Scholar] [CrossRef]
- Sousa, P.J.; Pinto, V.C.; Magalhães, V.H.; Rodrigues, R.O.; Sousa, P.C.; Minas, G. Development of highly sensitive temperature microsensors for localized measurements. Appl. Sci. 2021, 11, 3864. [Google Scholar] [CrossRef]
- Fernandes, L.A.; Hafliger, P.; Azadmehr, M.; Johannessen, E. Design and characterization of an osmotic sensor for the detection of events associated with dehydration and overhydration. IEEE J. Transl. Eng. Health Med. 2013, 1, 2700309. [Google Scholar] [CrossRef]
- Kincses, A.; Vigh, J.P.; Petrovszki, D.; Valkai, S.; Kocsis, A.E.; Walter, F.R.; Lin, H.Y.; Jan, J.S.; Deli, M.A.; Dér, A. The Use of Sensors in Blood-Brain Barrier-on-a-Chip Devices: Current Practice and Future Directions. Biosensors 2023, 13, 357. [Google Scholar] [CrossRef]
- Mir, M.; Palma-Florez, S.; Lagunas, A.; López-Martínez, M.J.; Samitier, J. Biosensors integration in blood–brain barrier-on-a-chip: Emerging platform for monitoring neurodegenerative diseases. ACS Sens. 2022, 7, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Deli, M.A.; Porkoláb, G.; Kincses, A.; Mészáros, M.; Szecskó, A.; Kocsis, A.E.; Vigh, J.P.; Valkai, S.; Veszelka, S.; Walter, F.R.; et al. Lab-on-a-chip models of the blood–brain barrier: Evolution, problems, perspectives. Lab Chip 2024, 24, 1030–1063. [Google Scholar] [CrossRef] [PubMed]
- Su, S.H.; Song, Y.; Stephens, A.; Situ, M.; McCloskey, M.C.; McGrath, J.L.; Andjelkovic, A.V.; Singer, B.H.; Kurabayashi, K. A tissue chip with integrated digital immunosensors: In situ brain endothelial barrier cytokine secretion monitoring. Biosens. Bioelectron. 2023, 224, 115030. [Google Scholar] [CrossRef]
- Yang, X.; Zhang, Z.; Wu, Y.; Wang, H.; Yun, Y.; Sun, Y.; Xie, H.; Bogdanov, B.; Senyushkin, P.; Chi, J.; et al. Printed Divisional Optical Biochip for Multiplex Visualizable Exosome Analysis at Point-of-Care. Adv. Mater. 2024, 36, 2304935. [Google Scholar] [CrossRef]


