
Chromatin Structure and Dynamics: Focus on Neuronal Differentiation and Pathological Implication


Abstract
:1. Introduction
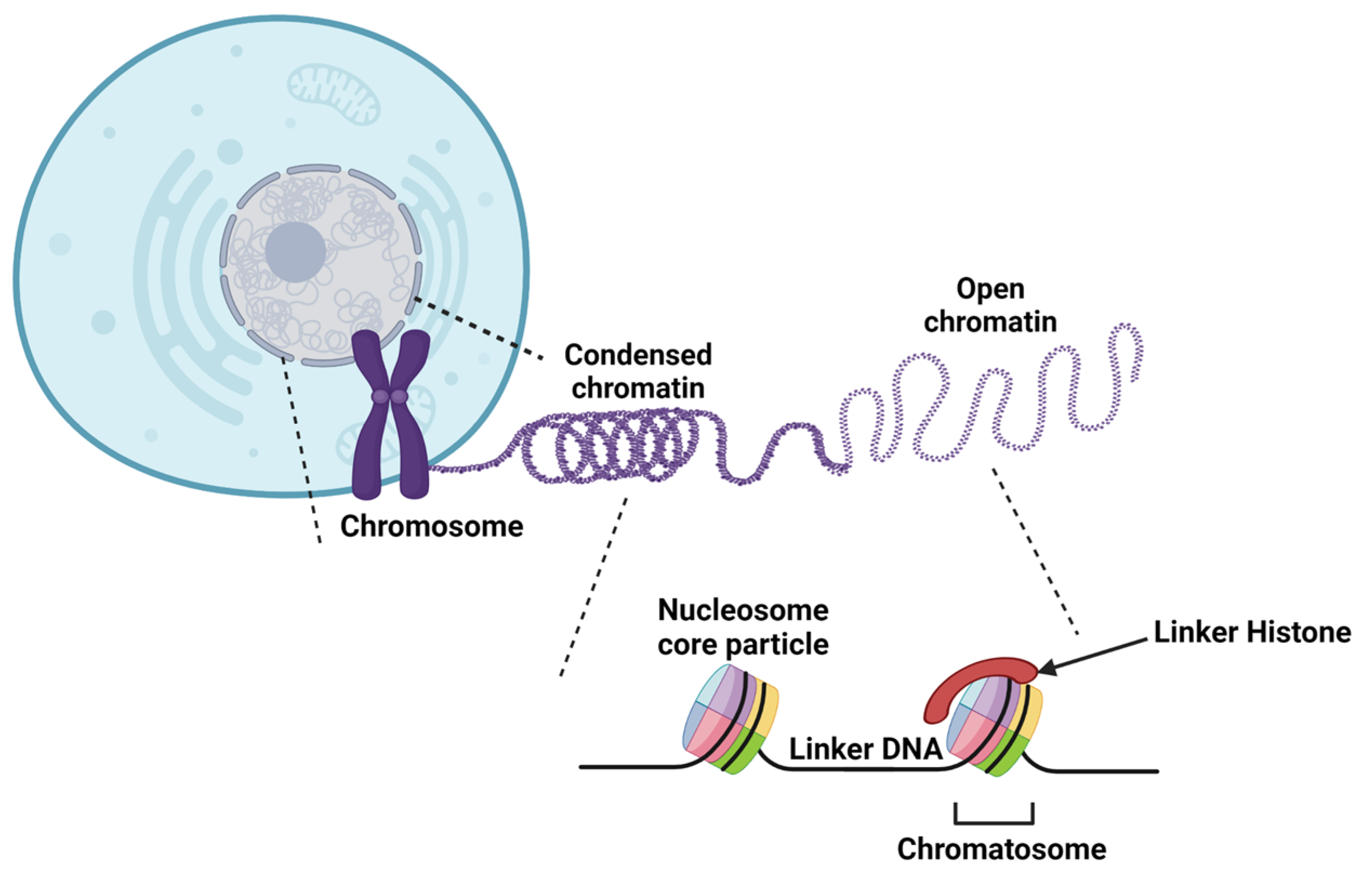
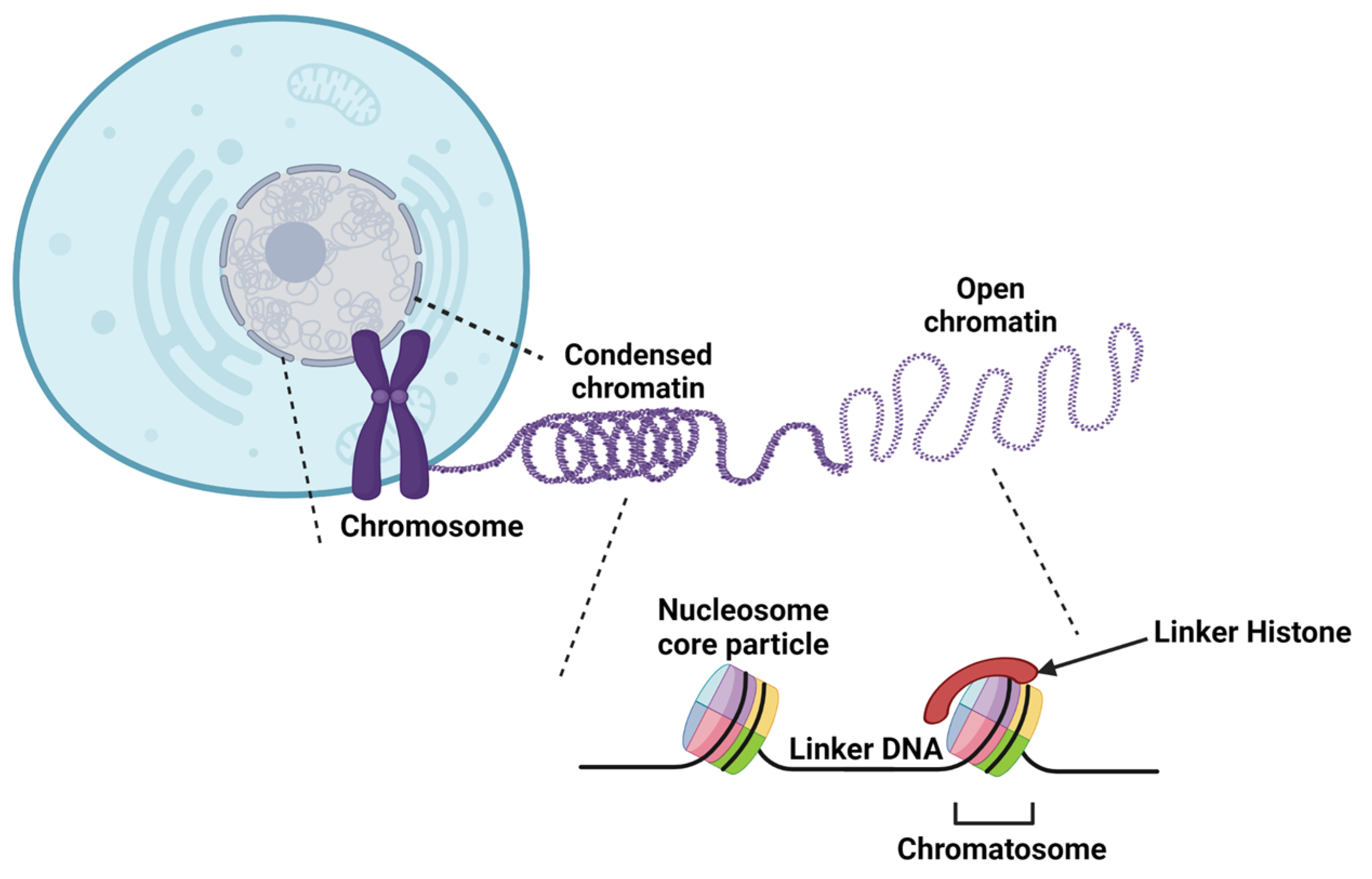
2. Chromatin Structure
2.1. Heterochromatin and Euchromatin
2.2. DNA Compaction
2.3. Histone Structure and Variants
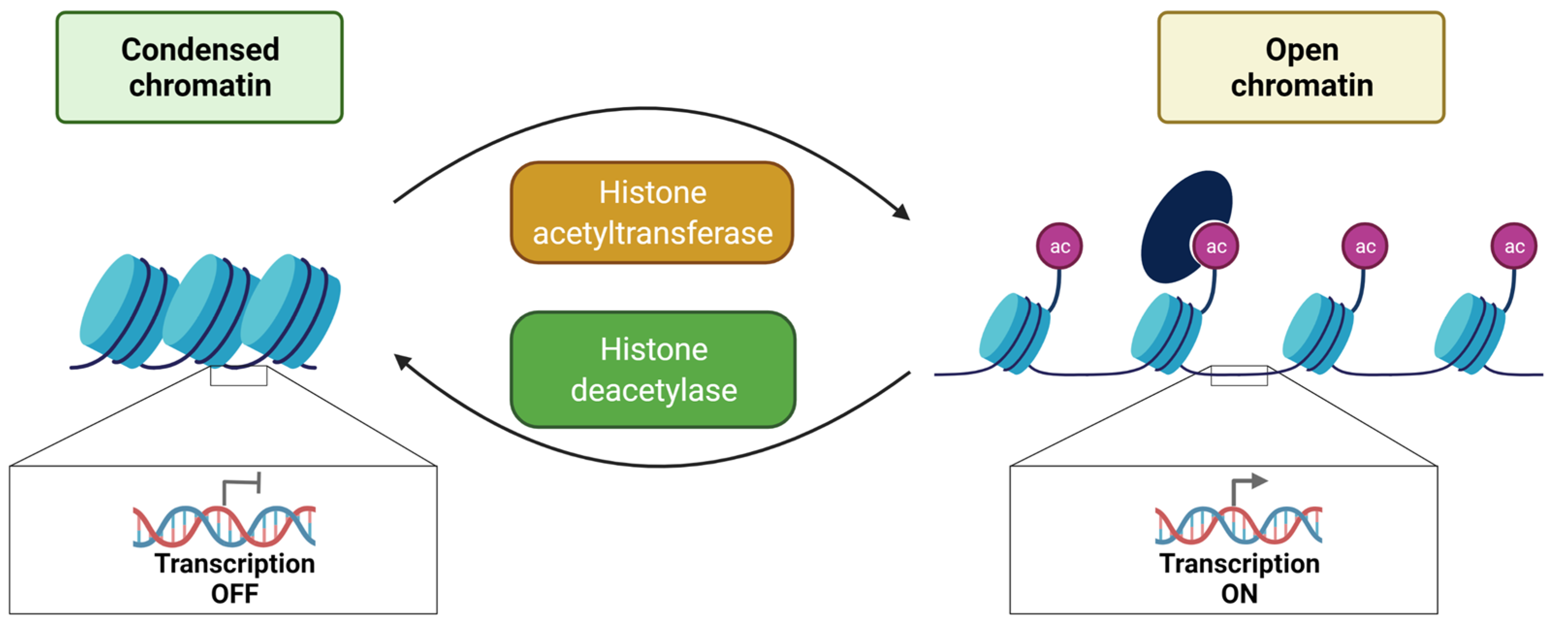
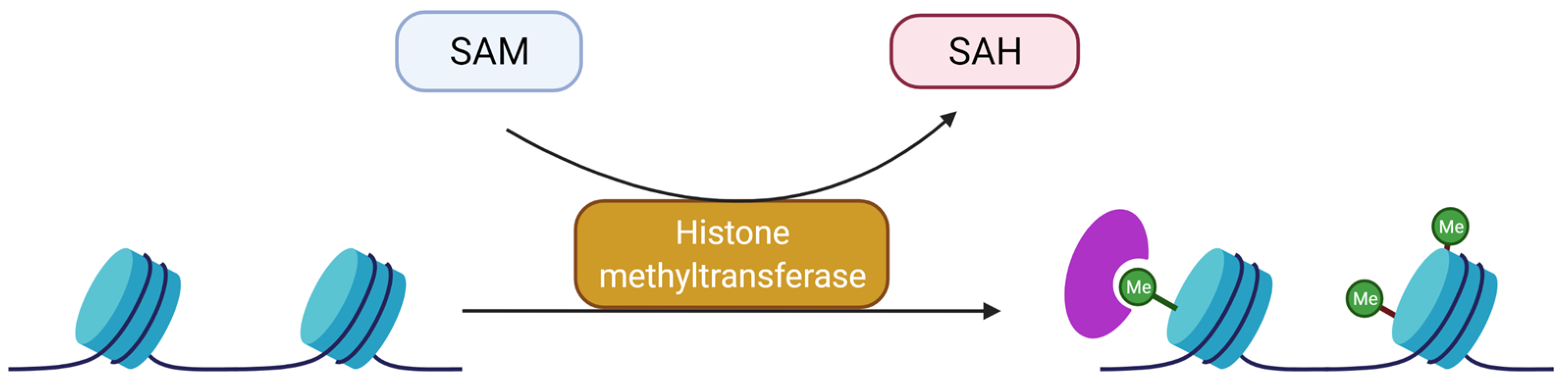
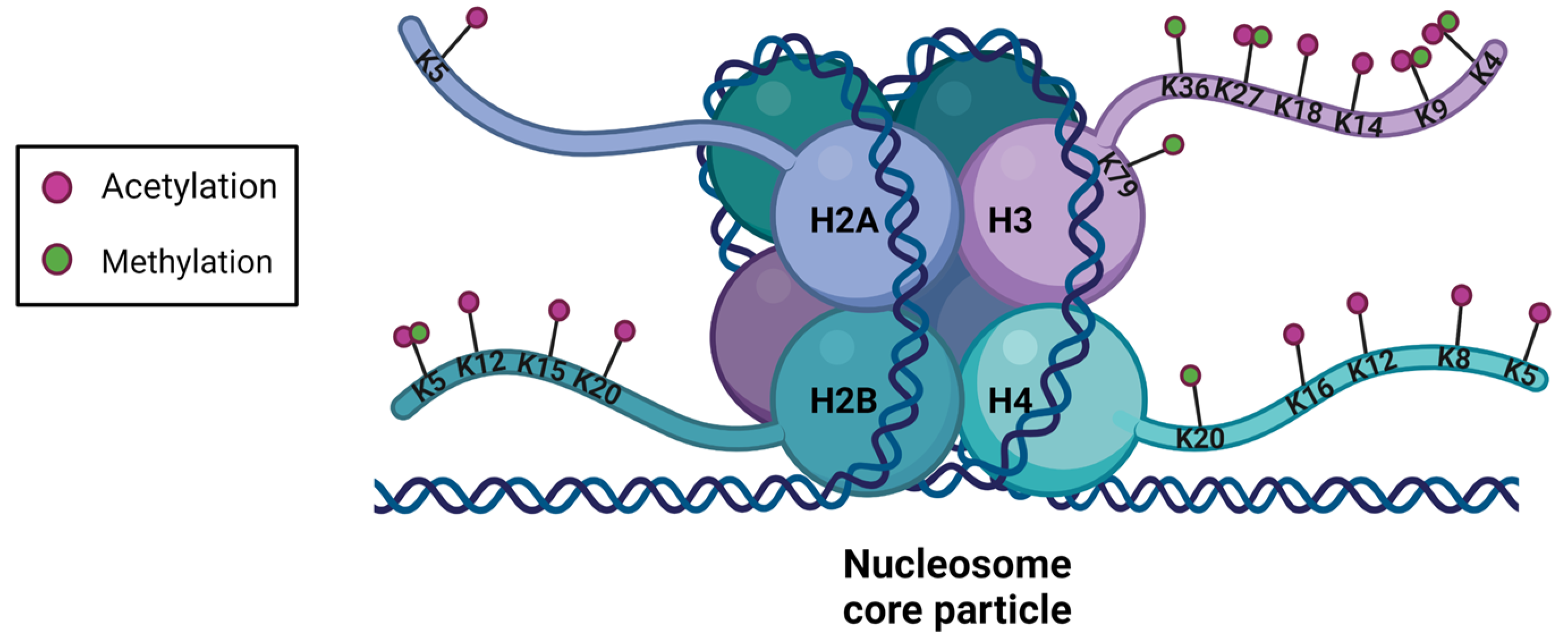
3. Histone Post-Translational Modifications
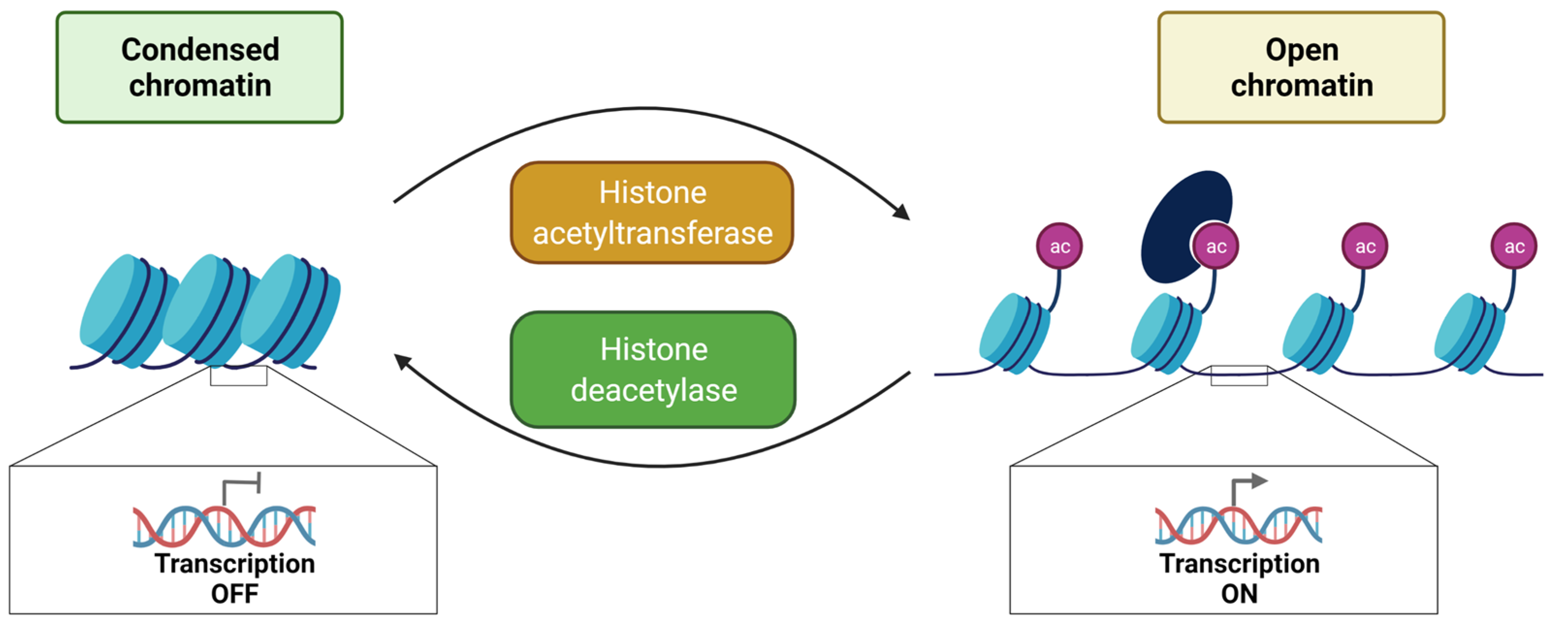
3.1. Histone Acetylation and Deacetylation
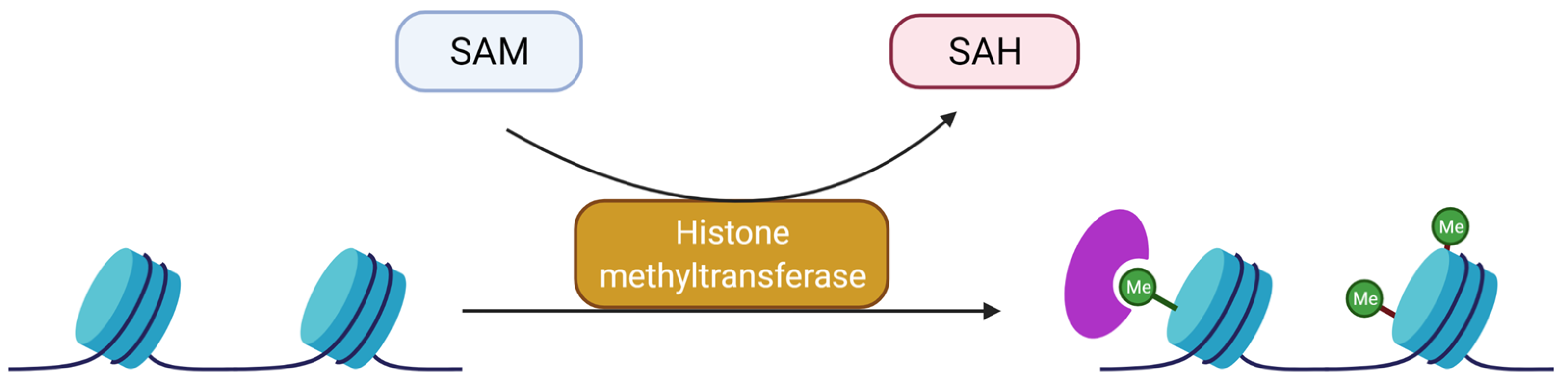
3.2. Methylation and Demethylation of Histones
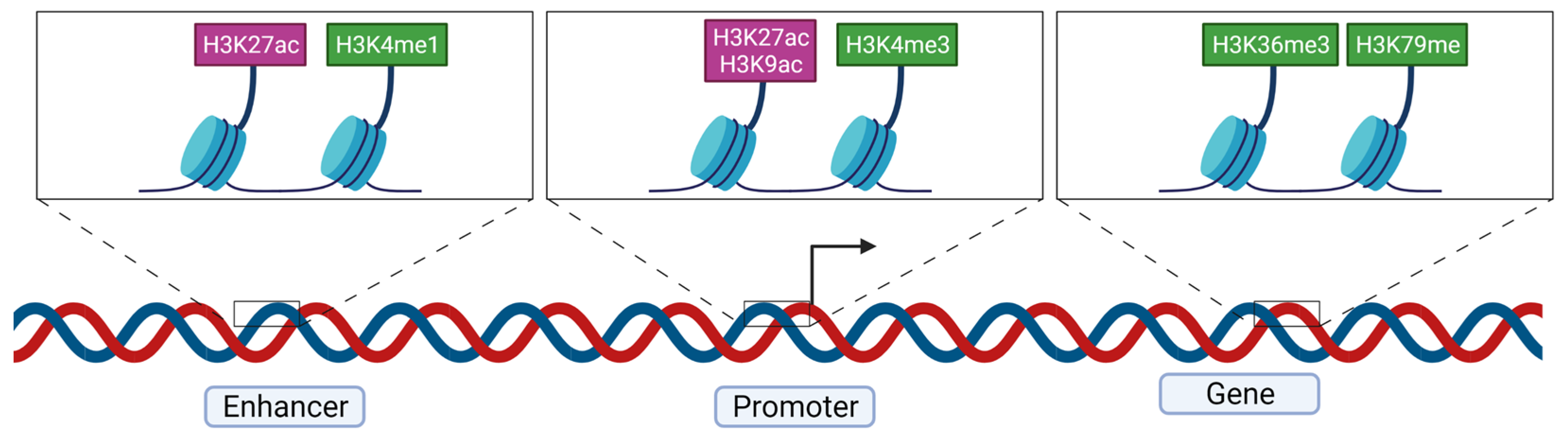
3.3. Main Active and Repressive Histone Marks
3.3.1. Repressive Marks
3.3.2. Active Marks
4. Histone Modification Analysis Techniques
4.1. ATAC-Seq Method
4.2. Targeted Chromatin Mapping Methods
4.2.1. ChIP-Seq
4.2.2. CUT and RUN
4.2.3. CUT and TAG
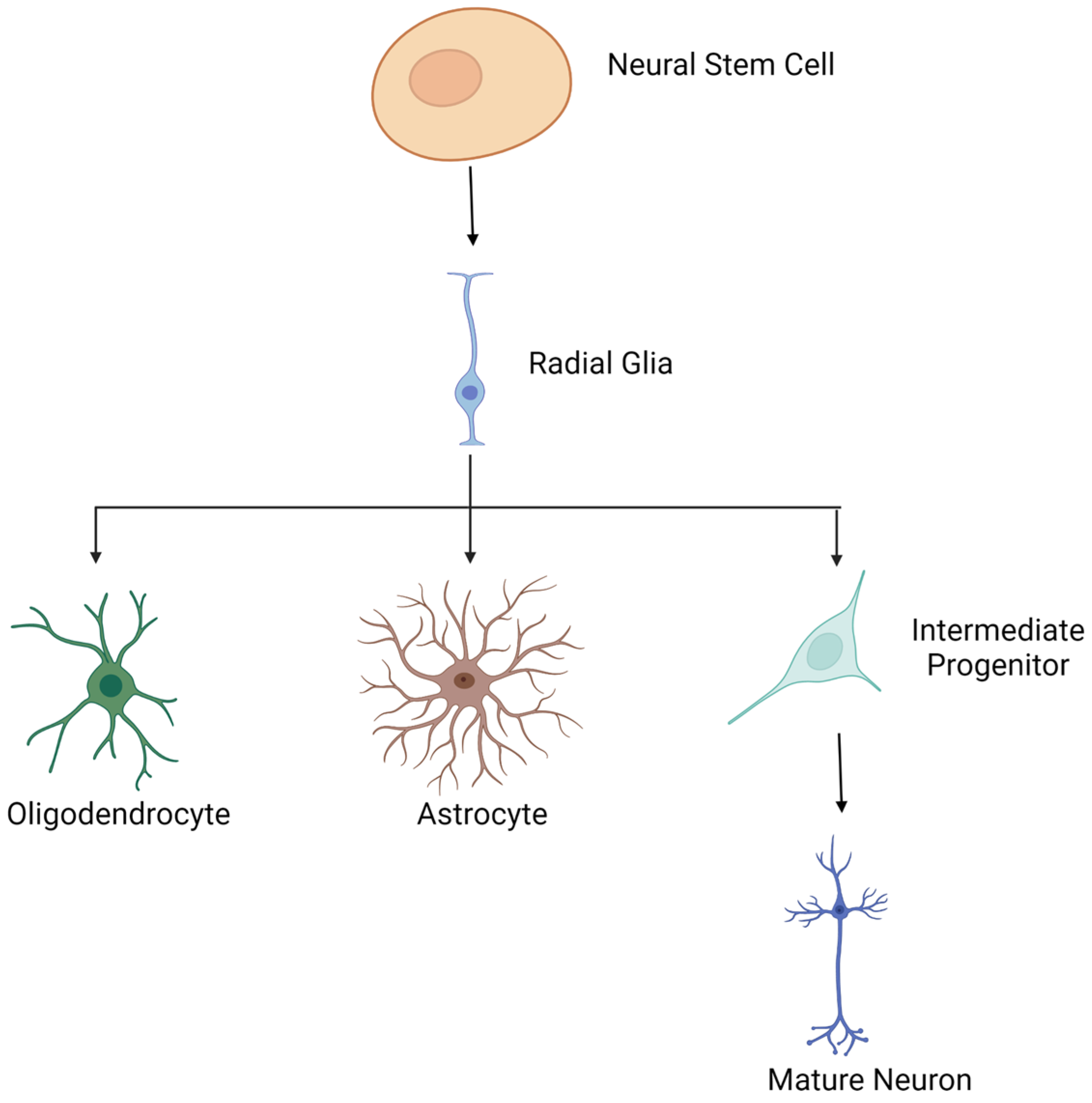
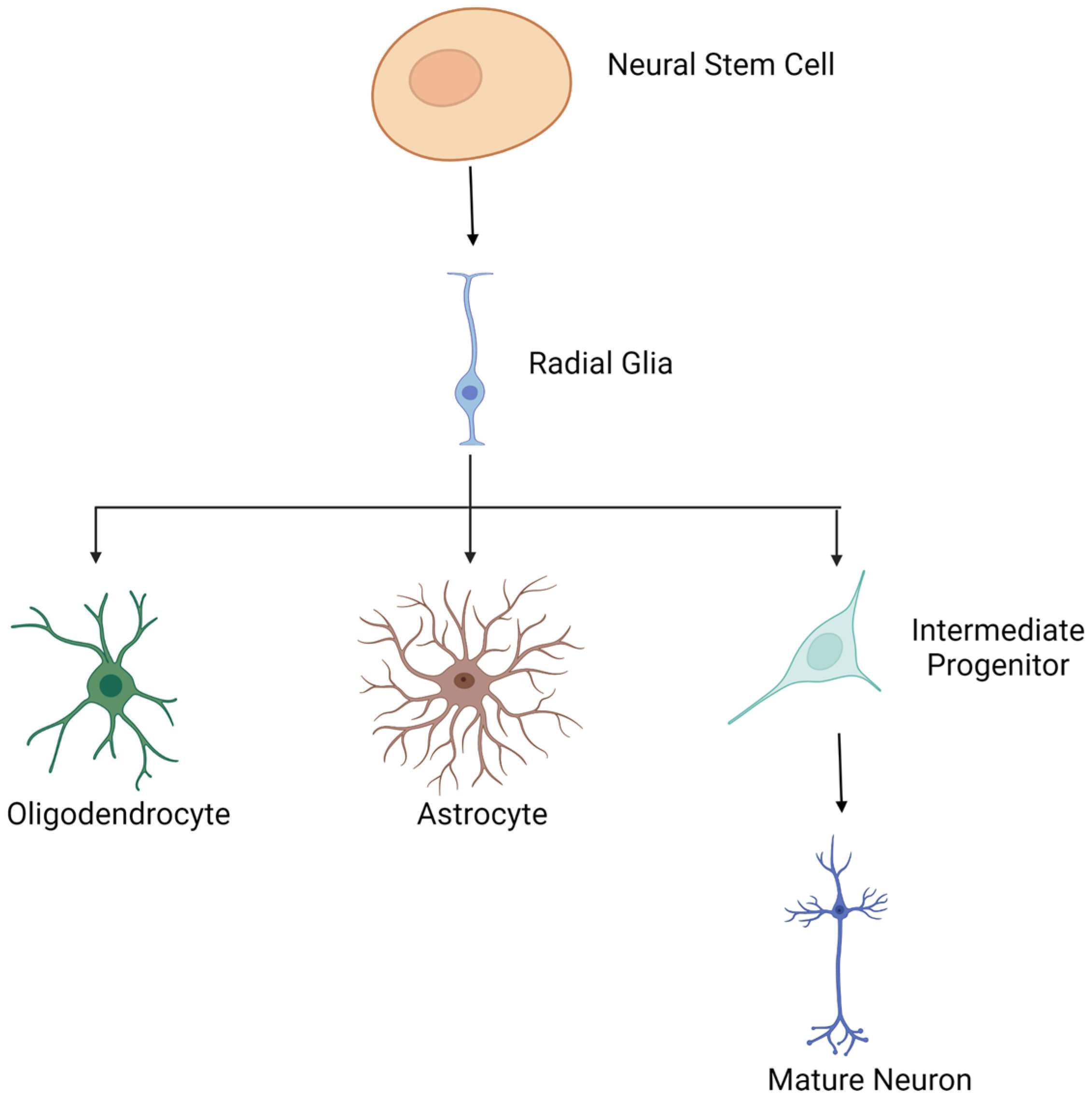
5. Histones and Neuronal Differentiation
5.1. Overview of Neuronal Differentiation
5.2. Models for Studying Histone Modifications during Neuronal Differentiation
5.3. Histone Changes
5.3.1. Role of Histone Acetylation and Deacetylation
5.3.2. Role of Methylation and Demethylation
5.3.3. Modifications of 3D Architecture of Chromatin during Neuronal Differentiation
6. Pathological Involvement of the Epigenetic Machinery
Chromatinopathies and Neurodevelopment
7. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Gross, D.S.; Chowdhary, S.; Anandhakumar, J.; Kainth, A.S. Chromatin. Curr. Biol. 2015, 25, R1158–R1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waddington, C.H. The Epigenotype. 1942. Int. J. Epidemiol. 2012, 41, 10–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahrner, J.A.; Bjornsson, H.T. Mendelian Disorders of the Epigenetic Machinery: Tipping the Balance of Chromatin States. Annu. Rev. Genom. Hum. Genet. 2014, 15, 269–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Ming, G.; Song, H. Adult Neurogenesis in the Mammalian Central Nervous System. Annu. Rev. Neurosci. 2005, 28, 223–250. [Google Scholar] [CrossRef]
- Matsuda, T.; Irie, T.; Katsurabayashi, S.; Hayashi, Y.; Nagai, T.; Hamazaki, N.; Adefuin, A.M.D.; Miura, F.; Ito, T.; Kimura, H.; et al. Pioneer Factor NeuroD1 Rearranges Transcriptional and Epigenetic Profiles to Execute Microglia-Neuron Conversion. Neuron 2019, 101, 472–485.e7. [Google Scholar] [CrossRef] [Green Version]
- Yu, I.T.; Park, J.-Y.; Kim, S.H.; Lee, J.; Kim, Y.-S.; Son, H. Valproic Acid Promotes Neuronal Differentiation by Induction of Proneural Factors in Association with H4 Acetylation. Neuropharmacology 2009, 56, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Hershey, A.D.; Chase, M. Independent Functions of Viral Protein and Nucleic Acid in Growth of Bacteriophage. J. Gen. Physiol. 1952, 36, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Chromosomal DNA and Its Packaging in the Chromatin Fiber. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Woodcock, C.L.; Ghosh, R.P. Chromatin Higher-Order Structure and Dynamics. Cold Spring Harb. Perspect. Biol. 2010, 2, a000596. [Google Scholar] [CrossRef] [PubMed]
- Ehrenhofer-Murray, A.E. Chromatin Dynamics at DNA Replication, Transcription and Repair. Eur. J. Biochem. 2004, 271, 2335–2349. [Google Scholar] [CrossRef]
- Passarge, E. Emil Heitz and the Concept of Heterochromatin: Longitudinal Chromosome Differentiation Was Recognized Fifty Years Ago. Am. J. Hum. Genet. 1979, 31, 106–115. [Google Scholar]
- Murakami, Y. Phosphorylation of Repressive Histone Code Readers by Casein Kinase 2 Plays Diverse Roles in Heterochromatin Regulation. J. Biochem. 2019, 166, 3–6. [Google Scholar] [CrossRef]
- Machida, S.; Takizawa, Y.; Ishimaru, M.; Sugita, Y.; Sekine, S.; Nakayama, J.; Wolf, M.; Kurumizaka, H. Structural Basis of Heterochromatin Formation by Human HP1. Mol. Cell 2018, 69, 385–397.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnstone, S.E.; Reyes, A.; Qi, Y.; Adriaens, C.; Hegazi, E.; Pelka, K.; Chen, J.H.; Zou, L.S.; Drier, Y.; Hecht, V.; et al. Large-Scale Topological Changes Restrain Malignant Progression in Colorectal Cancer. Cell 2020, 182, 1474–1489.e23. [Google Scholar] [CrossRef] [PubMed]
- Solovei, I.; Kreysing, M.; Lanctôt, C.; Kösem, S.; Peichl, L.; Cremer, T.; Guck, J.; Joffe, B. Nuclear Architecture of Rod Photoreceptor Cells Adapts to Vision in Mammalian Evolution. Cell 2009, 137, 356–368. [Google Scholar] [CrossRef] [Green Version]
- Jost, D.; Vaillant, C. Epigenomics in 3D: Importance of Long-Range Spreading and Specific Interactions in Epigenomic Maintenance. Nucleic Acids Res. 2018, 46, 2252–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falk, M.; Feodorova, Y.; Naumova, N.; Imakaev, M.; Lajoie, B.R.; Leonhardt, H.; Joffe, B.; Dekker, J.; Fudenberg, G.; Solovei, I.; et al. Heterochromatin Drives Compartmentalization of Inverted and Conventional Nuclei. Nature 2019, 570, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, C.L.; Frado, L.L.; Hatch, C.L.; Ricciardiello, L. Fine Structure of Active Ribosomal Genes. Chromosoma 1976, 58, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D. Chromatin Structure: A Repeating Unit of Histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef]
- Finch, J.T.; Lutter, L.C.; Rhodes, D.; Brown, R.S.; Rushton, B.; Levitt, M.; Klug, A. Structure of Nucleosome Core Particles of Chromatin. Nature 1977, 269, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal Structure of the Nucleosome Core Particle at 2.8 Å Resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Arents, G.; Burlingame, R.W.; Wang, B.C.; Love, W.E.; Moudrianakis, E.N. The Nucleosomal Core Histone Octamer at 3.1 A Resolution: A Tripartite Protein Assembly and a Left-Handed Superhelix. Proc. Natl. Acad. Sci. USA 1991, 88, 10148–10152. [Google Scholar] [CrossRef] [Green Version]
