Mycalamide A Shows Cytotoxic Properties and Prevents EGF-Induced Neoplastic Transformation through Inhibition of Nuclear Factors

 ,
, 

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abbreviations
| AP-1 | activator protein-1 |
| EGF | epidermal growth factor |
| FBS | fetal bovine serum |
| IC50 | inhibition concentration 50% |
| INCC50 | inhibition of number of colonies formed in soft agar concentration 50% |
| MAPK | mitogen activated protein kinases |
| MTS | 5-(3-carboxymethoxyphenyl)-2-(4,5-dimethylthiazolyl)-3-(4-sulfophenyl) tetrazolium, inner salt |
| NF-κB | nuclear factor kappa B |
| PI | propidium iodide |
1. Introduction
2. Results and Discussion
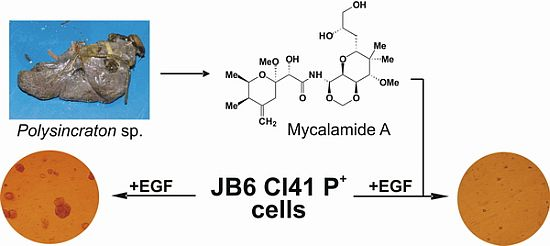
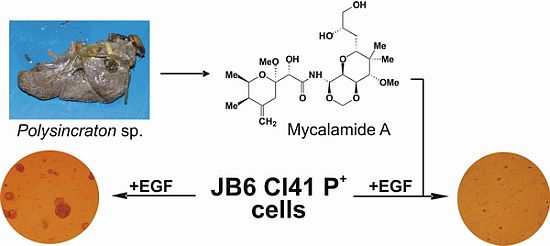
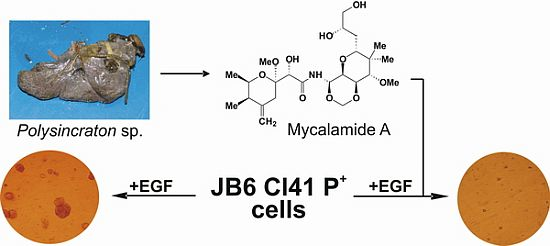
2.1. Isolation and Structural Identification of Mycalamide A from Ascidian Polysincraton sp.
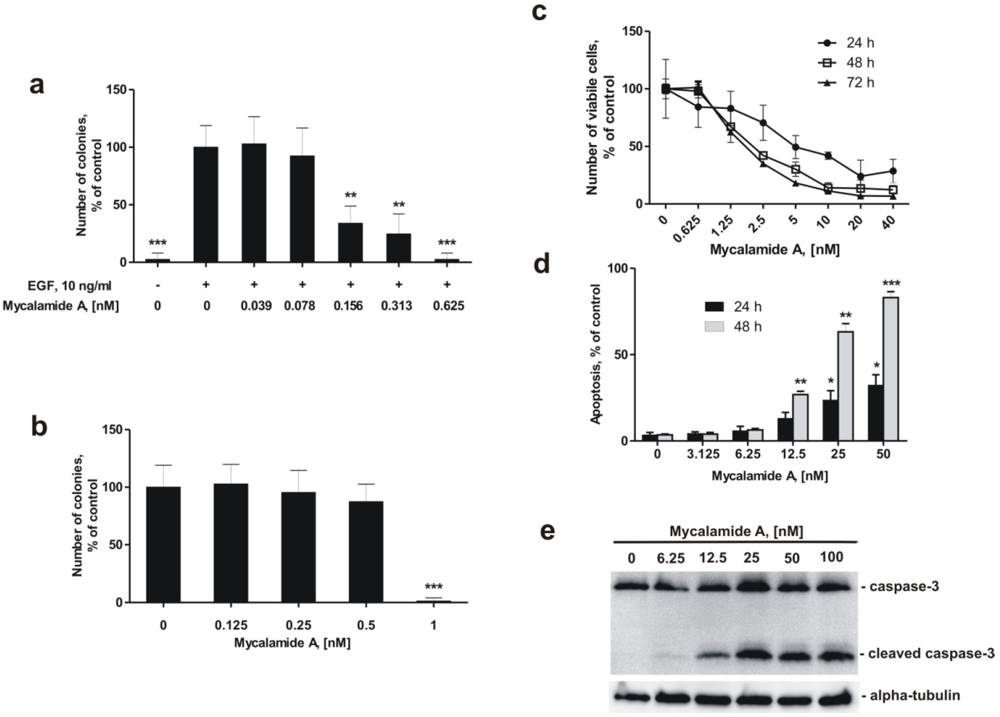
2.2. Mycalamide A Prevents EGF-Induced Transformation of JB6 Cl41 P+ Cells and Colony Growth of HeLa Cancer Cells
2.3. Mycalamide A Induces Apoptosis of JB6 Cl41 P+ Cells

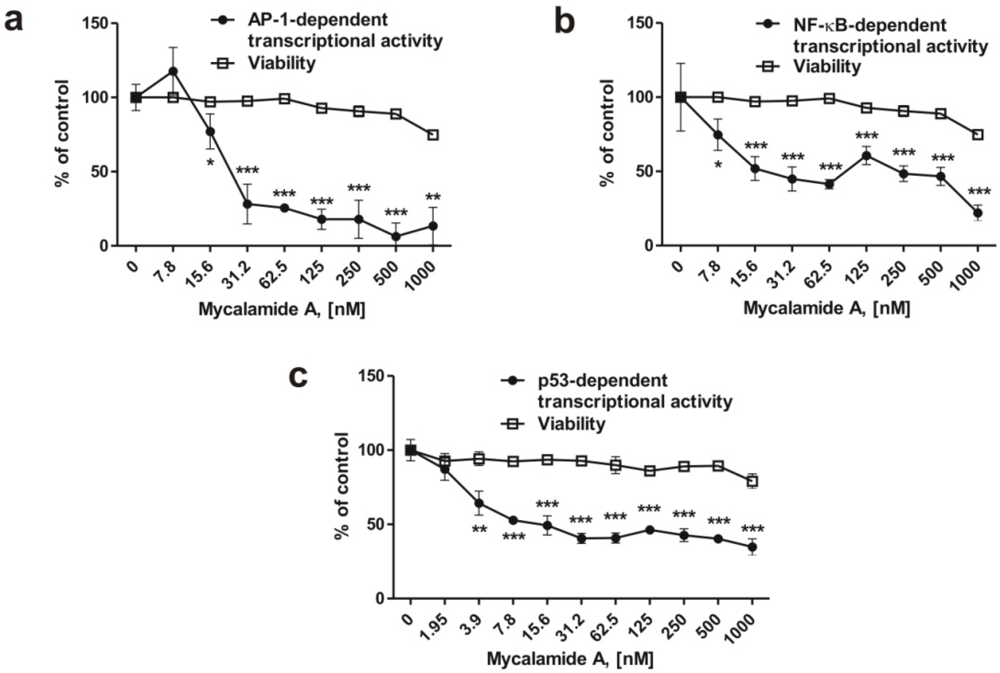
2.4. Mycalamide A Inhibits AP-1-, NF-κB-, and p53-Dependent Transcriptional Activity in JB6 Cells

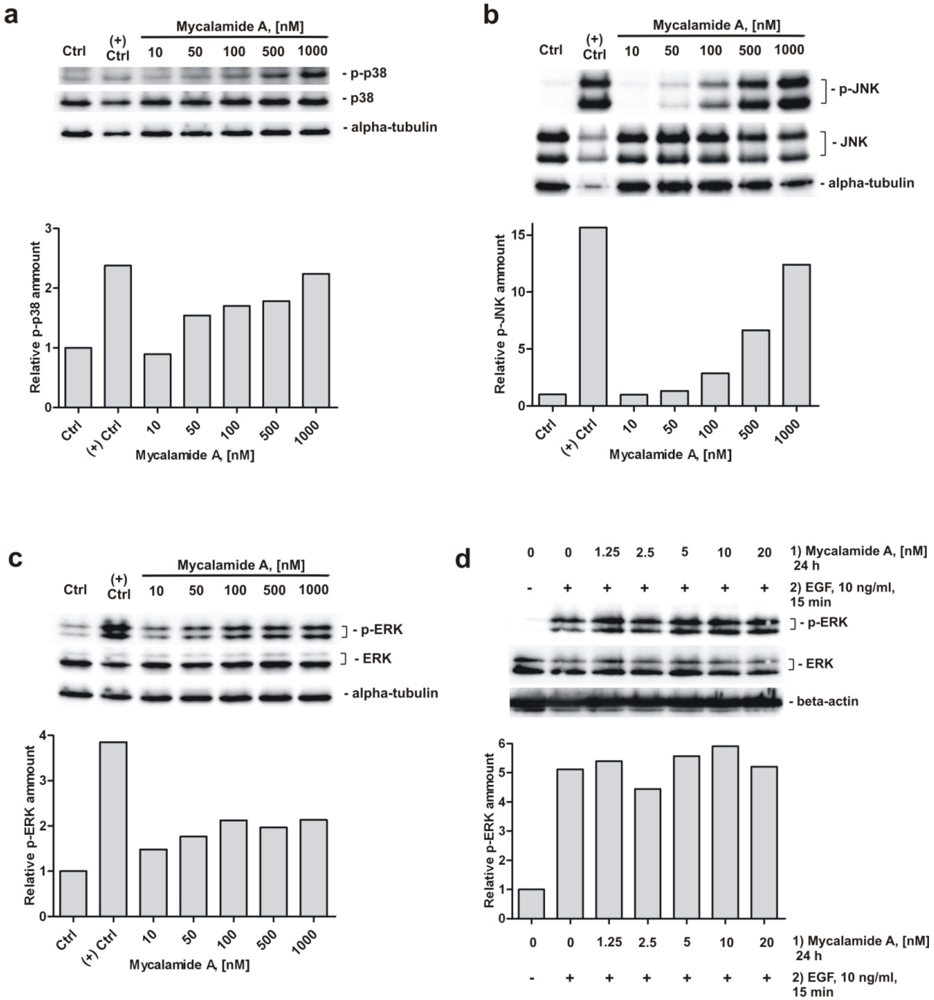
2.5. Analysis of Phosphorylation of MAPK p38, JNK, and ERK under Mycalamide A Treatment

3. Experimental Section
3.1. Reagents and Antibodies
3.2. Animal Material
3.3. Isolation of Mycalamide A
3.4. Cell Culture
3.5. Cell Proliferation Assay
3.6. Cytotoxicity Assay (MTS Test)
3.7. Detection of Apoptosis
3.8. Anchorage-Independent Neoplastic Transformation or Colony Growth Assay
3.9. Protein Preparation and Western Blotting
3.10. Determination of the Effect of Mycalamide A on the Basal Transcriptional Activity of AP-1, NF-κB or p53
4. Conclusions
Acknowledgments
Supplementary Files
References