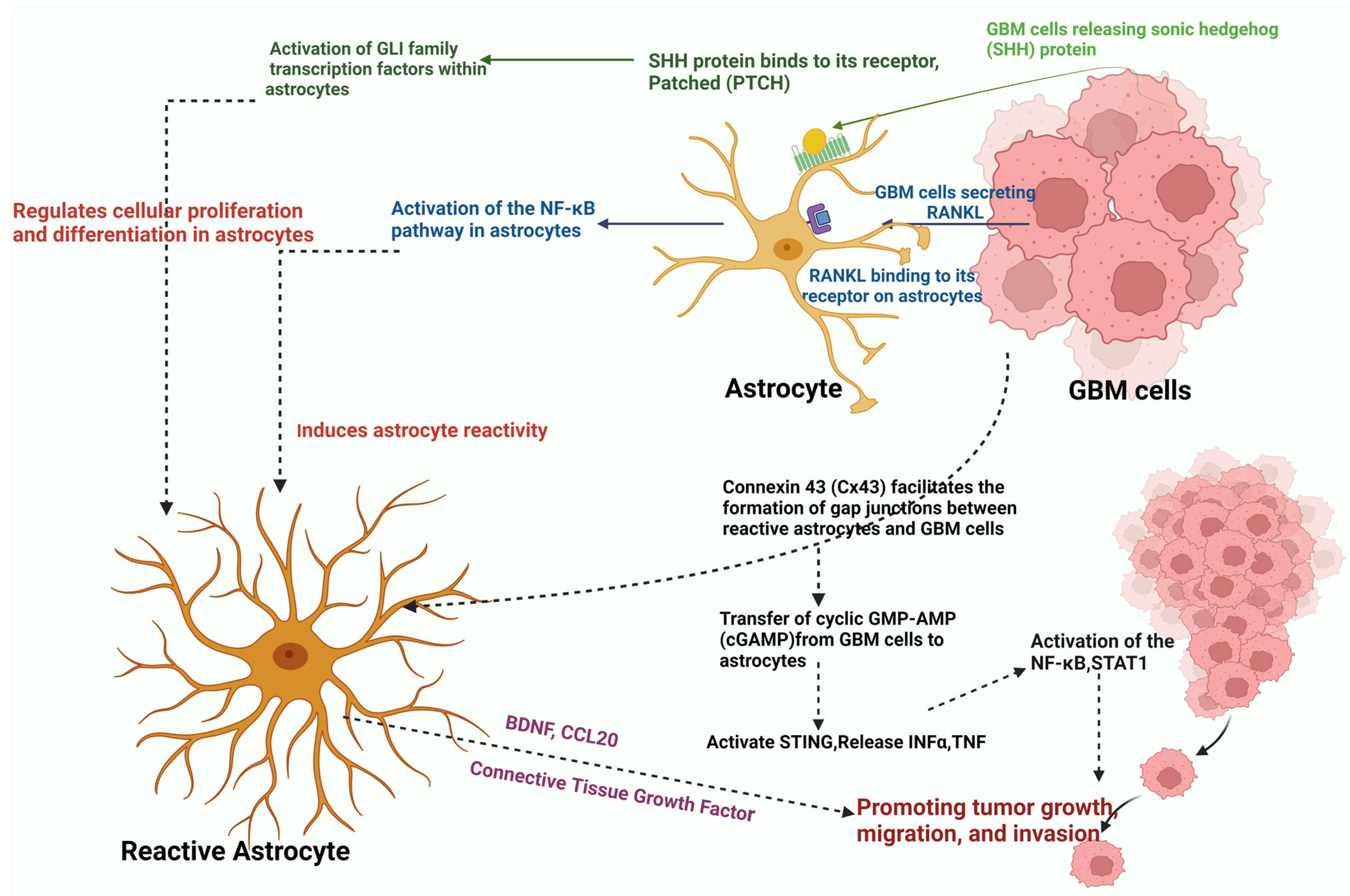
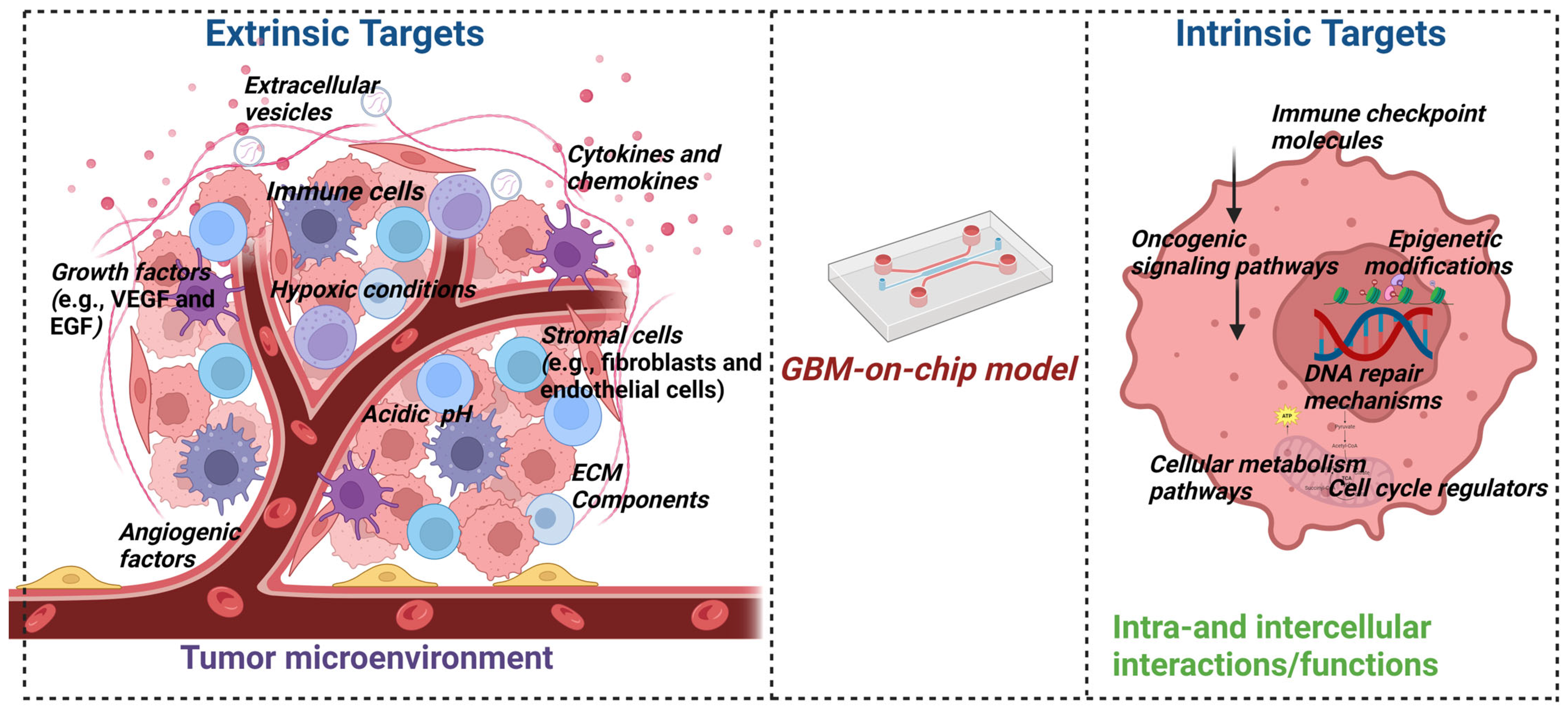
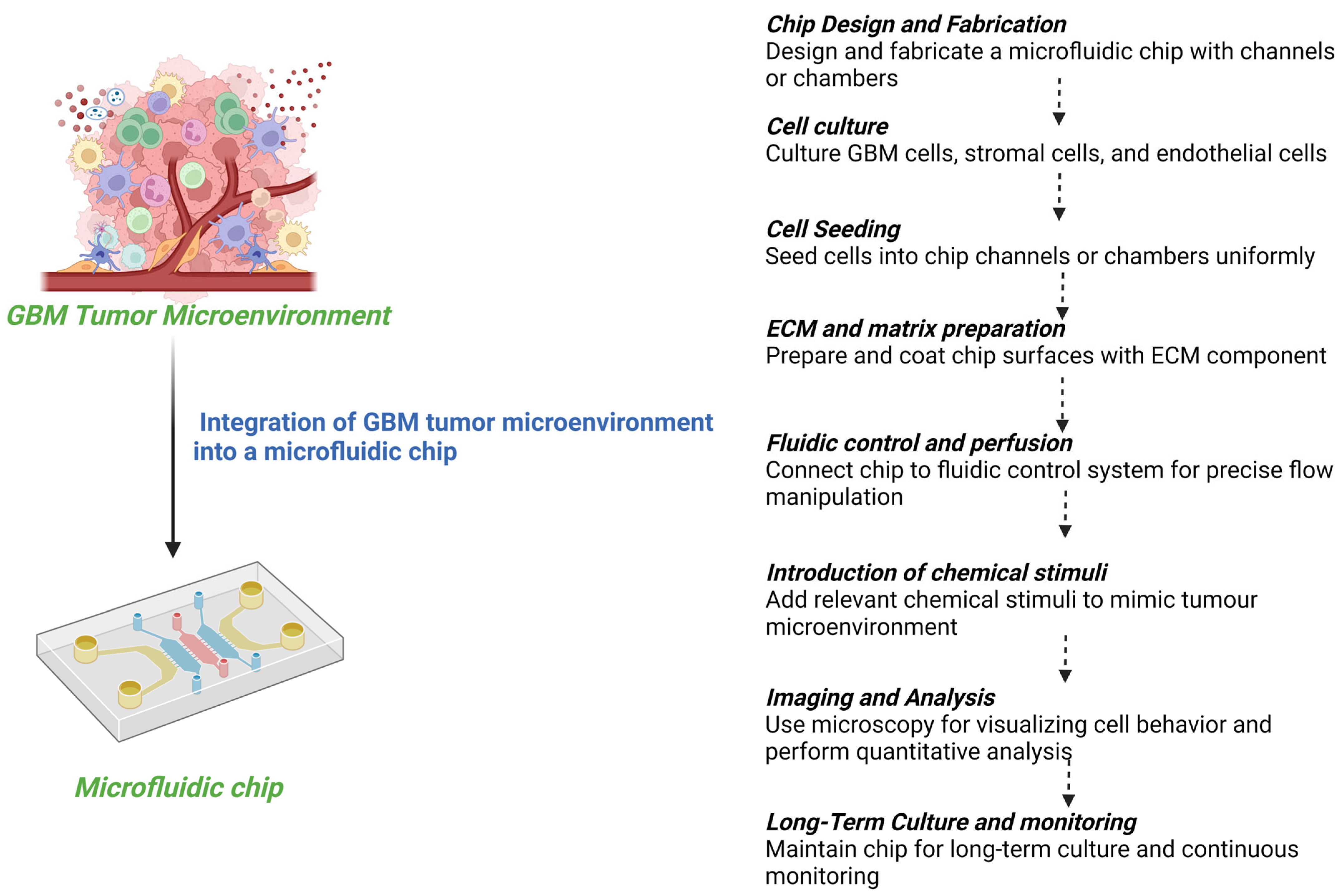

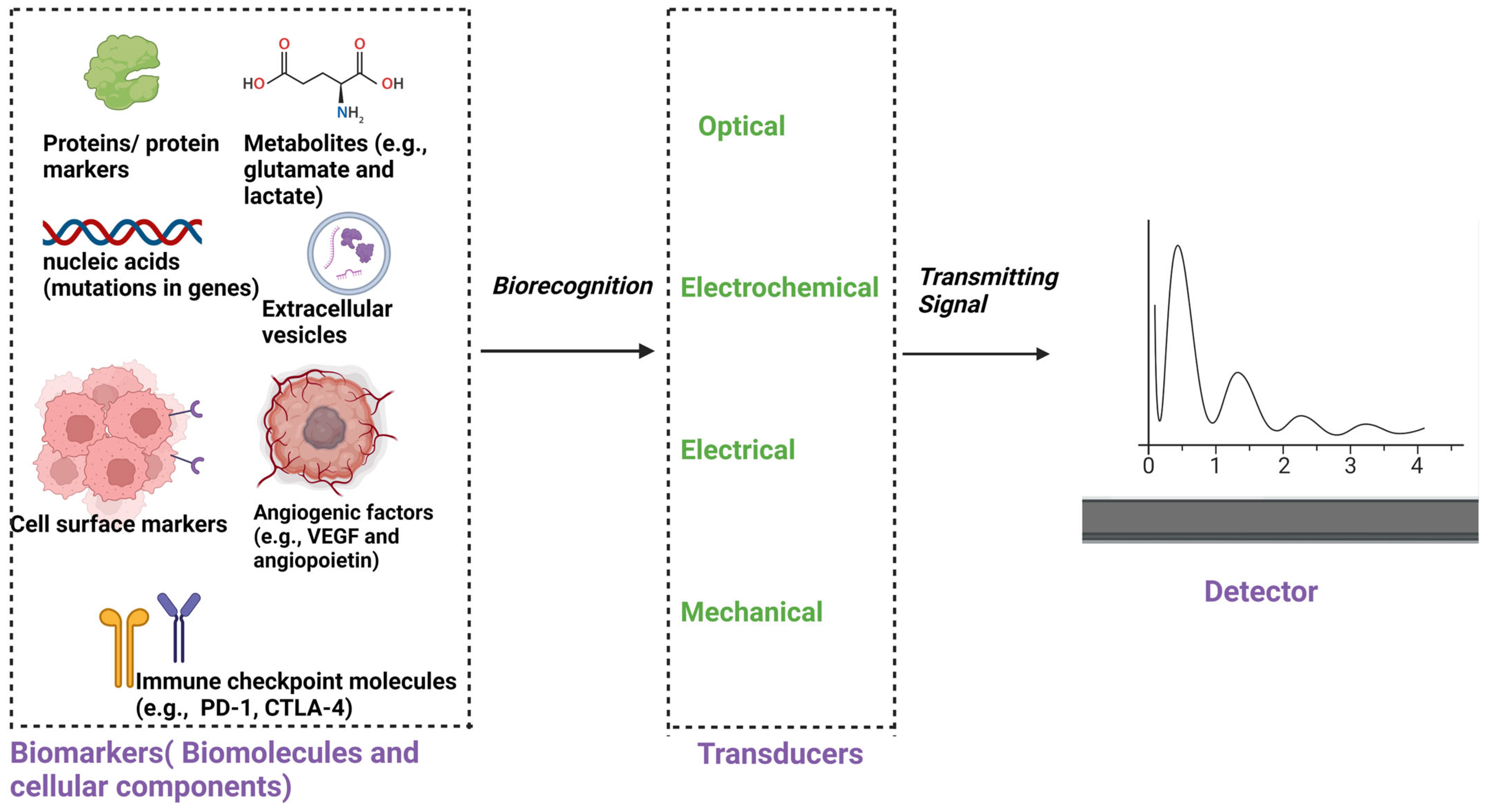


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ECM Component | Role in GBM Pathogenesis | References |
|---|---|---|
| Hyaluronic acid (HA) | Elevated expression levels correlate inversely with glioma patient prognosis. | [45] |
| Fibronectin | Enhances adherence of glioma stem-like cells, promotes glioma progression and immunosuppression. | [54,55,56,57,58,59,60,61] |
| Tenascin-C (TN-C) | Facilitates glioma cell invasion and migration through the ECM, modulated by the interleukin-33 (IL-33)-ST2-NFκB pathway. | [42,48,49,50,51,52,53] |
| Laminins | Promote glioma cell adhesion and invasion, implicated in glioma progression. | [64,65,66,67] |
| Collagen | Upregulated in GBMs, correlated with poor progression-free survival and overall survival, modulated by various receptors and pathways. | [69,70,71,72,73,74,75] |
| Matrix metalloproteinases (MMPs) | Degrade ECM proteins, promote cell migration, associated with glioma grading and development. | [76,77,78,79,80,81,82,83] |
| Other ECM proteins | Drives tumor progression by modulating Notch and NF-κB signaling pathways, correlates with poor patient survival. | [62,63] |
| Immune Cell Type | Role in GBM Microenvironment | Mechanisms/Functions | Targeted Therapeutic Approaches |
|---|---|---|---|
| Microglia | Predominant immune cell populations within the glioma microenvironment. Promote glioma proliferation and invasion [86]. |
| |
| Glioma-associated macrophages (GAMs) | Constitute up to 30–40% of the bulk tumor mass in GBM. Exert pro-tumorigenic activities by releasing various factors [86,87]. |
|
|