- White, C.L.; Suto, R.K.; Luger, K. Structure of the Yeast Nucleosome Core Particle Reveals Fundamental Changes in Internucleosome Interactions. EMBO J. 2001, 20, 5207–5218. [Google Scholar] [CrossRef] [PubMed]
- Kalashnikova, A.A.; Porter-Goff, M.E.; Muthurajan, U.M.; Luger, K.; Hansen, J.C. The Role of the Nucleosome Acidic Patch in Modulating Higher Order Chromatin Structure. J. R. Soc. Interface 2013, 10, 20121022. [Google Scholar] [CrossRef] [Green Version]
- Davey, C.A.; Sargent, D.F.; Luger, K.; Maeder, A.W.; Richmond, T.J. Solvent Mediated Interactions in the Structure of the Nucleosome Core Particle at 1.9Å Resolution††We Dedicate This Paper to the Memory of Max Perutz Who Was Particularly Inspirational and Supportive to T.J.R. in the Early Stages of This Study. J. Mol. Biol. 2002, 319, 1097–1113. [Google Scholar] [CrossRef]
- Simpson, R.T. Structure of the Chromatosome, a Chromatin Particle Containing 160 Base Pairs of DNA and All the Histones. Biochemistry 1978, 17, 5524–5531. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Feng, C.; Gao, T.; Mu, G.; Zhu, W.; Yang, Y. Linker Histone in Diseases. Int. J. Biol. Sci. 2017, 13, 1008–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fyodorov, D.V.; Zhou, B.-R.; Skoultchi, A.I.; Bai, Y. Emerging Roles of Linker Histones in Regulating Chromatin Structure and Function. Nat. Rev. Mol. Cell Biol. 2018, 19, 192–206. [Google Scholar] [CrossRef]
- Szerlong, H.J.; Herman, J.A.; Krause, C.M.; DeLuca, J.G.; Skoultchi, A.; Winger, Q.A.; Prenni, J.E.; Hansen, J.C. Proteomic Characterization of the Nucleolar Linker Histone H1 Interaction Network. J. Mol. Biol. 2015, 427, 2056–2071. [Google Scholar] [CrossRef] [Green Version]
- Finch, J.T.; Klug, A. Solenoidal Model for Superstructure in Chromatin. Proc. Natl. Acad. Sci. USA 1976, 73, 1897–1901. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; McGeehan, J.E.; Travers, A. A Metastable Structure for the Compact 30-Nm Chromatin Fibre. FEBS Lett. 2016, 590, 935–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, B.J.; Waxman, D.J. Computational Prediction of CTCF/Cohesin-Based Intra-TAD Loops That Insulate Chromatin Contacts and Gene Expression in Mouse Liver. eLife 2018, 7, e34077. [Google Scholar] [CrossRef]
- Gurard-Levin, Z.A.; Quivy, J.-P.; Almouzni, G. Histone Chaperones: Assisting Histone Traffic and Nucleosome Dynamics. Annu. Rev. Biochem. 2014, 83, 487–517. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, M.; Imam, N.; Verma, K.; Patel, A.K. Chromatin Remodelers: We Are the Drivers!! Nucleus 2016, 7, 388–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arents, G.; Moudrianakis, E.N. Topography of the Histone Octamer Surface: Repeating Structural Motifs Utilized in the Docking of Nucleosomal DNA. Proc. Natl. Acad. Sci. USA 1993, 90, 10489–10493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishnan, V.; Finch, J.T.; Graziano, V.; Lee, P.L.; Sweet, R.M. Crystal Structure of Globular Domain of Histone H5 and Its Implications for Nucleosome Binding. Nature 1993, 362, 219–223. [Google Scholar] [CrossRef] [PubMed]
- Marzluff, W.F.; Sanders, L.A.; Miller, D.M.; McCarty, K.S. Two Chemically and Metabolically Distinct Forms of Calf Thymus Histone F3. J. Biol. Chem. 1972, 247, 2026–2033. [Google Scholar] [CrossRef]
- Maze, I.; Noh, K.-M.; Soshnev, A.A.; Allis, C.D. Every Amino Acid Matters: Essential Contributions of Histone Variants to Mammalian Development and Disease. Nat. Rev. Genet. 2014, 15, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Vafa, O.; Shelby, R.D.; Sullivan, K.F. CENP-A Associated Complex Satellite DNA in the Kinetochore of the Indian Muntjac. Chromosoma 1999, 108, 367–374. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; Robert McDonald, E., III; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Gong, F.; Chiu, L.-Y.; Cox, B.; Aymard, F.; Clouaire, T.; Leung, J.W.; Cammarata, M.; Perez, M.; Agarwal, P.; Brodbelt, J.S.; et al. Screen Identifies Bromodomain Protein ZMYND8 in Chromatin Recognition of Transcription-Associated DNA Damage That Promotes Homologous Recombination. Genes Dev. 2015, 29, 197–211. [Google Scholar] [CrossRef] [Green Version]
- Gong, F.; Clouaire, T.; Aguirrebengoa, M.; Legube, G.; Miller, K.M. Histone Demethylase KDM5A Regulates the ZMYND8–NuRD Chromatin Remodeler to Promote DNA Repair. J. Cell Biol. 2017, 216, 1959–1974. [Google Scholar] [CrossRef] [Green Version]
- Kumbhar, R.; Sanchez, A.; Perren, J.; Gong, F.; Corujo, D.; Medina, F.; Devanathan, S.K.; Xhemalce, B.; Matouschek, A.; Buschbeck, M.; et al. Poly(ADP-Ribose) Binding and MacroH2A Mediate Recruitment and Functions of KDM5A at DNA Lesions. J. Cell Biol. 2021, 220, e202006149. [Google Scholar] [CrossRef]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. ACETYLATION AND METHYLATION OF HISTONES AND THEIR POSSIBLE ROLE IN THE REGULATION OF RNA SYNTHESIS. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutter, A.; Hayes, J.J. A Brief Review of Nucleosome Structure. FEBS Lett. 2015, 589, 2914–2922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef] [Green Version]
- Shiio, Y.; Eisenman, R.N. Histone Sumoylation Is Associated with Transcriptional Repression. Proc. Natl. Acad. Sci. USA 2003, 100, 13225–13230. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Zamudio, R.; Ha, H.C. Histone ADP-Ribosylation Facilitates Gene Transcription by Directly Remodeling Nucleosomes. Mol. Cell. Biol. 2012, 32, 2490–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagiwara, T.; Nakashima, K.; Hirano, H.; Senshu, T.; Yamada, M. Deimination of Arginine Residues in Nucleophosmin/B23 and Histones in HL-60 Granulocytes. Biochem. Biophys. Res. Commun. 2002, 290, 979–983. [Google Scholar] [CrossRef]
- Cuthbert, G.L.; Daujat, S.; Snowden, A.W.; Erdjument-Bromage, H.; Hagiwara, T.; Yamada, M.; Schneider, R.; Gregory, P.D.; Tempst, P.; Bannister, A.J.; et al. Histone Deimination Antagonizes Arginine Methylation. Cell 2004, 118, 545–553. [Google Scholar] [CrossRef] [Green Version]
