Dithiolopyrrolone Natural Products: Isolation, Synthesis and Biosynthesis


Abstract
:1. Introduction


{kind=link}
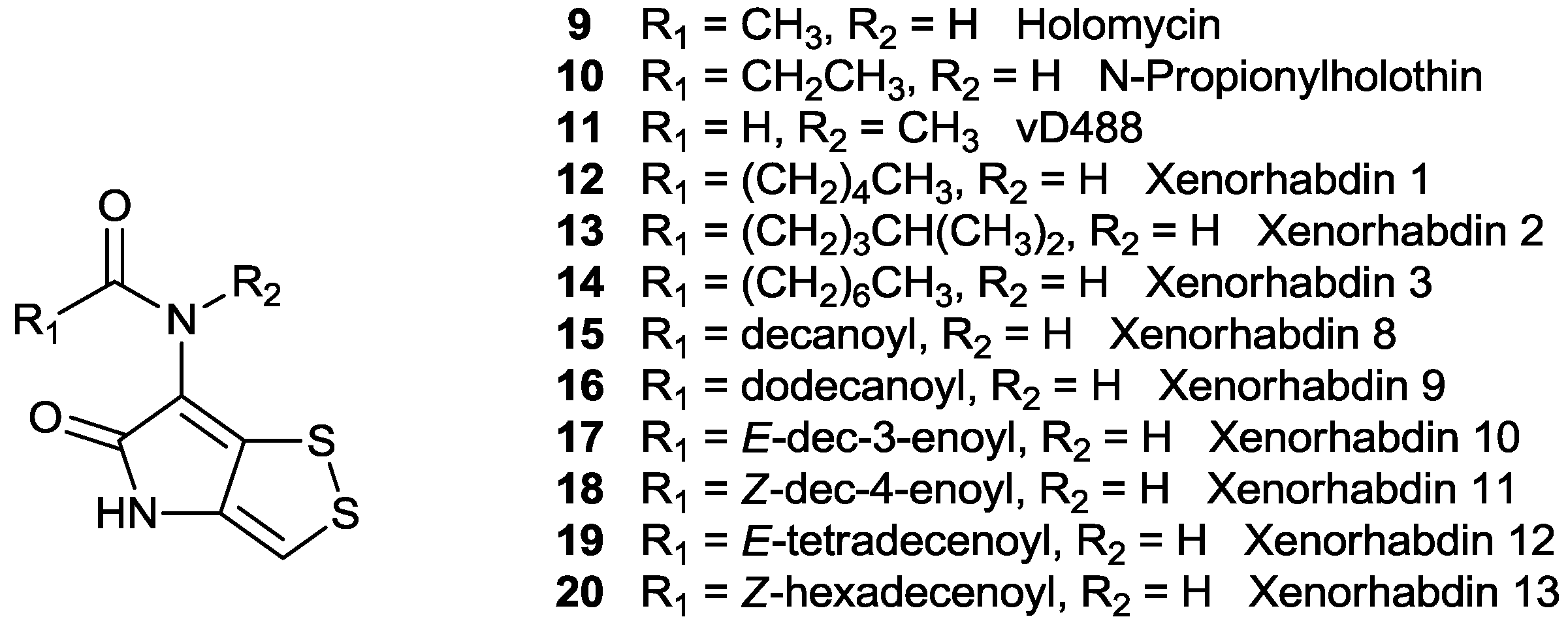{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
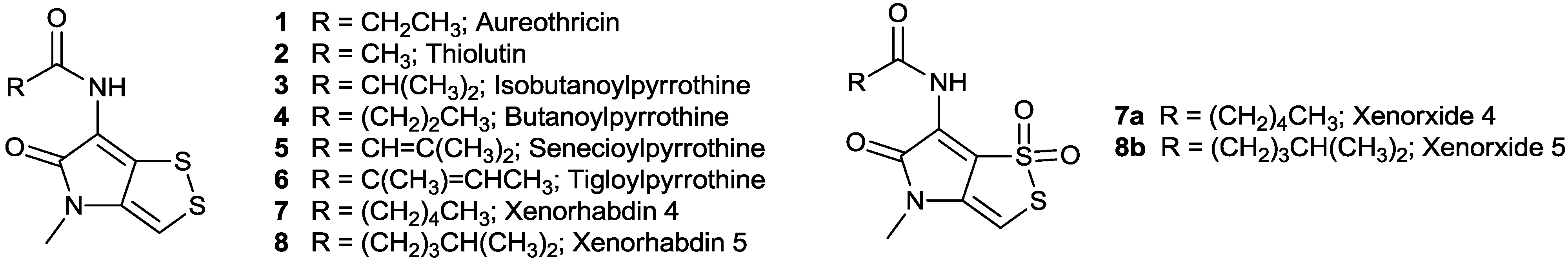
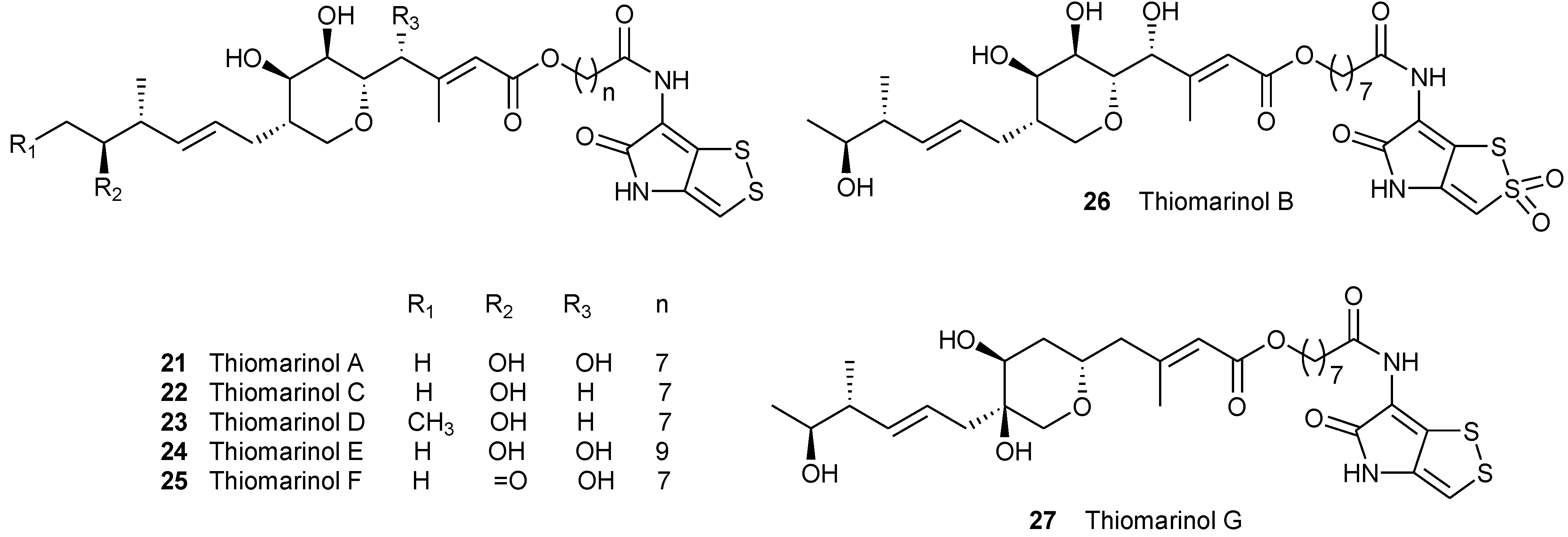
| NO. | Name | Structure | Source | Ref. | ||
|---|---|---|---|---|---|---|
| R1 | R2 | R3 | ||||
| 1 | Aureothricin | CH3CH2CO | H | CH3 | Streptomyces sp. 26A | [5] |
| 2 | Thiolutin | CH3CO | H | CH3 | Streptomyces albus | [6] |
| 3 | Isobutanoylpyrrothine | (CH3)2CHCO | H | CH3 | Saccharothrix algeriensis | [8] |