| Neutrophils | Subset of myeloid-derived suppressor cells exhibiting pro-tumorigenic properties. Contribute to tumor initiation, proliferation, and dissemination [113,114]. | ||
| Dendritic cells | Orchestrate the activation and regulation of immune effector cells. Likely participate in recognizing and presenting tumor antigens [121,123]. |
| |
| Natural killer (NK) cells | Exhibit innate anti-tumor activity. Sparse presence in the glioma microenvironment [132]. |
|
|
| Lymphocytes | Scarcity of infiltration in gliomas. CD8+ T cells associated with an improved prognosis [85,132]. |
|
| Neural Component | Role in GBM Microenvironment | Mechanisms/Interactions | References |
|---|---|---|---|
| Astrocytes | Disruption of blood–brain barrier integrity, secretion of RANKL, activation of SHH pathway, production of CTGF, release of CCL20. |
| [148,149,150,151,152,153] |
| Neurons | Regulation of PD-L1 signaling pathway, promotion of glioblastoma cell survival and proliferation. |
| [157,158] |
| Oligodendrocytes | Upregulation of GBM invasiveness through the angiopoietin-2 signaling pathway, facilitation of glioblastoma migration and invasion. |
| [159,160] |
| Paracrine interactions | Exchange of BDNF and NLGN3-mediated signals, regulation of BDNF expression by miRNAs, promotion of glioblastoma growth and survival. |
| [161,162,163,164,165] |
| Aspect | Hypoxia | Acidosis |
|---|---|---|
| Definition |
| |
| Etiology |
| |
| Cellular effects |
|
|
| Molecular mechanisms |
| |
| Therapeutic implications |
| |
| Clinical trials |
|
|
| Promising outcomes |
|
|
| Biosensors | Methods | Advantages | Disadvantages | Applications |
|---|---|---|---|---|
| Soft lithography | Microcontact printing (µCP) |
|
|
|
| Molding-based soft lithography |
|
|
| |
| Micromolding in capillaries (MIMIC) |
|
|
| |
| Embossing and replica molding |
|
|
| |
| 3D bioprinting | Extrusion-based bioprinting |