- Okanishi, H.; Kim, K.; Masui, R.; Kuramitsu, S. Lysine Propionylation Is a Prevalent Post-Translational Modification in Thermus Thermophilus. Mol. Cell. Proteom. MCP 2014, 13, 2382–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone Hypercitrullination Mediates Chromatin Decondensation and Neutrophil Extracellular Trap Formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Strahl, B.D.; Allis, C.D. The Language of Covalent Histone Modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Karlić, R.; Chung, H.-R.; Lasserre, J.; Vlahovicek, K.; Vingron, M. Histone Modification Levels Are Predictive for Gene Expression. Proc. Natl. Acad. Sci. USA 2010, 107, 2926–2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallah, M.S.; Szarics, D.; Robson, C.M.; Eubanks, J.H. Impaired Regulation of Histone Methylation and Acetylation Underlies Specific Neurodevelopmental Disorders. Front. Genet. 2021, 11, 613098. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Berger, S.L.; Cote, J.; Dent, S.; Jenuwien, T.; Kouzarides, T.; Pillus, L.; Reinberg, D.; Shi, Y.; Shiekhattar, R.; et al. New Nomenclature for Chromatin-Modifying Enzymes. Cell 2007, 131, 633–636. [Google Scholar] [CrossRef] [Green Version]
- Alaskhar Alhamwe, B.; Khalaila, R.; Wolf, J.; von Bülow, V.; Harb, H.; Alhamdan, F.; Hii, C.S.; Prescott, S.L.; Ferrante, A.; Renz, H.; et al. Histone Modifications and Their Role in Epigenetics of Atopy and Allergic Diseases. Allergy Asthma Clin. Immunol. Off. J. Can. Soc. Allergy Clin. Immunol. 2018, 14, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.; Rao, C.M. Epigenetic Tools (The Writers, The Readers and The Erasers) and Their Implications in Cancer Therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef] [PubMed]
- Borun, T.W.; Pearson, D.; Paik, W.K. Studies of Histone Methylation during the HeLa S-3 Cell Cycle. J. Biol. Chem. 1972, 247, 4288–4298. [Google Scholar] [CrossRef]
- Paik, W.K.; Kim, S. Enzymatic Demethylation of Calf Thymus Histones. Biochem. Biophys. Res. Commun. 1973, 51, 781–788. [Google Scholar] [CrossRef]
- Murray, K. The Occurrence of Epsilon-N-Methyl Lysine in Histones. Biochemistry 1964, 3, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Shi, Y. Histone Methylation: A Dynamic Mark in Health, Disease and Inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.-K.; Huang, Y.-Q.; Zou, Y.; Zheng, X.-K.; Ma, X.-D. Silencing of LSD1 Gene Modulates Histone Methylation and Acetylation and Induces the Apoptosis of JeKo-1 and MOLT-4 Cells. Int. J. Mol. Med. 2017, 40, 319–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franci, G.; Ciotta, A.; Altucci, L. The Jumonji Family: Past, Present and Future of Histone Demethylases in Cancer. Biomol. Concepts 2014, 5, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The Molecular Hallmarks of Epigenetic Control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Lachat, C.; Boyer-Guittaut, M.; Peixoto, P.; Hervouet, E. Epigenetic Regulation of EMT (Epithelial to Mesenchymal Transition) and Tumor Aggressiveness: A View on Paradoxical Roles of KDM6B and EZH2. Epigenomes 2018, 3, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margueron, R.; Reinberg, D. The Polycomb Complex PRC2 and Its Mark in Life. Nature 2011, 469, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sun, Z.; Jia, J.; Du, T.; Zhang, N.; Tang, Y.; Fang, Y.; Fang, D. Overview of Histone Modification. Adv. Exp. Med. Biol. 2021, 1283, 1–16. [Google Scholar] [CrossRef]
- Fischle, W.; Tseng, B.S.; Dormann, H.L.; Ueberheide, B.M.; Garcia, B.A.; Shabanowitz, J.; Hunt, D.F.; Funabiki, H.; Allis, C.D. Regulation of HP1-Chromatin Binding by Histone H3 Methylation and Phosphorylation. Nature 2005, 438, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Zegerman, P.; Partridge, J.F.; Miska, E.A.; Thomas, J.O.; Allshire, R.C.; Kouzarides, T. Selective Recognition of Methylated Lysine 9 on Histone H3 by the HP1 Chromo Domain. Nature 2001, 410, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Peters, A.H.F.M.; Mermoud, J.E.; O’Carroll, D.; Pagani, M.; Schweizer, D.; Brockdorff, N.; Jenuwein, T. Histone H3 Lysine 9 Methylation Is an Epigenetic Imprint of Facultative Heterochromatin. Nat. Genet. 2002, 30, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Rank, G.; Tan, Y.T.; Li, H.; Moritz, R.L.; Simpson, R.J.; Cerruti, L.; Curtis, D.J.; Patel, D.J.; Allis, C.D.; et al. PRMT5-Mediated Methylation of Histone H4R3 Recruits DNMT3A, Coupling Histone and DNA Methylation in Gene Silencing. Nat. Struct. Mol. Biol. 2009, 16, 304–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourmouli, N.; Jeppesen, P.; Mahadevhaiah, S.; Burgoyne, P.; Wu, R.; Gilbert, D.M.; Bongiorni, S.; Prantera, G.; Fanti, L.; Pimpinelli, S.; et al. Heterochromatin and Tri-Methylated Lysine 20 of Histone H4 in Animals. J. Cell Sci. 2004, 117, 2491–2501. [Google Scholar] [CrossRef] [Green Version]
- Local, A.; Huang, H.; Albuquerque, C.P.; Singh, N.; Lee, A.Y.; Wang, W.; Wang, C.; Hsia, J.E.; Shiau, A.K.; Ge, K.; et al. Identification of H3K4me1-Associated Proteins at Mammalian Enhancers. Nat. Genet. 2018, 50, 73–82. [Google Scholar] [CrossRef]
- Kim, T.; Buratowski, S. Dimethylation of H3K4 by Set1 Recruits the Set3 Histone Deacetylase Complex to 5′ Transcribed Regions. Cell 2009, 137, 259–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruthenburg, A.J.; Allis, C.D.; Wysocka, J. Methylation of Lysine 4 on Histone H3: Intricacy of Writing and Reading a Single Epigenetic Mark. Mol. Cell 2007, 25, 15–30. [Google Scholar] [CrossRef]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Kheradpour, P.; Zhang, Z.; Heravi-Moussavi, A.; Liu, Y.; Amin, V.; et al. Integrative Analysis of 111 Reference Human Epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karmodiya, K.; Krebs, A.R.; Oulad-Abdelghani, M.; Kimura, H.; Tora, L. H3K9 and H3K14 Acetylation Co-Occur at Many Gene Regulatory Elements, While H3K14ac Marks a Subset of Inactive Inducible Promoters in Mouse Embryonic Stem Cells. BMC Genom. 2012, 13, 424. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Wang, H.; Ng, H.H.; Erdjument-Bromage, H.; Tempst, P.; Struhl, K.; Zhang, Y. Methylation of H3-Lysine 79 Is Mediated by a New Family of HMTases without a SET Domain. Curr. Biol. 2002, 12, 1052–1058. [Google Scholar] [CrossRef] [Green Version]