| 4 | Butanoylpyrrothine | CH3(CH2)2CO | H | CH3 | Saccharothrix algeriensis | [9,10] |
| 5 | Senecioylpyrrothine | (CH3)2C=CHCO | H | CH3 | Saccharothrix algeriensis | [9,10] |
| 6 | Tigloylpyrrothine | (CH3)CH=C(CH3)CO | H | CH3 | Saccharothrix algeriensis | [9,10] |
| 7 | Xenorhabdin 4 | CH3(CH2)4CO | H | CH3 | Xenorhabdus nematophilus XQ1 (ATCC 39497) | [11] |
| 8 | Xenorhabdin 5 | (CH3)2CH(CH2)3CO | H | CH3 | Xenorhabdus nematophilus XQ1 (ATCC 39497) | [11] |
| 9 | Holomycin | CH3CO | H | H | Streptomyces griseus (NRRL 2764) | [12] |
| 10 | N-Propanoylholothine | CH3CH2CO | H | H | Streptomyces sp. P662 | [13] |
| 11 | vD844 | CHO | CH3 | H | Actinomycete sp. | [14] |
| 12 | Xenorhabdin 1 | CH3(CH2)4CO | H | H | Xenorhabdus nematophilus XQ1 (ATCC 39497) | [11] |
| 13 | Xenorhabdin 2 | (CH3)2CH(CH2)3CO | H | H | Xenorhabdus nematophilus XQ1 (ATCC 39497) | [11] |
| 14 | Xenorhabdin 3 | CH3(CH2)6CO | H | H | Xenorhabdus nematophilus XQ1 (ATCC 39497) | [11] |
| 15 | Xenorhabdin 8 | decanoyl | H | H | Pseudoalteromonas sp. SANK 73390 | [15] |
| 16 | Xenorhabdin 9 | dodecanoyl | H | H | Pseudoalteromonas sp. SANK 73390 | [15] |