|
|
|
| Inkjet-based bioprinting |
|
|
| |
| Laser-based bioprinting |
|
|
|
| Platform | Characteristics | Advantages | Disadvantages |
|---|---|---|---|
| Microfluidic cell culture platforms |
|
|
|
| 3D microfluidic culture systems |
|
|
|
| Hydrogel-based microfluidic platforms |
|
|
|
| organ-on-chip systems |
|
|
|
| Microscale tissue engineering platforms |
|
|
|
| Droplet-based microfluidic platforms |
|
|
|
| 3D bioprinting platforms |
|
|
|
| Biosensing Detection Method | Principle | Targets | Advantages | Disadvantages |
|---|---|---|---|---|
| Electrochemical biosensors [294,295,296,297] | Detection based on electrical signal changes includes alterations in current, voltage, impedance, or capacitance, depending on the nature of the electrochemical transduction mechanism resulting from analyte binding to bioreceptors immobilized on electrodes. | Enzymes, DNA, antibodies, whole cells. |
|
|
| Optical biosensors [293,307] | Detection relies on optical signals generated by interactions between target analytes and recognition elements. These biosensors can be categorized into label-free and label-based models. Label-free models rely on inherent material properties for detection, such as light scattering and SPR. In contrast, label-based models utilize labeling reagents to produce detectable optical signals, such as fluorescence. | Enzymes, antibodies, antigens, nucleic acids, whole cells, tissues. |
|
|
| Electrical biosensors [315] | Detection based on changes in the electrical properties of sensing elements upon exposure to analytes. | Voltage changes, electrical impedance. |
|
|
| Mechanical biosensors [292] | Detection based on mechanical changes induced by analyte binding or cellular activities. | Mechanical stress. |
|
|
| Biosensor | Analyte | Main Function | Type | Reference | |
|---|---|---|---|---|---|