- Ng, H.H.; Ciccone, D.N.; Morshead, K.B.; Oettinger, M.A.; Struhl, K. Lysine-79 of Histone H3 Is Hypomethylated at Silenced Loci in Yeast and Mammalian Cells: A Potential Mechanism for Position-Effect Variegation. Proc. Natl. Acad. Sci. USA 2003, 100, 1820–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, K.; Tellier, M.; Murphy, S. DOT1L and H3K79 Methylation in Transcription and Genomic Stability. Biomolecules 2018, 8, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, M.A.; Wu, X.; Li, A.X.; Hahn, T.; Pfeifer, G.P. Relationship between Gene Body DNA Methylation and Intragenic H3K9me3 and H3K36me3 Chromatin Marks. PLoS ONE 2011, 6, e18844. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Ma, Y.; Huang, M.; Liang, W.; Zhao, X.; Li, Q.; Wang, S.; Hu, Z.; He, L.; Gao, T.; et al. Asymmetrical Arginine Dimethylation of Histone H4 by 8-Oxog/OGG1/PRMT1 Is Essential for Oxidative Stress-Induced Transcription Activation. Free Radic. Biol. Med. 2021, 164, 175–186. [Google Scholar] [CrossRef]
- Völker-Albert, M.C.; Schmidt, A.; Forne, I.; Imhof, A. Analysis of Histone Modifications by Mass Spectrometry. Curr. Protoc. Protein Sci. 2018, 92, e54. [Google Scholar] [CrossRef] [PubMed]
- Zee, B.M.; Young, N.L.; Garcia, B.A. Quantitative Proteomic Approaches to Studying Histone Modifications. Curr. Chem. Genomics 2011, 5, 106–114. [Google Scholar] [CrossRef] [Green Version]
- O’Geen, H.; Echipare, L.; Farnham, P.J. Using ChIP-Seq Technology to Generate High-Resolution Profiles of Histone Modifications. Methods Mol. Biol. Clifton NJ 2011, 791, 265–286. [Google Scholar] [CrossRef] [Green Version]
- Buenrostro, J.; Wu, B.; Chang, H.; Greenleaf, W. ATAC-Seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr. Protoc. Mol. Biol. 2015, 109, 21–29. [Google Scholar] [CrossRef]
- Skene, P.J.; Henikoff, J.G.; Henikoff, S. Targeted in Situ Genome-Wide Profiling with High Efficiency for Low Cell Numbers. Nat. Protoc. 2018, 13, 1006–1019. [Google Scholar] [CrossRef] [PubMed]
- Kaya-Okur, H.S.; Wu, S.J.; Codomo, C.A.; Pledger, E.S.; Bryson, T.D.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. CUT&Tag for Efficient Epigenomic Profiling of Small Samples and Single Cells. Nat. Commun. 2019, 10, 1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goryshin, I.Y.; Reznikoff, W.S. Tn5 in Vitro Transposition*. J. Biol. Chem. 1998, 273, 7367–7374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, F.; Powell, D.R.; Curtis, D.J.; Wong, N.C. From Reads to Insight: A Hitchhiker’s Guide to ATAC-Seq Data Analysis. Genome Biol. 2020, 21, 22. [Google Scholar] [CrossRef] [PubMed]
- Skene, P.J.; Henikoff, S. An Efficient Targeted Nuclease Strategy for High-Resolution Mapping of DNA Binding Sites. eLife 2017, 6, e21856. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Mortazavi, A. Integrating ChIP-Seq with Other Functional Genomics Data. Brief. Funct. Genom. 2018, 17, 104–115. [Google Scholar] [CrossRef]
- Thibodeau, A.; Khetan, S.; Eroglu, A.; Tewhey, R.; Stitzel, M.L.; Ucar, D. CoRE-ATAC: A Deep Learning Model for the Functional Classification of Regulatory Elements from Single Cell and Bulk ATAC-Seq Data. PLoS Comput. Biol. 2021, 17, e1009670. [Google Scholar] [CrossRef]
- Martínez-Cerdeño, V.; Noctor, S.C. Neural Progenitor Cell Terminology. Front. Neuroanat. 2018, 12, 104. [Google Scholar] [CrossRef]
- Agirman, G.; Broix, L.; Nguyen, L. Cerebral Cortex Development: An Outside-in Perspective. FEBS Lett. 2017, 591, 3978–3992. [Google Scholar] [CrossRef] [Green Version]
- Noctor, S.C.; Martínez-Cerdeño, V.; Ivic, L.; Kriegstein, A.R. Cortical Neurons Arise in Symmetric and Asymmetric Division Zones and Migrate through Specific Phases. Nat. Neurosci. 2004, 7, 136–144. [Google Scholar] [CrossRef]
- Yao, B.; Christian, K.M.; He, C.; Jin, P.; Ming, G.; Song, H. Epigenetic Mechanisms in Neurogenesis. Nat. Rev. Neurosci. 2016, 17, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.S.; Hinds, J.W. Neurogenesis in the Adult Rat: Electron Microscopic Analysis of Light Radioautographs. Science 1977, 197, 1092–1094. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.S.; Wang, S.; Jiang, L.; Kang, J.; Benraiss, A.; Harrison-Restelli, C.; Fraser, R.A.; Couldwell, W.T.; Kawaguchi, A.; Okano, H.; et al. In Vitro Neurogenesis by Progenitor Cells Isolated from the Adult Human Hippocampus. Nat. Med. 2000, 6, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.A.; Su, Y.; Jimenez-Cyrus, D.; Patel, A.; Huang, N.; Morizet, D.; Lee, S.; Shah, R.; Ringeling, F.R.; Jain, R.; et al. A Common Embryonic Origin of Stem Cells Drives Developmental and Adult Neurogenesis. Cell 2019, 177, 654–668.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, G.J.; Zhou, Y.; Ito, S.; Bonaguidi, M.A.; Stein-O’Brien, G.; Kawasaki, N.; Modak, N.; Zhu, Y.; Ming, G.; Song, H. Latent Tri-Lineage Potential of Adult Hippocampal Neural Stem Cells Revealed by Nf1 Inactivation. Nat. Neurosci. 2015, 18, 1722–1724. [Google Scholar] [CrossRef] [Green Version]
- Bragado Alonso, S.; Reinert, J.K.; Marichal, N.; Massalini, S.; Berninger, B.; Kuner, T.; Calegari, F. An Increase in Neural Stem Cells and Olfactory Bulb Adult Neurogenesis Improves Discrimination of Highly Similar Odorants. EMBO J. 2019, 38, e98791. [Google Scholar] [CrossRef]
- Lin, L.; Yuan, J.; Sander, B.; Golas, M.M. In Vitro Differentiation of Human Neural Progenitor Cells Into Striatal GABAergic Neurons. Stem Cells Transl. Med. 2015, 4, 775–788. [Google Scholar] [CrossRef]
- Gantner, C.W.; Cota-Coronado, A.; Thompson, L.H.; Parish, C.L. An Optimized Protocol for the Generation of Midbrain Dopamine Neurons under Defined Conditions. STAR Protoc. 2020, 1, 100065. [Google Scholar] [CrossRef]
