
Transient Exposure of Endothelial Cells to Doxorubicin Leads to Long-Lasting Vascular Endothelial Growth Factor Receptor 2 Downregulation



Abstract
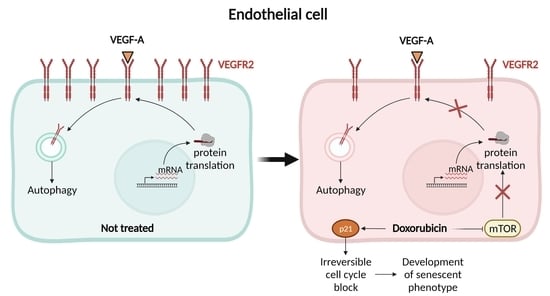
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Chemicals
2.2. Cell Death Assay
2.3. β-Galactosidase Assay
2.4. Real-Time Quantitative PCR
2.5. Immunoblotting
2.6. Statistical Analysis
3. Results
3.1. Transient Exposure of ECs to Doxorubicin Triggers Cellular Senescence That Is Sustained after Dox Removal
3.2. Transient Exposure of ECs to Doxorubicin Leads to Reduced VEGFR2 Level
3.3. Doxorubicin Enhances Autophagy in ECs
3.4. Doxorubicin Inhibits Global Protein Synthesis through the mTOR Axis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nebigil, C.G.; Desaubry, L. Updates in anthracycline-mediated cardiotoxicity. Front. Pharmacol. 2018, 9, 1262. [Google Scholar] [CrossRef] [Green Version]
- Wallace, K.B.; Sardao, V.A.; Oliveira, P.J. Mitochondrial determinants of doxorubicin-induced cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Cappetta, D.; Rossi, F.; Piegari, E.; Quaini, F.; Berrino, L.; Urbanek, K.; De Angelis, A. Doxorubicin targets multiple players: A new view of an old problem. Pharmacol. Res. 2018, 127, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, E.L.; Sidaway, J.E.; Cross, M.J. Cardiotoxic drugs Herceptin and doxorubicin inhibit cardiac microvascular endothelial cell barrier formation resulting in increased drug permeability. Biol. Open 2016, 5, 1362–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahn, S.K.; Hennicke, T.; Kassack, M.U.; Drews, L.; Reichert, A.S.; Fritz, G. Distinct influence of the anthracycline derivative doxorubicin on the differentiation efficacy of mESC-derived endothelial progenitor cells. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118711. [Google Scholar] [CrossRef]
- Sun, Z.; Schriewer, J.; Tang, M.; Marlin, J.; Taylor, F.; Shohet, R.V.; Konorev, E.A. The TGF-β pathway mediates doxorubicin effects on cardiac endothelial cells. J. Mol. Cell. Cardiol. 2016, 90, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Luu, A.Z.; Luu, V.Z.; Chowdhury, B.; Kosmopoulos, A.; Pan, Y.; Al-Omran, M.; Quan, A.; Teoh, H.; Hess, D.A.; Verma, S. Loss of endothelial cell-specific autophagy-related protein 7 exacerbates doxorubicin-induced cardiotoxicity. Biochem. Biophys. Rep. 2021, 25, 100926. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bove, A.M.; Simone, G.; Ma, B. Molecular bases of VEGFR-2-mediated physiological function and pathological role. Front. Cell Dev. Biol. 2020, 8, 599281. [Google Scholar] [CrossRef]
- Kivela, R.; Hemanthakumar, K.A.; Vaparanta, K.; Robciuc, M.; Izumiya, Y.; Kidoya, H.; Takakura, N.; Peng, X.; Sawyer, D.B.; Elenius, K.; et al. Endothelial cells regulate physiological cardiomyocyte growth via VEGFR2-mediated paracrine signaling. Circulation 2019, 139, 2570–2584. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.A.; Fearnley, G.W.; Tomlinson, D.C.; Harrison, M.A.; Ponnambalam, S. The cellular response to vascular endothelial growth factors requires co-ordinated signal transduction, trafficking and proteolysis. Biosci. Rep. 2015, 35, e00253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, M.R.; Ho, S.H.; Owen, D.A.; Tai, I.T. Inhibition of VEGF induces cellular senescence in colorectal cancer cells. Int. J. Cancer 2011, 129, 2115–2123. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mizushima, N. LC3- and p62-based biochemical methods for the analysis of autophagy progression in mammalian cells. Methods 2015, 75, 13–18. [Google Scholar] [CrossRef]
- Bruns, A.F.; Herbert, S.P.; Odell, A.F.; Jopling, H.M.; Hooper, N.M.; Zachary, I.C.; Walker, J.H.; Ponnambalam, S. Ligand-stimulated VEGFR2 signaling is regulated by co-ordinated trafficking and proteolysis. Traffic 2010, 11, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Aviner, R. The science of puromycin: From studies of ribosome function to applications in biotechnology. Comput. Struct. Biotechnol. J. 2020, 18, 1074–1083. [Google Scholar] [CrossRef] [PubMed]