| Electrochemical | Glucose, lactate | Continuous monitoring of glucose consumption and lactate secretion | Colon cancer microtissues | [298] | |
| Oxygen, lactate, and glucose | Monitoring of energy metabolites and culture conditions in 3D cell cultures | Matrix-based, heterogeneous 3D cultures | [299] | ||
| Creatine kinase (CK)-MB biomarker | Monitoring biomarkers secreted from damaged cardiac tissue | Heart-on-a-chip platform | [302] | ||
| Heat shock protein 70 (HSP-70) biomarker | Immobilizing antiHSP70 antibodies on SPE | OOC platform | [303] | ||
| Interleukin 6 (IL-6) and tumor necrosis factor α (TNF-α) | Detection of IL-6 and TNF-α following electrical stimulation of skeletal muscle tissue | Integrated platform for muscle cell stimulation | [304] | ||
| Biochemical signals (nitric oxide, reactive oxygen species) | Monitoring biochemical signals during vascular mechanotransduction in real-time | Vascular chip model | [305] | ||
| Lactate dehydrogenase (LDH) | Measurement of LDH concentration, indicating cytotoxicity and cell damage | [306] | |||
| Optical | Tissue responses | Non-invasive monitoring of tissue-level remodeling | Synovium-on-a-chip | [307] | |
| Insulin secretion in response to glucose stimulation | Real-time detection of insulin secretion | Islets-on-a-chip | [311] | ||
| Oxygen consumption | Monitoring O2 consumption under electrical stimuli | SpheroFlow HoC system | [312] | ||
| Oxygen consumption | Monitoring O2 consumption to evaluate cardiac metabolism | Co-culture of mesenchymal stem cells and cardiac tissues | [314] | ||
| Electrical | TEER | Measuring TEER and cell layer capacitance | Human lung airway chip; human gut chip | [316] | |
| TEER | Real-time measurement for BBB-on-a-chip | hiPSC-derived blood–brain barrier chips | [317] | ||
| TEER | Monitoring reliably in the presence of microbubbles | Intestinal barrier-on-chip | [318] | ||