- Shi, Y.; Kirwan, P.; Livesey, F.J. Directed Differentiation of Human Pluripotent Stem Cells to Cerebral Cortex Neurons and Neural Networks. Nat. Protoc. 2012, 7, 1836–1846. [Google Scholar] [CrossRef]
- Valencia, A.M.; Pașca, S.P. Chromatin Dynamics in Human Brain Development and Disease. Trends Cell Biol. 2022, 32, 98–101. [Google Scholar] [CrossRef]
- Qiao, Y.; Wang, R.; Yang, X.; Tang, K.; Jing, N. Dual Roles of Histone H3 Lysine 9 Acetylation in Human Embryonic Stem Cell Pluripotency and Neural Differentiation. J. Biol. Chem. 2015, 290, 2508–2520. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Tapiola, T.; Korhonen, P.; Suuronen, T. Neuronal Apoptosis Induced by Histone Deacetylase Inhibitors. Brain Res. Mol. Brain Res. 1998, 61, 203–206. [Google Scholar] [CrossRef]
- Zhang, X.; He, X.; Li, Q.; Kong, X.; Ou, Z.; Zhang, L.; Gong, Z.; Long, D.; Li, J.; Zhang, M.; et al. PI3K/AKT/MTOR Signaling Mediates Valproic Acid-Induced Neuronal Differentiation of Neural Stem Cells through Epigenetic Modifications. Stem Cell Rep. 2017, 8, 1256–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, S.; Vinnakota, R.; Kumar, J.; Kale, V.; Limaye, L. Improved Neural Differentiation of Normal and Abnormal Induced Pluripotent Stem Cell Lines in the Presence of Valproic Acid. J. Tissue Eng. Regen. Med. 2019, 13, 1482–1496. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Tang, Y.; Liu, H.; Guo, F.; Ni, J.; Le, W. Suppression of Histone Deacetylation Promotes the Differentiation of Human Pluripotent Stem Cells towards Neural Progenitor Cells. BMC Biol. 2014, 12, 95. [Google Scholar] [CrossRef]
- Ferrari, F.; Arrigoni, L.; Franz, H.; Izzo, A.; Butenko, L.; Trompouki, E.; Vogel, T.; Manke, T. DOT1L-Mediated Murine Neuronal Differentiation Associates with H3K79me2 Accumulation and Preserves SOX2-Enhancer Accessibility. Nat. Commun. 2020, 11, 5200. [Google Scholar] [CrossRef] [PubMed]
- Efroni, S.; Duttagupta, R.; Cheng, J.; Dehghani, H.; Hoeppner, D.J.; Dash, C.; Bazett-Jones, D.P.; Le Grice, S.; McKay, R.D.G.; Buetow, K.H.; et al. Global Transcription in Pluripotent Embryonic Stem Cells. Cell Stem Cell 2008, 2, 437–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meshorer, E.; Yellajoshula, D.; George, E.; Scambler, P.J.; Brown, D.T.; Misteli, T. Hyperdynamic Plasticity of Chromatin Proteins in Pluripotent Embryonic Stem Cells. Dev. Cell 2006, 10, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Roidl, D.; Hacker, C. Histone Methylation during Neural Development. Cell Tissue Res. 2014, 356, 539–552. [Google Scholar] [CrossRef]
- Thiel, G. How Sox2 Maintains Neural Stem Cell Identity. Biochem. J. 2013, 450, e1–e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milne, T.A.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.D.; Hess, J.L. MLL Targets SET Domain Methyltransferase Activity to Hox Gene Promoters. Mol. Cell 2002, 10, 1107–1117. [Google Scholar] [CrossRef]
- Lee, M.G.; Villa, R.; Trojer, P.; Norman, J.; Yan, K.-P.; Reinberg, D.; Croce, L.D.; Shiekhattar, R. Demethylation of H3K27 Regulates Polycomb Recruitment and H2A Ubiquitination. Science 2007, 318, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.A.; Huang, Y.-C.; Swigut, T.; Mirick, A.L.; Garcia-Verdugo, J.M.; Wysocka, J.; Ernst, P.; Alvarez-Buylla, A. Chromatin Remodelling Factor Mll1 Is Essential for Neurogenesis from Postnatal Neural Stem Cells. Nature 2009, 458, 529–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-C.; Shih, H.-Y.; Lin, S.-J.; Chiu, C.-C.; Ma, T.-L.; Yeh, T.-H.; Cheng, Y.-C. The Epigenetic Factor Kmt2a/Mll1 Regulates Neural Progenitor Proliferation and Neuronal and Glial Differentiation. Dev. Neurobiol. 2015, 75, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Sugimoto, K.; Nozaki, M.; Ueda, J.; Ohta, T.; Ohki, M.; Fukuda, M.; Takeda, N.; Niida, H.; Kato, H.; et al. G9a Histone Methyltransferase Plays a Dominant Role in Euchromatic Histone H3 Lysine 9 Methylation and Is Essential for Early Embryogenesis. Genes Dev. 2002, 16, 1779–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tachibana, M.; Sugimoto, K.; Fukushima, T.; Shinkai, Y. SET Domain-Containing Protein, G9a, Is a Novel Lysine-Preferring Mammalian Histone Methyltransferase with Hyperactivity and Specific Selectivity to Lysines 9 and 27 of Histone H3 *. J. Biol. Chem. 2001, 276, 25309–25317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, S.E.; Campbell, R.D.; Sanderson, C.M. Novel NG36/G9a Gene Products Encoded within the Human and Mouse MHC Class III Regions. Mamm. Genome 2001, 12, 916–924. [Google Scholar] [CrossRef]
- Fiszbein, A.; Giono, L.E.; Quaglino, A.; Berardino, B.G.; Sigaut, L.; von Bilderling, C.; Schor, I.E.; Steinberg, J.H.E.; Rossi, M.; Pietrasanta, L.I.; et al. Alternative Splicing of G9a Regulates Neuronal Differentiation. Cell Rep. 2016, 14, 2797–2808. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhou, W.; Li, X.; Gao, M.; Ji, S.; Tian, W.; Ji, G.; Du, J.; Hao, A. SOX19b Regulates the Premature Neuronal Differentiation of Neural Stem Cells through EZH2-Mediated Histone Methylation in Neural Tube Development of Zebrafish. Stem Cell Res. Ther. 2019, 10, 389. [Google Scholar] [CrossRef]
- Chathoth, K.T.; Zabet, N.R. Chromatin Architecture Reorganization during Neuronal Cell Differentiation in Drosophila Genome. Genome Res. 2019, 29, 613–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arzate-Mejía, R.G.; Recillas-Targa, F.; Corces, V.G. Developing in 3D: The Role of CTCF in Cell Differentiation. Dev. Camb. Engl. 2018, 145, dev137729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, R.P.; Meyer, B.J. Spatial Organization of Chromatin: Emergence of Chromatin Structure During Development. Annu. Rev. Cell Dev. Biol. 2021, 37, 199–232. [Google Scholar] [CrossRef] [PubMed]