- Kuzmich, A.S.; Fedorov, S.N.; Shastina, V.V.; Shubina, L.K.; Radchenko, O.S.; Balaneva, N.N.; Zhidkov, M.E.; Park, J.I.; Kwak, J.Y.; Stonik, V.A. The anticancer activity of 3- and 10-bromofascaplysins is mediated by caspase-8, -9, -3-dependent apoptosis. Bioorg. Med. Chem. 2010, 18, 3834–3840. [Google Scholar]
- Dyshlovoy, S.; Fedorov, S.; Shubina, L.; Honecker, F.; Stonik, V. Anticancer activity of aaptamine and its derivatives isolated from marine Vietnamese sponge Aaptos sp. Ann. Oncol. 2011, 22, 33–33. [Google Scholar]
- Fedorov, S.; Dyshlovoy, S.; Monastyrnaya, M.; Shubina, L.; Leychenko, E.; Kozlovskaya, E.; Jin, J.O.; Kwak, J.Y.; Bode, A.M.; Dong, Z.G.; et al. The anticancer effects of actinoporin RTX-Afrom the sea anemone Heteractis crispa (=Radianthus macrodactylus). Toxicon 2010, 55, 811–817. [Google Scholar] [CrossRef]
- Shubina, L.K.; Makarieva, T.N.; Dyshlovoy, S.A.; Fedorov, S.N.; Dmitrenok, P.S.; Stonik, V.A. Three new aaptamines from the marine sponge Aaptos sp. and their proapoptotic properties. Nat. Prod. Commun. 2010, 5, 1881–1884. [Google Scholar]
- Fedorov, S.N.; Makarieva, T.N.; Guzii, A.G.; Shubina, L.K.; Kwak, J.Y.; Stonik, V.A. Marine two-headed sphingolipid-like compound rhizochalin inhibits EGF-induced transformation of JB6 P+ Cl41 cells. Lipids 2009, 44, 777–785. [Google Scholar]
- Fedorov, S.N.; Shubina, L.K.; Kicha, A.A.; Ivanchina, N.V.; Kwak, J.Y.; Jin, J.O.; Bode, A.M.; Dong, Z.G.; Stonik, V.A. Proapoptotic and anticarcinogenic activities of leviusculoside G from the starfish Henricia leviuscula and probable molecular mechanism. Nat. Prod. Commun. 2008, 3, 1575–1580. [Google Scholar]
- Lee, N.Y.; Ermakova, S.P.; Zvyagintseva, T.N.; Kang, K.W.; Dong, Z.; Choi, H.S. Inhibitory effects of fucoidan on activation of epidermal growth factor receptor and cell transformation in JB6 Cl41 cells. Food Chem. Toxicol. 2008, 46, 1793–1800. [Google Scholar] [CrossRef]
- Bhatnagar, I.; Kim, S.K. Marine antitumor drugs: Status, shortfalls and strategies. Mar. Drugs 2010, 8, 2702–2720. [Google Scholar] [CrossRef]
- Perry, N.B.; Blunt, J.W.; Munro, M.H.G.; Pannell, L.K. Mycalamide A, an antiviral compound from a New Zealand sponge of the genus Mycale. J. Am. Chem. Soc. 1988, 110, 4850–4851. [Google Scholar]