| 17 | Xenorhabdin 10 | E-dec-3-enoyl | H | H | Pseudoalteromonas sp. SANK 73390 | [15] |
| 18 | Xenorhabdin 11 | Z-dec-4-enoyl | H | H | Pseudoalteromonas sp. SANK 73390 | [15] |
| 19 | Xenorhabdin 12 | E-tetradecenoyl | H | H | Pseudoalteromonas sp. SANK 73390 | [15] |
| 20 | Xenorhabdin 13 | Z-hexadecenoyl | H | H | Pseudoalteromonas sp. SANK 73390 | [15] |
| 21 | Thiomarinol A | Marinolic acids A | H | H | Pseudoalteromonas sp. SANK 73390 | [16] |
| 22 | Thiomarinol B | Marinolic acids B | H | H | Pseudoalteromonas sp. SANK 73390 | [17] |
| 23 | Thiomarinol C | Marinolic acids C | H | H | Pseudoalteromonas sp. SANK 73390 | [17] |
| 24 | Thiomarinol D | Marinolic acids D | H | H | Pseudoalteromonas sp. SANK 73390 | [18] |
| 25 | Thiomarinol E | Marinolic acids E | H | H | Pseudoalteromonas sp. SANK 73390 | [18] |
| 26 | Thiomarinol F | Marinolic acids F | H | H | Pseudoalteromonas sp. SANK 73390 | [18] |
| 27 | Thiomarinol G | Marinolic acids G | H | H | Pseudoalteromonas sp. SANK 73390 | [18] |
2. Isolation and Characterization
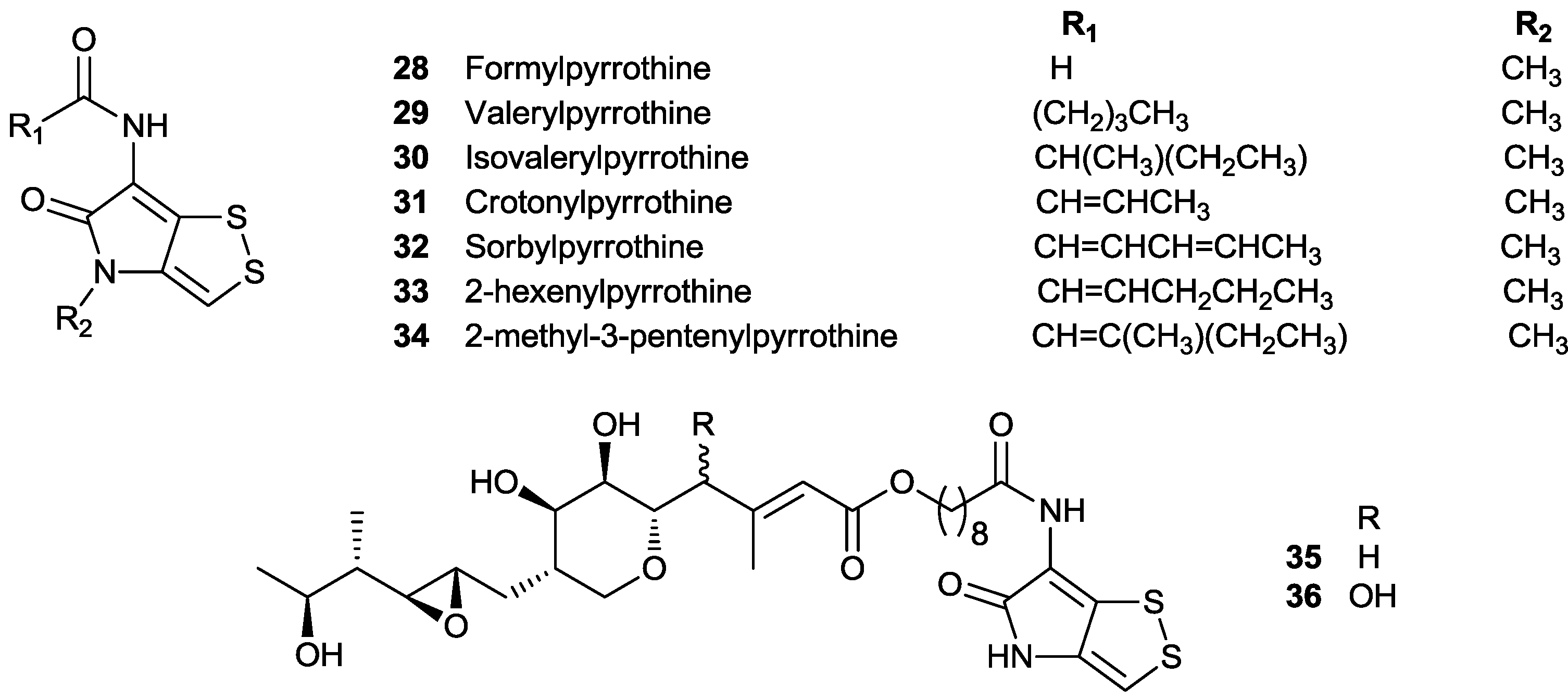
2.1. N-Methyl, N-Acylpyrrothine (Thiolutin-Type) Derivatives

2.2. N-Acylpyrrothine (Holomycin Type) Derivatives