- Kishi, Y.; Gotoh, Y. Regulation of Chromatin Structure During Neural Development. Front. Neurosci. 2018, 12, 874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonev, B.; Mendelson Cohen, N.; Szabo, Q.; Fritsch, L.; Papadopoulos, G.L.; Lubling, Y.; Xu, X.; Lv, X.; Hugnot, J.-P.; Tanay, A.; et al. Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell 2017, 171, 557–572.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tjong, H.; Li, W.; Kalhor, R.; Dai, C.; Hao, S.; Gong, K.; Zhou, Y.; Li, H.; Zhou, X.J.; Le Gros, M.A.; et al. Population-Based 3D Genome Structure Analysis Reveals Driving Forces in Spatial Genome Organization. Proc. Natl. Acad. Sci. USA 2016, 113, E1663–E1672. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Zhang, B. Predicting three-dimensional genome organization with chromatin states. PLoS Comput. Biol. 2019, 15, e1007024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Y.; Reyes, A.; Johnstone, S.E.; Aryee, M.J.; Bernstein, B.E.; Zhang, B. Data-Driven Polymer Model for Mechanistic Exploration of Diploid Genome Organization. Biophys. J. 2020, 119, 1905–1916. [Google Scholar] [CrossRef]
- Bartsch, O.; Schmidt, S.; Richter, M.; Morlot, S.; Seemanová, E.; Wiebe, G.; Rasi, S. DNA Sequencing of CREBBP Demonstrates Mutations in 56% of Patients with Rubinstein-Taybi Syndrome (RSTS) and in Another Patient with Incomplete RSTS. Hum. Genet. 2005, 117, 485–493. [Google Scholar] [CrossRef]
- Grangeia, A.; Leão, M.; Moura, C.P. Wiedemann-Steiner Syndrome in Two Patients from Portugal. Am. J. Med. Genet. A 2020, 182, 25–28. [Google Scholar] [CrossRef]
- Stadelmaier, R.T.; Kenna, M.A.; Barrett, D.; Mullen, T.E.; Bodamer, O.; Agrawal, P.B.; Robson, C.D.; Wojcik, M.H. Neuroimaging in Kabuki Syndrome and Another KMT2D-Related Disorder. Am. J. Med. Genet. A 2021, 185, 3770–3783. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, J.A.; Bjornsson, H.T. Mendelian Disorders of the Epigenetic Machinery: Postnatal Malleability and Therapeutic Prospects. Hum. Mol. Genet. 2019, 28, R254–R264. [Google Scholar] [CrossRef] [PubMed]
- Sadikovic, B.; Levy, M.A.; Kerkhof, J.; Aref-Eshghi, E.; Schenkel, L.; Stuart, A.; McConkey, H.; Henneman, P.; Venema, A.; Schwartz, C.E.; et al. Clinical Epigenomics: Genome-Wide DNA Methylation Analysis for the Diagnosis of Mendelian Disorders. Genet. Med. 2021, 23, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Cerrato, F.; Sparago, A.; Ariani, F.; Brugnoletti, F.; Calzari, L.; Coppedè, F.; De Luca, A.; Gervasini, C.; Giardina, E.; Gurrieri, F.; et al. DNA Methylation in the Diagnosis of Monogenic Diseases. Genes 2020, 11, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris-Rosendahl, D.J.; Crocq, M.-A. Neurodevelopmental Disorders—the History and Future of a Diagnostic Concept. Dialogues Clin. Neurosci. 2020, 22, 65–72. [Google Scholar] [CrossRef]
- Harris, J.C. Animal models of neurodevelopmental disorders with behavioral phenotypes. Curr. Opin. Psychiatry 2021, 34, 87–93. [Google Scholar] [CrossRef]
- Zhang, L.; Pilarowski, G.; Pich, E.M.; Nakatani, A.; Dunlop, J.; Baba, R.; Matsuda, S.; Daini, M.; Hattori, Y.; Matsumoto, S.; et al. Inhibition of KDM1A Activity Restores Adult Neurogenesis and Improves Hippocampal Memory in a Mouse Model of Kabuki Syndrome. Mol. Ther. Methods Clin. Dev. 2021, 20, 779–791. [Google Scholar] [CrossRef]
- Garg, S.K.; Lioy, D.T.; Cheval, H.; McGann, J.C.; Bissonnette, J.M.; Murtha, M.J.; Foust, K.D.; Kaspar, B.K.; Bird, A.; Mandel, G. Systemic Delivery of MeCP2 Rescues Behavioral and Cellular Deficits in Female Mouse Models of Rett Syndrome. J. Neurosci. Off. J. Soc. Neurosci. 2013, 33, 13612–13620. [Google Scholar] [CrossRef] [Green Version]
- Achilly, N.P.; Wang, W.; Zoghbi, H.Y. Presymptomatic Training Mitigates Functional Deficits in Rett Syndrome Mice. Nature 2021, 592, 596–600. [Google Scholar] [CrossRef]
- Alarcón, J.M.; Malleret, G.; Touzani, K.; Vronskaya, S.; Ishii, S.; Kandel, E.R.; Barco, A. Chromatin Acetylation, Memory, and LTP Are Impaired in CBP+/− Mice: A Model for the Cognitive Deficit in Rubinstein-Taybi Syndrome and Its Amelioration. Neuron 2004, 42, 947–959. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Atalaya, J.P.; Gervasini, C.; Mottadelli, F.; Spena, S.; Piccione, M.; Scarano, G.; Selicorni, A.; Barco, A.; Larizza, L. Histone Acetylation Deficits in Lymphoblastoid Cell Lines from Patients with Rubinstein–Taybi Syndrome. J. Med. Genet. 2012, 49, 66–74. [Google Scholar] [CrossRef]
- Zhao, X.; Bhattacharyya, A. Human Models Are Needed for Studying Human Neurodevelopmental Disorders. Am. J. Hum. Genet. 2018, 103, 829–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, B. Mouse Models and Induced Pluripotent Stem Cells in Researching Psychiatric Disorders. Stem Cell Investig. 2017, 4, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alari, V.; Russo, S.; Terragni, B.; Ajmone, P.F.; Sironi, A.; Catusi, I.; Calzari, L.; Concolino, D.; Marotta, R.; Milani, D.; et al. IPSC-Derived Neurons of CREBBP- and EP300-Mutated Rubinstein-Taybi Syndrome Patients Show Morphological Alterations and Hypoexcitability. Stem Cell Res. 2018, 30, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Calzari, L.; Barcella, M.; Alari, V.; Braga, D.; Muñoz-Viana, R.; Barlassina, C.; Finelli, P.; Gervasini, C.; Barco, A.; Russo, S.; et al. Transcriptome Analysis of IPSC-Derived Neurons from Rubinstein-Taybi Patients Reveals Deficits in Neuronal Differentiation. Mol. Neurobiol. 2020, 57, 3685–3701. [Google Scholar] [CrossRef]
- Varderidou-Minasian, S.; Hinz, L.; Hagemans, D.; Posthuma, D.; Altelaar, M.; Heine, V.M. Quantitative Proteomic Analysis of Rett IPSC-Derived Neuronal Progenitors. Mol. Autism 2020, 11, 38. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral Organoids Model Human Brain Development and Microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Lancaster, M.A.; Castanon, R.; Nery, J.R.; Knoblich, J.A.; Ecker, J.R. Cerebral Organoids Recapitulate Epigenomic Signatures of the Human Fetal Brain. Cell Rep. 2016, 17, 3369–3384. [Google Scholar] [CrossRef] [Green Version]