- Harvey, R.F.; Pöyry, T.A.A.; Stoneley, M.; Willis, A.E. Signaling from mTOR to eIF2α mediates cell migration in response to the chemotherapeutic doxorubicin. Sci. Signal. 2019, 12, aaw6763. [Google Scholar] [CrossRef] [PubMed]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the regulation of cellular senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [Green Version]
- Gewirtz, D.A. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem. Pharmacol. 1999, 57, 727–741. [Google Scholar] [CrossRef]
- Chiusa, M.; Hool, S.L.; Truetsch, P.; Djafarzadeh, S.; Jakob, S.M.; Seifriz, F.; Scherer, S.J.; Suter, T.M.; Zuppinger, C.; Zbinden, S. Cancer therapy modulates VEGF signaling and viability in adult rat cardiac microvascular endothelial cells and cardiomyocytes. J. Mol. Cell. Cardiol. 2012, 52, 1164–1175. [Google Scholar] [CrossRef]
- Bent, E.H.; Gilbert, L.A.; Hemann, M.T. A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes Dev. 2016, 30, 1811–1821. [Google Scholar] [CrossRef] [Green Version]
- Horton, L.E.; Bushell, M.; Barth-Baus, D.; Tilleray, V.J.; Clemens, M.J.; Hensold, J.O. p53 activation results in rapid dephosphorylation of the eIF4E-binding protein 4E-BP1, inhibition of ribosomal protein S6 kinase and inhibition of translation initiation. Oncogene 2002, 21, 5325–5334. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Xu, Y.; Wen, G.Z.; Wang, Q.; Yuan, S.M. Rapamycin suppresses angiogenesis and lymphangiogenesis in melanoma by downregulating VEGF-A/VEGFR-2 and VEGF-C/VEGFR-3 expression. OncoTargets Ther. 2019, 12, 4643–4654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ash, D.; Sudhahar, V.; Youn, S.W.; Okur, M.N.; Das, A.; O’Bryan, J.P.; McMenamin, M.; Hou, Y.; Kaplan, J.H.; Fukai, T.; et al. The P-type ATPase transporter ATP7A promotes angiogenesis by limiting autophagic degradation of VEGFR2. Nat. Commun. 2021, 12, 3091. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Jiang, L.; Li, Q.; Liu, X.; Zhang, T.; Yang, G.; Zhang, C.; Wang, N.; Sun, X.; Jiang, L. Pyrroloquinoline quinine ameliorates doxorubicin-induced autophagy-dependent apoptosis via lysosomal-mitochondrial axis in vascular endothelial cells. Toxicology 2019, 425, 152238. [Google Scholar] [CrossRef]
- Park, M.; Kim, J.Y.; Kim, J.; Lee, J.-H.; Kwon, Y.-G.; Kim, Y.-M. Low-dose metronomic doxorubicin inhibits mobilization and differentiation of endothelial progenitor cells through REDD1-mediated VEGFR-2 downregulation. BMB Rep. 2021, 54, 470–475. [Google Scholar] [CrossRef]
- Hemanthakumar, K.A.; Kivela, R. Angiogenesis and angiocrines regulating heart growth. Vasc. Biol. 2020, 2, R93–R104. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Graziani, S.; Scorrano, L.; Pontarin, G. Transient Exposure of Endothelial Cells to Doxorubicin Leads to Long-Lasting Vascular Endothelial Growth Factor Receptor 2 Downregulation. Cells 2022, 11, 210. https://doi.org/10.3390/cells11020210
Graziani S, Scorrano L, Pontarin G. Transient Exposure of Endothelial Cells to Doxorubicin Leads to Long-Lasting Vascular Endothelial Growth Factor Receptor 2 Downregulation. Cells. 2022; 11(2):210. https://doi.org/10.3390/cells11020210
Chicago/Turabian StyleGraziani, Silvia, Luca Scorrano, and Giovanna Pontarin. 2022. "Transient Exposure of Endothelial Cells to Doxorubicin Leads to Long-Lasting Vascular Endothelial Growth Factor Receptor 2 Downregulation" Cells 11, no. 2: 210. https://doi.org/10.3390/cells11020210
APA StyleGraziani, S., Scorrano, L., & Pontarin, G. (2022). Transient Exposure of Endothelial Cells to Doxorubicin Leads to Long-Lasting Vascular Endothelial Growth Factor Receptor 2 Downregulation. Cells, 11(2), 210. https://doi.org/10.3390/cells11020210