2.3. Thiomarinols, PKS/NRPS Hybrid Antibiotic Natural Products

3. Bioactivities and Possible Mode of Action
| Organism | Thiolutin | Holomycin | Thiomarinol | |
|---|---|---|---|---|
| MIC (μg/mL)/IC50 (μM) | ||||
| G+ | Bacilius coagulans CIP 6625 | <0.2 | NC | NC |
| Bacillus subtilis ATCC 6633 | 2 | NC | NC | |
| Microcoecus leteus ATCC 9314 | <0.2 | NC | NC | |
| Staphylococcus aureus | 20 | 4 | <0.01 | |
| G− | Klebsiella pneumonia | 1 | 8 | 0.78 |
| Escherichia coli | >100 | <2 | 3.13 | |
| Salmonella enteric | >100 | NC | NC | |
| Pseudomanas aeruginosa | >100 | 64 | 0.39 | |
| Proteus mirabilis | NC | 4 | NC | |
| Haemophilus influenza | NC | <0.3 | NC | |
| Fungi | Mucor ramannianus NRRL 1829 | 10 | NC | NC |
| Penicillium sp. | 20 | NC | NC | |
| Alternaria sp. | 20 | NC | NC | |
| Fusarium | <40 | NC | NC | |
| Candida albicans | 20 | NC | NC | |
| Yeast | Saccharamyces cerevisiae | 10 | NA | NC |
| HUVEC | VTN | 0.83 | NC | NC |
| FN | 0.16 | NC | NC | |
| COL | 0.48 | NC | NC | |
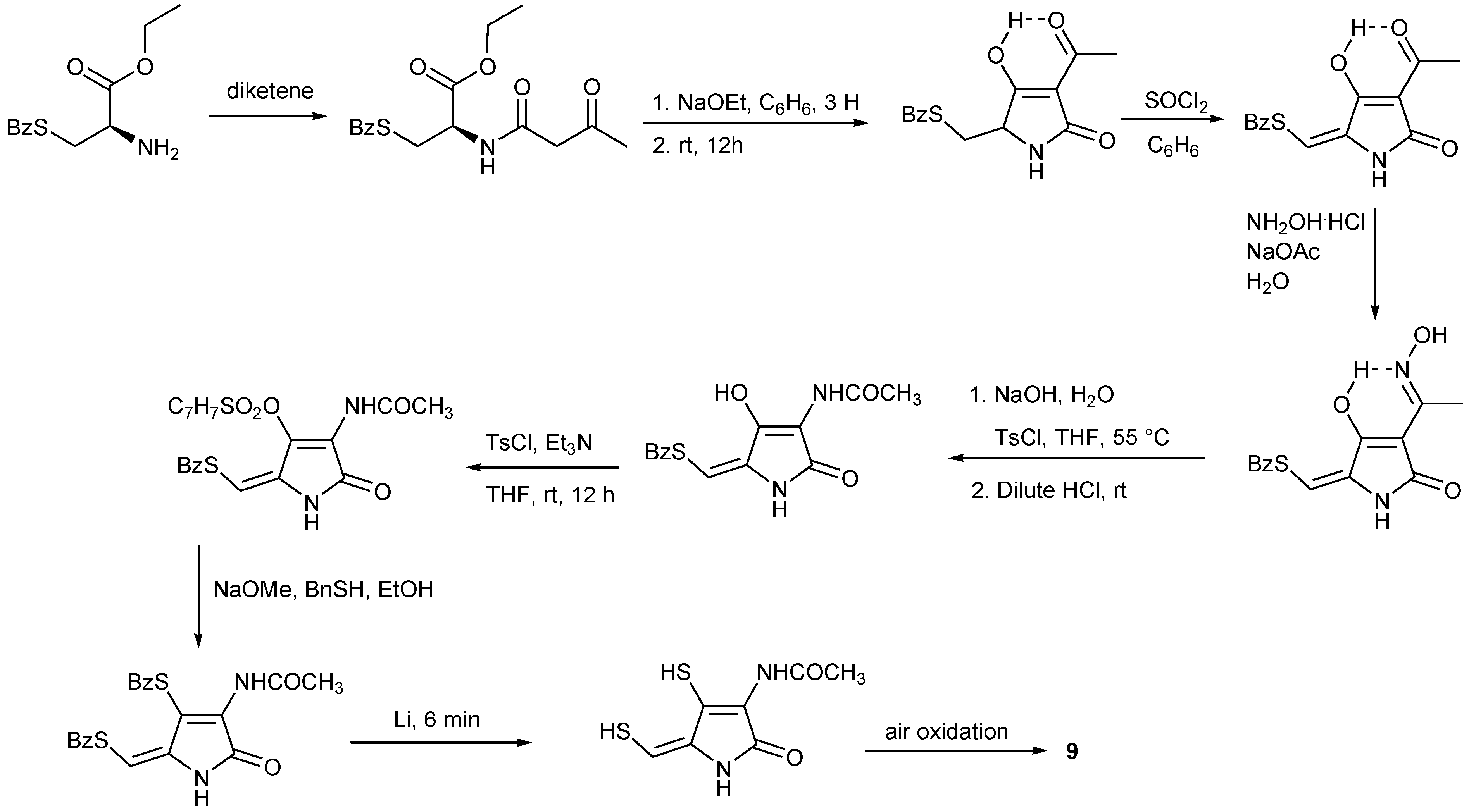
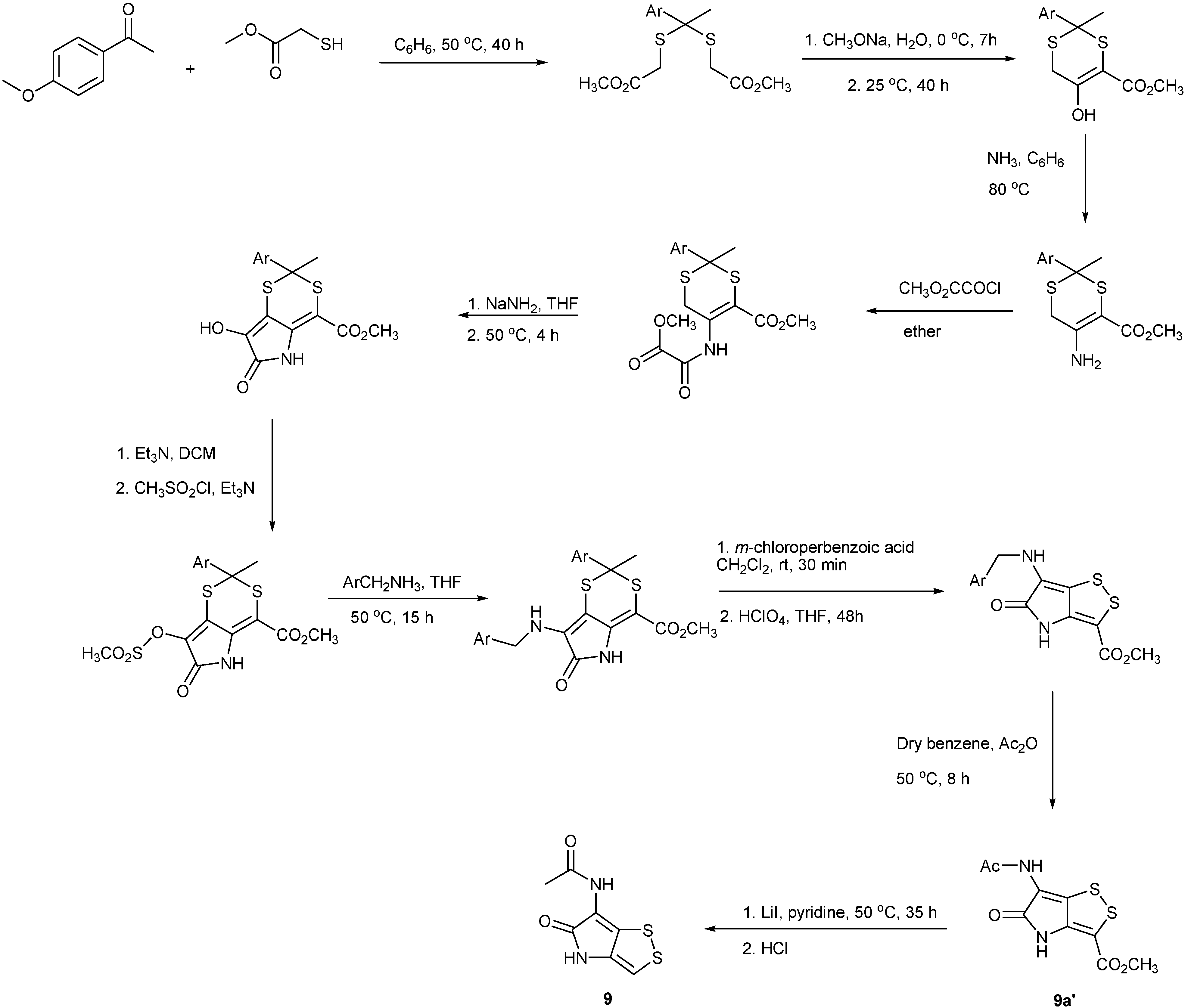
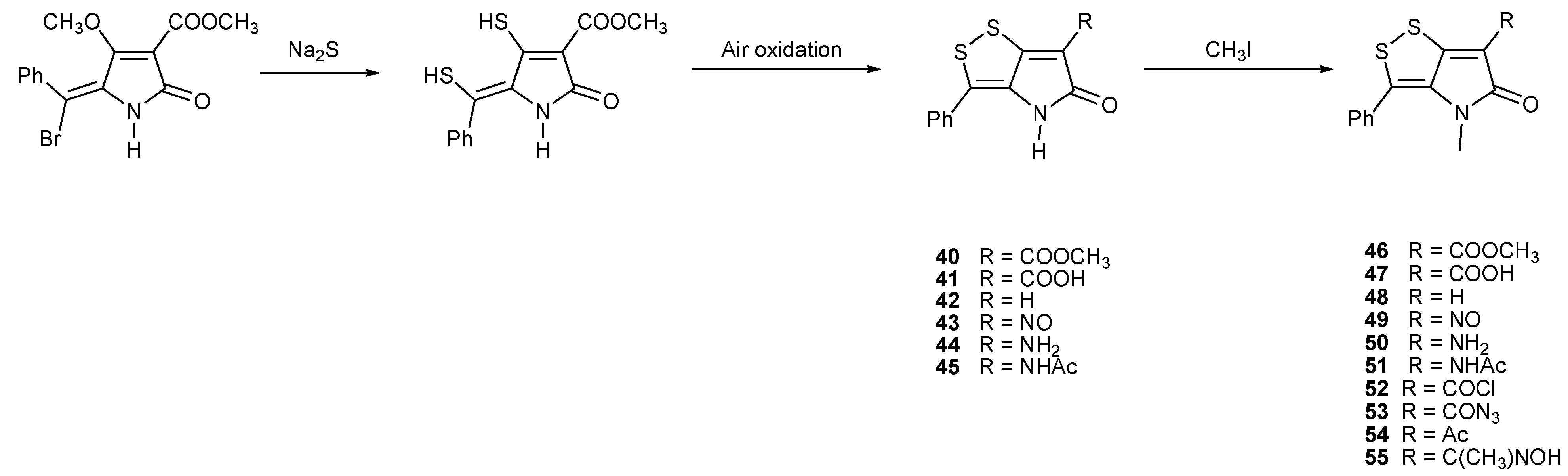
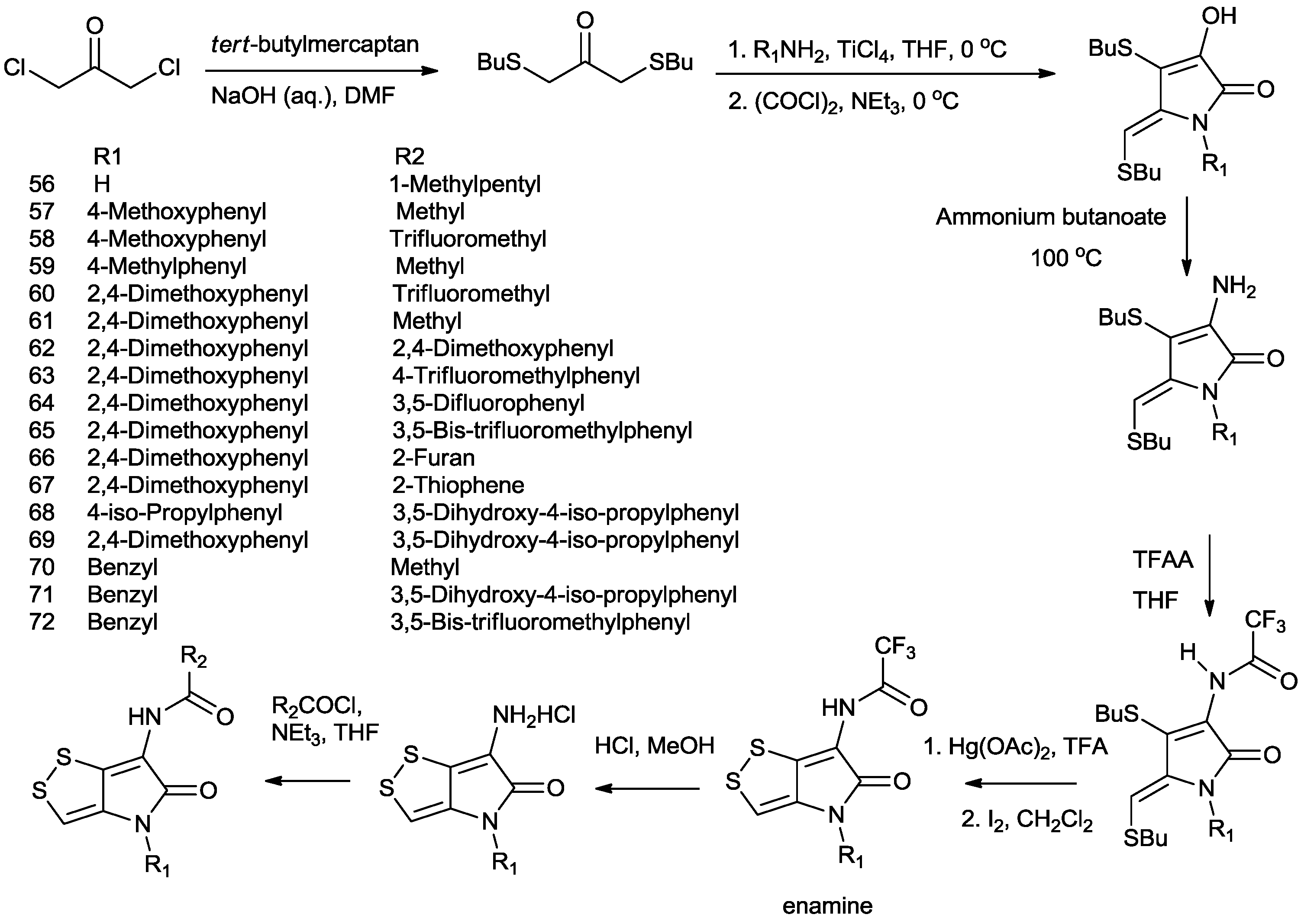
4. Total Synthesis of Dithiolopyrrolones