- Camp, J.G.; Badsha, F.; Florio, M.; Kanton, S.; Gerber, T.; Wilsch-Bräuninger, M.; Lewitus, E.; Sykes, A.; Hevers, W.; Lancaster, M.; et al. Human Cerebral Organoids Recapitulate Gene Expression Programs of Fetal Neocortex Development. Proc. Natl. Acad. Sci. USA 2015, 112, 15672–15677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, A.R.; Fernandes, T.G.; Vaz, S.H.; Silva, T.P.; Bekman, E.P.; Xapelli, S.; Duarte, S.; Ghazvini, M.; Gribnau, J.; Muotri, A.R.; et al. Modeling Rett Syndrome With Human Patient-Specific Forebrain Organoids. Front. Cell Dev. Biol. 2020, 8, 610427. [Google Scholar] [CrossRef]
- Negri, G.; Magini, P.; Milani, D.; Crippa, M.; Biamino, E.; Piccione, M.; Sotgiu, S.; Perrìa, C.; Vitiello, G.; Frontali, M.; et al. Exploring by Whole Exome Sequencing Patients with Initial Diagnosis of Rubinstein–Taybi Syndrome: The Interconnections of Epigenetic Machinery Disorders. Hum. Genet. 2019, 138, 257–269. [Google Scholar] [CrossRef]
- Van Gils, J.; Magdinier, F.; Fergelot, P.; Lacombe, D. Rubinstein-Taybi Syndrome: A Model of Epigenetic Disorder. Genes 2021, 12, 968. [Google Scholar] [CrossRef] [PubMed]
- Haghshenas, S.; Bhai, P.; Aref-Eshghi, E.; Sadikovic, B. Diagnostic Utility of Genome-Wide DNA Methylation Analysis in Mendelian Neurodevelopmental Disorders. Int. J. Mol. Sci. 2020, 21, 9303. [Google Scholar] [CrossRef] [PubMed]
- Kerkhof, J.; Squeo, G.M.; McConkey, H.; Levy, M.A.; Piemontese, M.R.; Castori, M.; Accadia, M.; Biamino, E.; Della Monica, M.; Di Giacomo, M.C.; et al. DNA Methylation Episignature Testing Improves Molecular Diagnosis of Mendelian Chromatinopathies. Genet. Med. 2022, 24, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.A.; McConkey, H.; Kerkhof, J.; Barat-Houari, M.; Bargiacchi, S.; Biamino, E.; Bralo, M.P.; Cappuccio, G.; Ciolfi, A.; Clarke, A.; et al. Novel Diagnostic DNA Methylation Episignatures Expand and Refine the Epigenetic Landscapes of Mendelian Disorders. HGG Adv. 2022, 3, 100075. [Google Scholar] [CrossRef] [PubMed]
- Aref-Eshghi, E.; Schenkel, L.C.; Lin, H.; Skinner, C.; Ainsworth, P.; Paré, G.; Siu, V.; Rodenhiser, D.; Schwartz, C.; Sadikovic, B. Clinical Validation of a Genome-Wide DNA Methylation Assay for Molecular Diagnosis of Imprinting Disorders. J. Mol. Diagn. 2017, 19, 848–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alari, V.; Scalmani, P.; Ajmone, P.F.; Perego, S.; Avignone, S.; Catusi, I.; Lonati, P.A.; Borghi, M.O.; Finelli, P.; Terragni, B.; et al. Histone Deacetylase Inhibitors Ameliorate Morphological Defects and Hypoexcitability of IPSC-Neurons from Rubinstein-Taybi Patients. Int. J. Mol. Sci. 2021, 22, 5777. [Google Scholar] [CrossRef] [PubMed]
- Bjornsson, H.T.; Benjamin, J.S.; Zhang, L.; Weissman, J.; Gerber, E.E.; Chen, Y.-C.; Vaurio, R.G.; Potter, M.C.; Hansen, K.D.; Dietz, H.C. Histone Deacetylase Inhibition Rescues Structural and Functional Brain Deficits in a Mouse Model of Kabuki Syndrome. Sci. Transl. Med. 2014, 6, 256ra135. [Google Scholar] [CrossRef] [Green Version]
- Foust, K.D.; Nurre, E.; Montgomery, C.L.; Hernandez, A.; Chan, C.M.; Kaspar, B.K. Intravascular AAV9 Preferentially Targets Neonatal-Neurons and Adult-Astrocytes in CNS. Nat. Biotechnol. 2009, 27, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Sztainberg, Y.; Wang, Q.; Bajikar, S.S.; Trostle, A.J.; Wan, Y.-W.; Jafar-nejad, P.; Rigo, F.; Liu, Z.; Tang, J.; et al. Antisense Oligonucleotide Therapy in a Humanized Mouse Model of MECP2 Duplication Syndrome. Sci. Transl. Med. 2021, 13, eaaz7785. [Google Scholar] [CrossRef] [PubMed]
- Gadalla, K.K.; Bailey, M.E.; Spike, R.C.; Ross, P.D.; Woodard, K.T.; Kalburgi, S.N.; Bachaboina, L.; Deng, J.V.; West, A.E.; Samulski, R.J.; et al. Improved Survival and Reduced Phenotypic Severity Following AAV9/MECP2 Gene Transfer to Neonatal and Juvenile Male Mecp2 Knockout Mice. Mol. Ther. 2013, 21, 18–30. [Google Scholar] [CrossRef] [Green Version]
- Carrette, L.L.G.; Wang, C.-Y.; Wei, C.; Press, W.; Ma, W.; Kelleher, R.J.; Lee, J.T. A Mixed Modality Approach towards Xi Reactivation for Rett Syndrome and Other X-Linked Disorders. Proc. Natl. Acad. Sci. USA 2018, 115, E668–E675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ousterout, D.G.; Kabadi, A.M.; Thakore, P.I.; Majoros, W.H.; Reddy, T.E.; Gersbach, C.A. Multiplex CRISPR/Cas9-Based Genome Editing for Correction of Dystrophin Mutations That Cause Duchenne Muscular Dystrophy. Nat. Commun. 2015, 6, 6244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Mokhtari, R.; Pedrosa, E.; Kirschenbaum, M.; Bayrak, C.; Zheng, D.; Lachman, H.M. CRISPR/Cas9-Mediated Heterozygous Knockout of the Autism Gene CHD8 and Characterization of Its Transcriptional Networks in Cerebral Organoids Derived from IPS Cells. Mol. Autism 2017, 8, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.S.; Wu, H.; Krzisch, M.; Wu, X.; Graef, J.; Muffat, J.; Hnisz, D.; Li, C.H.; Yuan, B.; Xu, C.; et al. Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 2018, 172, 979–992.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, X.; Song, H.; Ming, G. Brain Organoids: Advances, Applications and Challenges. Development 2019, 146, dev166074. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Histone Acetyltransferase | TYPE B | TYPE A | ||||
| Cytoplasmic KAT Family | GNAT Family | P300/CBP Family | MYST Family | Transcription Factor Related Family | Nuclear Receptor Coactivator Family | |
| KAT1 (HAT1) | KAT2A (hGCN5) | KAT3A (CBP) | KAT5 (TIP60/PLIP) | KAT4 (TAF1) | KAT13A (SRC1) | |
| KAT2B (PCAF) | KAT3B (p300) | KAT6A (MOZ/MYST3) | KAT12 (TFIIIC90) | KAT13B (ACTR) | ||
| KAT9 (ELP3) | KAT6B (MORF/MYST4) | KAT13C (P160) | ||||
| KAT7 (HBO1/MYST2) | KAT13D (CLOCK) | |||||
| KAT8 (HMOF/MYST1) | ||||||
| Histone Deacetylase | Class I | Class II | Class III | Class IV | ||
| HDAC1 | HDAC4 | SIRT1 | HDAC11 | |||
| HDAC2 | HDAC5 | SIRT2 | ||||
| HDAC3 | HDAC6 | SIRT3 | ||||
| HDAC8 | HDAC7 | SIRT4 | ||||
| HDAC9 | SIRT5 | |||||
| HDAC10 | SIRT6 | |||||
| SIRT7 | ||||||
| Modified Histone Residue | Type of Post-Translational Modification | Target Amino Acid | Effect on Gene Expression |
|---|---|---|---|
| H2AS1 | Phosphorylation | Serine, threonine, tyrosine | Modulation of DNA compaction and interaction with other histone post-translational modifications |