- Hood, K.A.; West, L.M.; Northcote, P.T.; Berridge, M.V.; Miller, J.H. Induction of apoptosis by the marine sponge (Mycale) metabolites, mycalamide A and pateamine. Apoptosis 2001, 6, 207–219. [Google Scholar] [CrossRef]
- Burres, N.S.; Clement, J.J. Antitumor activity and mechanism of action of the novel marine natural products mycalamide-A and -B and onnamide. Cancer Res. 1989, 49, 2935–2940. [Google Scholar]
- Perry, N.B.; Blunt, J.W.; Munro, M.H.G.; Thompson, A.M. Antiviral and antitumor agents from a New Zealand sponge, Mycale sp. 2. Structures and solution conformations of mycalamides A and B. J. Org. Chem. 1990, 55, 223–227. [Google Scholar]
- West, L.M.; Northcote, P.T.; Hood, K.A.; Miller, J.H.; Page, M.J. Mycalamide D, a new cytotoxic amide from the New Zealand marine sponge Mycale species. J. Nat. Prod. 2000, 63, 707–709. [Google Scholar] [CrossRef]
- Simpson, J.S.; Garson, M.J.; Blunt, J.W.; Munro, M.H.G.; Hooper, J.N.A. Mycalamides C and D, cytotoxic compounds from the marine sponge Stylinos n. species. J. Nat. Prod. 2000, 63, 704–706. [Google Scholar] [CrossRef]
- Miller, J.H.; Singh, A.J.; Northcote, P.T. Microtubule-stabilizing drugs from marine sponges: Focus on peloruside A and zampanolide. Mar. Drugs 2010, 8, 1059–1079. [Google Scholar] [CrossRef]
- Ogawara, H.; Higashi, K.; Uchino, K.; Perry, N.B. Change of ras-transformed NRK-cells back to normal morphology by mycalamides A and B, antitumor agents from a marine sponge. Chem. Pharm. Bull. (Tokyo) 1991, 39, 2152–2154. [Google Scholar] [CrossRef]
- Galvin, F.; Freeman, G.J.; Raziwolf, Z.; Benacerraf, B.; Nadler, L.; Reiser, H. Effects of cyclosporin A, FK 506, and mycalamide A on the activation of murine CD4+ T cells by the murine B7 antigen. Eur. J. Immunol. 1993, 23, 283–286. [Google Scholar] [CrossRef]