| Resistivity | Real-time monitoring of resistivity changes | A breathing lung-on-chip | [319] | ||
| Electrical signals | Measuring the electrical activity from neurons | TFT systems applicable to neuron culture | [320] | ||
| Contractile force, electrical conductivity | Monitoring the electrical activity of cardiomyocytes | Multi-organ human-on-a-chip system | [321] | ||
| Multiple biosensors | Electrical/optical | Oxygen, TEER | Monitoring oxygen levels and TEER | HuMiX OOC systems | [322] |
| Electrical/electrochemical | Oxygen, ROS | Monitoring oxygen levels and oxidative stress | Gut-on-a-chip models | [324] | |
| Electrical/electrochemical | TEER, glucose levels, ROS | Assessment of TEER, glucose levels, and ROS | Distal tubule-on-a-chip models | [326] | |
| Electrochemical/electrical | pH, cell proliferation | Monitoring pH and cell proliferation | Colorectal Perfused Tumoroid Platforms | [327] | |
| Optical/electrochemical | pH, TEER, biomarkers | Assessment of pH, TEER, and biomarkers | Lung cancer on a chip model | [328] | |
| Optical/electrochemical | Various biomarkers | Comprehensive toxicity assessment | Cancer, heart, and liver models | [329] | |
| Other types of biosensors | Electrochemical stripping analysis sensors | TEER, ion levels | Real-time measurements of TEER and ion levels | Cell culture platform | [332] |
| Micro-pump sensor array (MPSA) system | Various tissue parameters | Support various tissue models, including liver, vascular, gastrointestinal, and kidney chips | Various tissue models | [333] | |
| Phosphorescent microprobes and electrochemical sensors | Glucose, lactate, oxygen | Real-time monitoring of mitochondrial respiration and metabolic shifts from oxidative phosphorylation to anaerobic glycolysis | Liver-on-chip | [334] | |