| H2AS139 | |||
| H2BS14 | |||
| H3T3 | |||
| H3T6 | |||
| H3S10 | |||
| H3T11 | |||
| H3S28 | |||
| H4S1 | |||
| H2AK119 | Ubiquitination | Lysine | Regulation of transcription initiation and elongation |
| H2BK120 | |||
| H2AK5 | Acetylation | Lysine | Decrease in histone/DNA interaction, chromatin is structurally loose, less compact and transcription is activated |
| H2BK5 | |||
| H2BK12 | |||
| H2BK15 | |||
| H2BK20 | |||
| H3K4 | |||
| H3K9 | |||
| H3K14 | |||
| H3K18 | |||
| H3K27 | |||
| H4K5 | |||
| H4K8 | |||
| H4K12 | |||
| H4K16 | |||
| H2BK5me1 | Methylation | Lysine and arginine | Transcription is activated |
| H3K4me1 | |||
| H3K4me2 | |||
| H3K4me3 | |||
| H3K9me1 | |||
| H3K27me1 | |||
| H3K36me3 | |||
| H3K79me1 | |||
| H4K20me1 | |||
| H3K9me2 | Methylation | Lysine | Transcription is inhibited |
| H3K9me3 | |||
| H3K27me3 | |||
| H3K79me3 | |||
| H3K18 | SUMOylation | Lysine | Competition with other lysine modifications. Decrease and stop of transcription |
| H4K12 |
| Gene Function | Gene | Disease |
|---|---|---|
| Writer Histone Acetyltransferase | CREBBP * | Rubinstein Taybi syndrome 1 |
| EP300 * | Rubinstein Taybi syndrome 2 | |
| KANSL1 | Koolen-De Vries syndrome | |
| KAT6A * | Mental retardation autosomal dominant (MRAD) 32 | |
| KAT6B * | Say-Barber-Biessecker-Young-Simpson syndrome/Genitopatellar syndrome | |
| Writer Histone Methyltransferase | ASH1L * | MRAD 52 |
| EHMT1 * | Kleefstra syndrome 1 | |
| EZH2 | Weaver syndrome | |
| KMT2A * | Wiedemann-Steiner syndrome | |
| KMT2B * | Childhood-onset dystonia 28 | |
| KMT2C * | Kleefstra syndrome 2 | |
| KMT2D * | Kabuki syndrome type 1 | |
| KMT2E * | KMT2E deficiency | |
| KMT5B | MRAD 51 | |
| NSD1 * | Sotos syndrome 1 | |
| NSD2 * | Wolf-Hirschhorn syndrome | |
| PRDM5 | Brittle cornea syndrome | |
| PRDM12 | Hereditary sensory and autonomic neuropathy 8 | |
| PRDM16 | Dilated cardiomyopathy | |
| SETD5 | MRAD 23 | |
| SETD2 | Luscan-Lumish syndrome | |
| SETD1B | SETD1B-related syndrome | |
| Writer DNA methyltransferase | DNMT1 * | AD cerebellar ataxia deafness and narcolepsy/Hereditary sensory neuropathy 1E |
| DNMT3A * | Tatton-Brown-Rahman syndrome/Microcephalic dwarfism | |
| DNMT3B * | Immunodeficiency-centromeric instability-facial anomalies syndrome | |
| Eraser Histone deacetylase | HDAC4 | Brachydactyly–mental retardation syndrome |
| HDAC6 | Chondrodysplasia with platyspondyly/distinctive brachydactyly/hydrocephaly and microphthalmia | |
| HDAC8 | Cornelia de Lange syndrome type 5 | |
| Eraser Histone demethylase | HR | Alopecia universalis/Atrichia with papular lesions/Hypotrichosis type 4 |
| KDM1A | Cleft palate, psychomotor retardation, and distinctive facial features | |
| KDM5B * | Mental retardation autosomal recessive 65 | |
| KDM5C * | Claes–Jensen X-linked mental retardation | |
| KDM6A | Kabuki syndrome type 2 | |
| KDM6B | KDM6B deficiency | |
| PHF8 * | Siderius X-linked mental retardation | |
| Remodeler | ARID1A | Mental retardation autosomal dominant 14 (Coffin–Siris syndrome) |
| ARID1B | Mental retardation autosomal dominant 12 (Coffin–Siris syndrome) | |
| ATRX * | α-thalassemia/mental retardation X-linked (ATRX) syndrome | |
| CHD1 * | Pilarowski–Bjornsson syndrome | |
| CHD2 * | Epileptic encephalopathy, childhood onset | |
| CHD3 * | Snijders Blok–Campeau syndrome | |
| CHD4 * | Sifrim–Hitz–Weiss syndrome | |
| CHD7 * | Charge syndrome/Hypogonadotropic hypogonadism | |
| CHD8 * | Autism susceptibility 8/overgrowth and ID | |
| SMARCA2 * | Nicolaides–Baraitser syndrome | |
| SMARCA4 * | Coffin-Siris syndrome 4/Rhabdoid tumor predisposition syndrome | |
| SRCAP | Floating–Harbor syndrome | |
| Reader | AIRE | Autoimmune polyendocrinopathy syndrome 1 |
| ALG13 | Eary infantile epileptic encephalopathy 36 | |
| ASXL1 | Bohring–Opitz syndrome | |
| ASXL2 | Shashi–Pena syndrome | |
| ASXL3 | Bainbridge–Ropers syndrome | |
| BPTF | Neurodevelopmental disorder with dysmorphic facies and distal limb anomalies | |
| BRPF1 | Intellectual developmental disorder with dysmorphic facies and ptosis | |
| BRWD3 | X-linked mental retardation 93 | |
| CBX2 | Sex reversal | |
| DPF2 | Coffin Siris 7 | |
| EED | Cohen–Gibson syndrome | |
| LBR | Pelger–Huet anomaly (PHA)/PHA with muskuloskeletal findings Greenberg skeletal dysplasia | |
| MBD5 | MRAD 1 | |
| MECP2 | Rett syndrome and related disorders | |
| MORC2 | Charcot–Marie–Tooth disease 2Z | |
| MSH6 | Hereditary nonpolyposis colorectal cancer 5/Mismatch repair cancer syndrome | |
| ORC1 | Meier–Gorlin syndrome 1 | |
| PHF6 | Borjeson–Forssman–Lehmann syndrome | |
| PHIP | Developmental delay, ID, obesity and dysmorphic features | |
| RAG2 | Omenn syndrome and severe combined immunodeficiency | |
| RAI1 | Smith–Magenis syndrome | |
| RERE | Neurodevelopmental disorder with or without other anomalies | |
| SMN1 | Spinal muscular atrophy | |
| SP110 | Hepatic venoocclusive disease and immune deficiency | |
| TAF1 | X-linked Mental retardation 33 | |
| TDRD7 | Cataract 36 | |
| ZMYND11 | MRAD 30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nothof, S.A.; Magdinier, F.; Van-Gils, J. Chromatin Structure and Dynamics: Focus on Neuronal Differentiation and Pathological Implication. Genes 2022, 13, 639. https://doi.org/10.3390/genes13040639
Nothof SA, Magdinier F, Van-Gils J. Chromatin Structure and Dynamics: Focus on Neuronal Differentiation and Pathological Implication. Genes. 2022; 13(4):639. https://doi.org/10.3390/genes13040639
Chicago/Turabian StyleNothof, Sophie A., Frédérique Magdinier, and Julien Van-Gils. 2022. "Chromatin Structure and Dynamics: Focus on Neuronal Differentiation and Pathological Implication" Genes 13, no. 4: 639. https://doi.org/10.3390/genes13040639
APA StyleNothof, S. A., Magdinier, F., & Van-Gils, J. (2022). Chromatin Structure and Dynamics: Focus on Neuronal Differentiation and Pathological Implication. Genes, 13(4), 639. https://doi.org/10.3390/genes13040639