- Barltrop, J.A.; Owen, T.C.; Cory, A.H.; Cory, J.G. 5-(3-carboxymethoxyphenyl)-2-(4,5-dimethylthiazolyl)-3-(4-sulfophenyl)tetrazolium, inner salt (MTS) and related analogs of 3-(4,5-dimethylthiazolyl)-2,5-diphenyltetrazolium bromide (MTT) reducing to purple water-soluble formazans as cell-viability indicators. Bioorg. Med. Chem. Lett. 1991, 1, 611–614. [Google Scholar] [CrossRef]
- Kellner, R.L.L. Suppression of pederin biosynthesis through antibiotic elimination of endosymbionts in Paederus sabaeus. J. Insect Physiol. 2001, 47, 475–483. [Google Scholar] [CrossRef]
- Piel, J. A polyketide synthase-peptide synthetase gene cluster from an uncultured bacterial symbiont of Paederus beetles. Proc. Natl. Acad. Sci. USA 2002, 99, 14002–14007. [Google Scholar] [CrossRef]
- Davis, A.R.; Butler, A.J.; Vanaltena, I. Settlement behaviour of ascidian larvae: Preliminary evidence for inhibition by sponge allelochemicals. Mar. Ecol. Prog. Ser. 1991, 72, 117–123. [Google Scholar] [CrossRef]
- Dong, Z.G.; Birrer, M.J.; Watts, R.G.; Matrisian, L.M.; Colburn, N.H. Blocking of tumor promoter-induced AP-1 activity inhibits induced transformation in JB6 mouse epidermal cells. Proc. Natl. Acad. Sci. USA 1994, 91, 609–613. [Google Scholar]
- Dong, Z.G.; Watts, R.G.; Sun, Y.; Zhan, S.N.; Colburn, N.H. Progressive elevation of AP-1 activity during preneoplastic-to neoplastic progression as modeled in mouse JB6 cell variants. Int. J. Oncol. 1995, 7, 359–364. [Google Scholar]
- Colburn, N.H.; Former, B.F.; Nelson, K.A.; Yuspa, S.H. Tumour promoter induces anchorage independence irreversibly. Nature 1979, 281, 589–591. [Google Scholar]
- Strickland, J.; Sun, Y.; Dong, Z.G.; Colburn, N.H. Grafting assay distinguishes promotion sensitive from promotion resistant JB6 cells. Carcinogenesis 1997, 18, 1135–1138. [Google Scholar] [CrossRef]
- He, Z.W.; Tang, F.Q.; Ermakova, S.; Li, M.; Zhao, Q.; Cho, Y.Y.; Ma, W.Y.; Choi, H.S.; Bode, A.M.; Yang, C.S.; et al. Fyn is a novel target of (−)-epigallocatechin gallate in the inhibition of JB6 Cl41 cell transformation. Mol. Carcinog. 2008, 47, 172–183. [Google Scholar] [CrossRef]