| Clark-type oxygen microsensors | Oxygen | Long-term oxygen monitoring, implementation of the Ross principle | Various | [335] | |
| Electrochemically dissolved oxygen sensors | Oxygen | Local online monitoring of oxygen concentrations | Liver-on-chip | [336] | |
| Microfluidic pH sensor-embedded chip | pH levels | Real-time pH measurement using micro-cantilever | Microfluidic chip | [337] | |
| Microfluidic system with light addressable sensor (LAPS) | Extracellular acidification | Real-time detection of acidification | Modular microfluidic system | [338] | |
| Smart temperature sensors | Temperature | Real-time temperature monitoring | Silicon-based organ-on-a-chip device | [339] | |
| Resistive temperature sensors (RTDs) | Temperature | Accurate temperature monitoring and regulation | Organ chips | [340] | |
| Osmotic hydration sensor | Osmotic pressure | Monitor osmotic pressure | Microfluidic systems | [341] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thenuwara, G.; Javed, B.; Singh, B.; Tian, F. Biosensor-Enhanced Organ-on-a-Chip Models for Investigating Glioblastoma Tumor Microenvironment Dynamics. Sensors 2024, 24, 2865. https://doi.org/10.3390/s24092865
Thenuwara G, Javed B, Singh B, Tian F. Biosensor-Enhanced Organ-on-a-Chip Models for Investigating Glioblastoma Tumor Microenvironment Dynamics. Sensors. 2024; 24(9):2865. https://doi.org/10.3390/s24092865
Chicago/Turabian StyleThenuwara, Gayathree, Bilal Javed, Baljit Singh, and Furong Tian. 2024. "Biosensor-Enhanced Organ-on-a-Chip Models for Investigating Glioblastoma Tumor Microenvironment Dynamics" Sensors 24, no. 9: 2865. https://doi.org/10.3390/s24092865
APA StyleThenuwara, G., Javed, B., Singh, B., & Tian, F. (2024). Biosensor-Enhanced Organ-on-a-Chip Models for Investigating Glioblastoma Tumor Microenvironment Dynamics. Sensors, 24(9), 2865. https://doi.org/10.3390/s24092865