- Huang, C.S.; Ma, W.Y.; Goranson, A.; Dong, Z.G. Resveratrol suppresses cell transformation and induces apoptosis through a p53-dependent pathway. Carcinogenesis 1999, 20, 237–242. [Google Scholar] [CrossRef]
- Bernstein, L.R.; Colburn, N.H. AP1/jun function is differentially induced in promotion-sensitive and resistant JB6 cells. Science 1989, 244, 566–569. [Google Scholar]
- Huang, C.S.; Ma, W.Y.; Young, M.R.; Colburn, N.; Dong, Z.G. Shortage of mitogen-activated protein kinase is responsible for resistance to AP-1 transactivation and transformation in mouse JB6 cells. Proc. Natl. Acad. Sci. USA 1998, 95, 156–161. [Google Scholar]
- Huang, C.S.; Ma, W.Y.; Dawson, M.I.; Rincon, M.; Flavell, R.A.; Dong, Z.G. Blocking activator protein-1 activity, but not activating retinoic acid response element, is required for the antitumor promotion effect of retinoic acid. Proc. Natl. Acad. Sci. USA 1997, 94, 5826–5830. [Google Scholar]
- Hsu, T.C.; Nair, R.; Tulsian, P.; Camalier, C.E.; Hegamyer, G.A.; Young, M.R.; Colburn, N.H. Transformation nonresponsive cells owe their resistance to lack of p65/nuclear factor-kappa B activation. Cancer Res. 2001, 61, 4160–4168. [Google Scholar]
- Li, J.J.; Westergaard, C.; Ghosh, P.; Colburn, N.H. Inhibitors of both nuclear factor-kappa Beta and activator protein-1 activation block the neoplastic transformation response. Cancer Res. 1997, 57, 3569–3576. [Google Scholar]
- Koch, S.; Mayer, F.; Honecker, F.; Schittenhelm, M.; Bokemeyer, C. Efficacy of cytotoxic agents used in the treatment of testicular germ cell tumours under normoxic and hypoxic conditions in vitro. Br. J. Cancer 2003, 89, 2133–2139. [Google Scholar] [CrossRef]
- Chinni, S.R.; Li, Y.W.; Upadhyay, S.; Koppolu, P.K.; Sarkar, F.H. Indole3-carbinol (I3C) induced cell growth inhibition, G1 cell cycle arrest and apoptosis in prostate cancer cells. Oncogene 2001, 20, 2927–2936. [Google Scholar] [CrossRef]
- Arakaki, N.; Toyofuku, A.; Emoto, Y.; Nagao, T.; Kuramoto, Y.; Shibata, H.; Higuti, T. Induction of G(1) cell cycle arrest in human umbilical vein endothelial cells by flavone’s inhibition of the extracellular signal regulated kinase cascade. Biochem. Cell Biol. 2004, 82, 583–588. [Google Scholar] [CrossRef]
- Regula, K.M.; Kirshenbaum, L.A. p53 activates the mitochondrial death pathway and apoptosis of ventricular myocytes independent of de novo gene transcription. J. Mol. Cell. Cardiol. 2001, 33, 1435–1445. [Google Scholar] [CrossRef]
- Marchenko, N.D.; Moll, U.M. The role of ubiquitination in the direct mitochondrial death program of p53. Cell Cycle 2007, 6, 1718–1723. [Google Scholar] [CrossRef]
- Fedorov, S.N.; Shubina, L.K.; Bode, A.M.; Stonik, V.A.; Dong, Z.G. Dactylone inhibits epidermal growth factor-induced transformation and phenotype expression of human cancer cells and induces G(1)-S arrest and apoptosis. Cancer Res. 2007, 67, 5914–5920. [Google Scholar]
- Fedorov, S.N.; Bode, A.M.; Stonik, V.A.; Gorshkova, I.A.; Schmid, P.C.; Radchenko, O.S.; Berdyshev, E.V.; Dong, Z.G. Marine alkaloid polycarpine and its synthetic derivative dimethylpolyearpine induce apoptosis in JB6 cells through p53-and caspase 3-dependent pathways. Pharm. Res. 2004, 21, 2307–2319. [Google Scholar]
- Lee, K.H.; Nishimura, S.; Matsunaga, S.; Fusetani, N.; Horinouchi, S.; Yoshida, M. Inhibition of protein synthesis and activation of stress-activated protein kinases by onnamide A and theopederin B, antitumor marine natural products. Cancer Sci. 2005, 96, 357–364. [Google Scholar] [CrossRef]
- Xia, Z.G.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar]
- Lewis, T.S.; Shapiro, P.S.; Ahn, N.G. Signal transduction through MAP kinase cascades. Adv.Cancer Res. 1998, 74, 49–139. [Google Scholar] [CrossRef]
- Glaesener, S.; Honecker, F.; Veltman, I.M.; Gillis, A.J.M.; Rohlfing, T.; Streichert, T.; Otto, B.; Brummendorf, T.H.; Looijenga, L.H.J.; Bokemeyer, C.; et al. Comparative proteome, transcriptome, and genome analysis of a gonadal and an extragonadal germ cell tumor cell. J. Proteome Res. 2008, 7, 3890–3899. [Google Scholar]
- Ummanni, R.; Mundt, F.; Pospisil, H.; Venz, S.; Scharf, C.; Barett, C.; Falth, M.; Kollermann, J.; Walther, R.; Schlomm, T.; et al. Identification of clinically relevant protein targets in prostate cancer with 2D-DIGE coupled mass spectrometry and systems biology network platform. PLos One 2011, 6. [Google Scholar] [CrossRef]
- Dyshlovoy, S.A.; Naeth, I.; Venz, S.; Preukschas, M.; Sievert, H.; Jacobsen, C.; Shubina, L.K.; Gesell Salazar, M.; Scharf, C.; Walther, R.; et al. Proteomic profiling of germ cell cancer cells treated with aaptamine, a marine alkaloid with antiproliferative activity. J. Proteome Res. 2012, 11, 2316–2330. [Google Scholar] [CrossRef]
- Samples Availability: Available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dyshlovoy, S.A.; Fedorov, S.N.; Kalinovsky, A.I.; Shubina, L.K.; Bokemeyer, C.; Stonik, V.A.; Honecker, F. Mycalamide A Shows Cytotoxic Properties and Prevents EGF-Induced Neoplastic Transformation through Inhibition of Nuclear Factors. Mar. Drugs 2012, 10, 1212-1224. https://doi.org/10.3390/md10061212
Dyshlovoy SA, Fedorov SN, Kalinovsky AI, Shubina LK, Bokemeyer C, Stonik VA, Honecker F. Mycalamide A Shows Cytotoxic Properties and Prevents EGF-Induced Neoplastic Transformation through Inhibition of Nuclear Factors. Marine Drugs. 2012; 10(6):1212-1224. https://doi.org/10.3390/md10061212
Chicago/Turabian StyleDyshlovoy, Sergey A., Sergey N. Fedorov, Anatoly I. Kalinovsky, Larisa K. Shubina, Carsten Bokemeyer, Valentin A. Stonik, and Friedemann Honecker. 2012. "Mycalamide A Shows Cytotoxic Properties and Prevents EGF-Induced Neoplastic Transformation through Inhibition of Nuclear Factors" Marine Drugs 10, no. 6: 1212-1224. https://doi.org/10.3390/md10061212
APA StyleDyshlovoy, S. A., Fedorov, S. N., Kalinovsky, A. I., Shubina, L. K., Bokemeyer, C., Stonik, V. A., & Honecker, F. (2012). Mycalamide A Shows Cytotoxic Properties and Prevents EGF-Induced Neoplastic Transformation through Inhibition of Nuclear Factors. Marine Drugs, 10(6), 1212-1224. https://doi.org/10.3390/md10061212