5. Biosynthesis of Dithiolopyrrolones
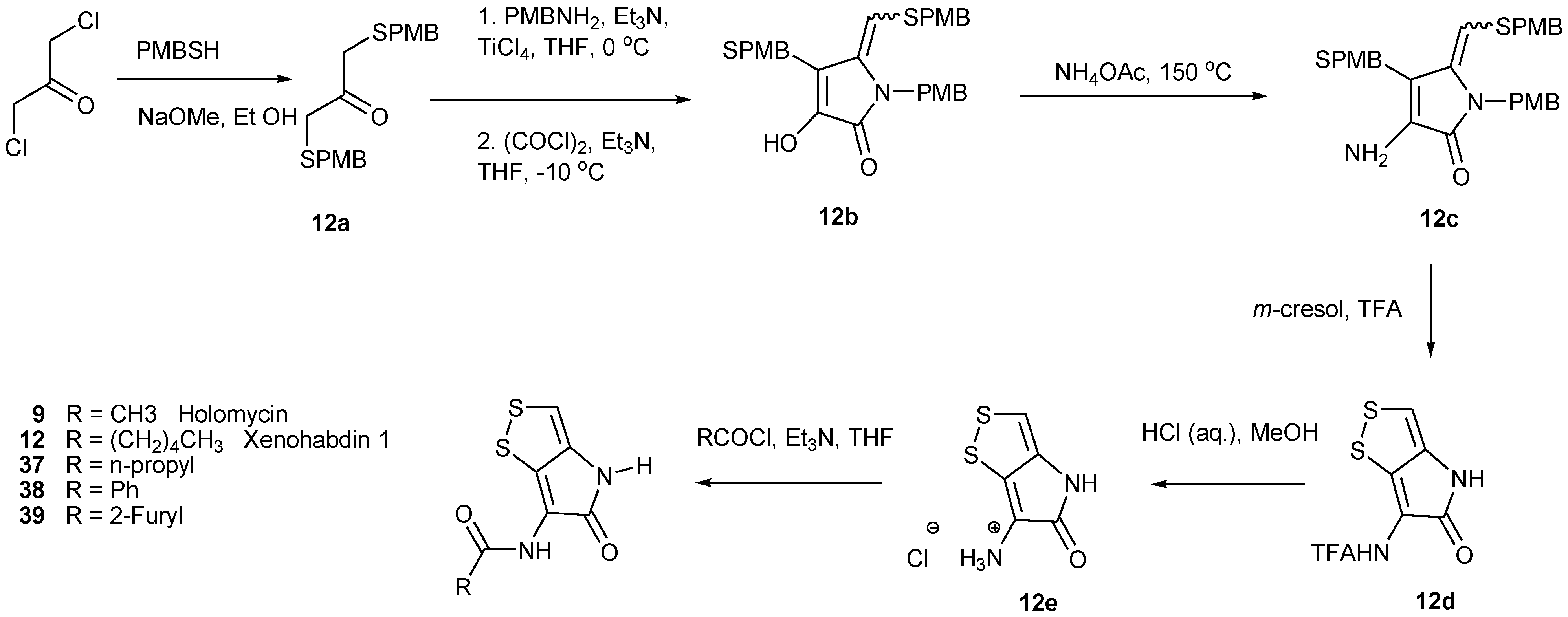
5.1. Precursor-Directed Biosynthesis (PDB) of Dithiolopyrrolones

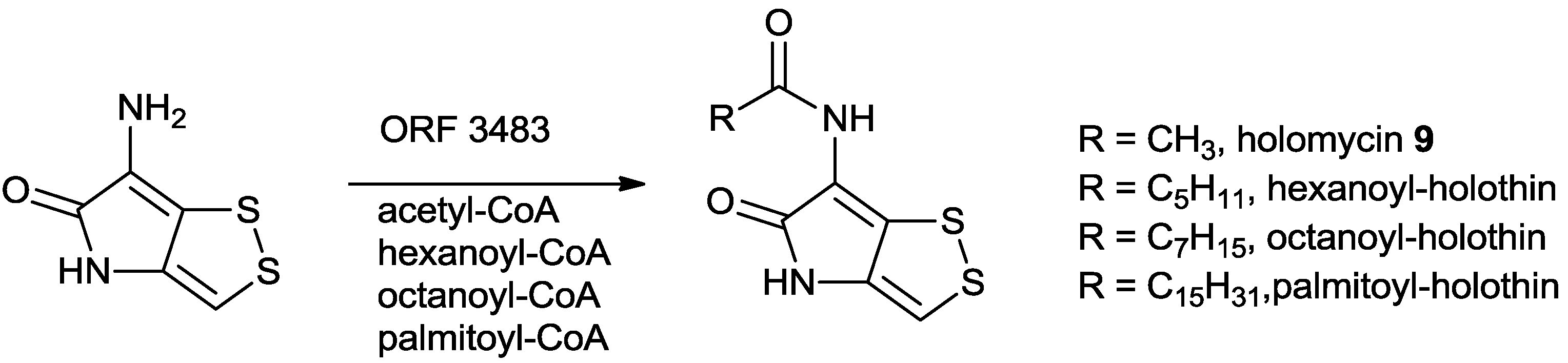
5.2. Biosynthesis of Dithiolopyrrolones
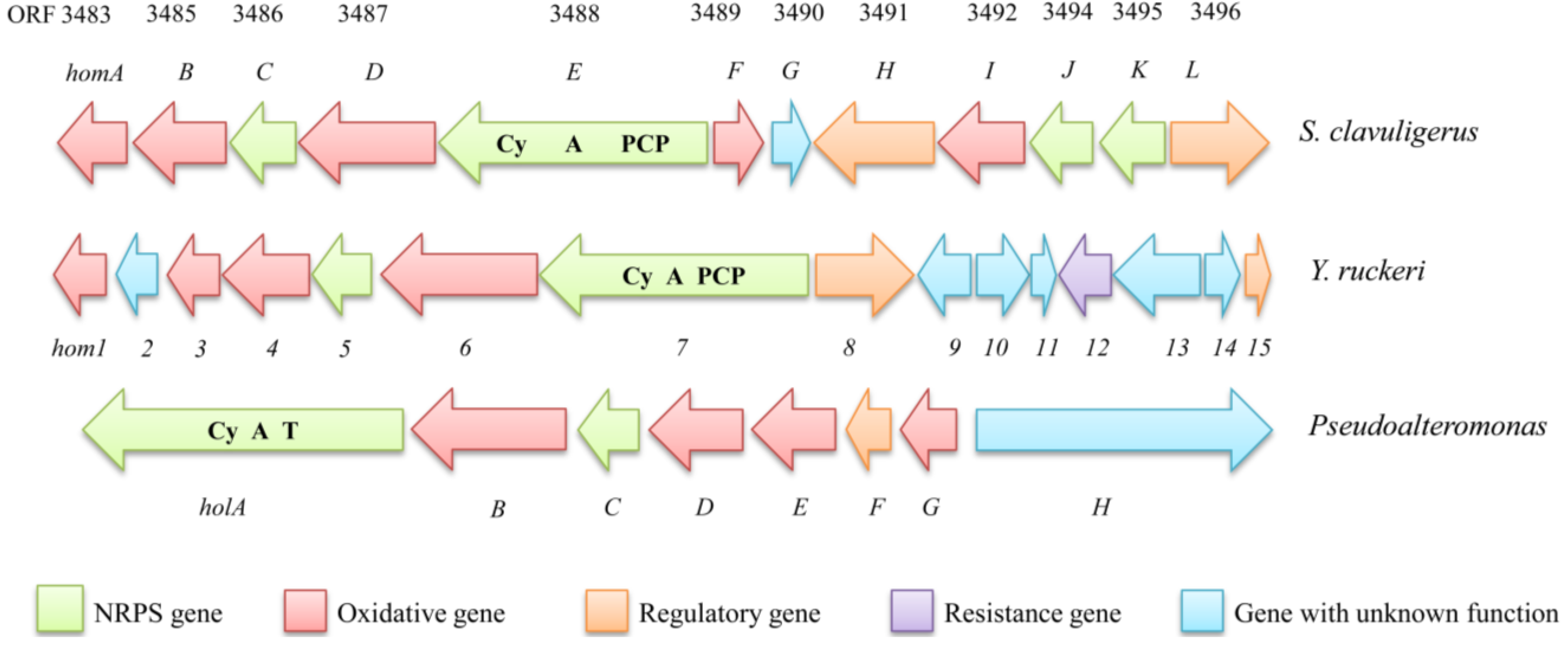
5.2.1. Identification of the Holomycin Gene Cluster in S. clavuligerus

| ORFs in S. clavuligerus [83] | Homolog in Y. ruckeri (Identity %) [38] | Homolog in Pseudoalteromonas (Identity %) [87] | Proposed Function |
|---|---|---|---|
| ORF3489(HlmF) | Hom1 (61%) | HolG (72%) | PPC-DC decarboxylase |
| ORF3490(HlmG) | Hom2(65%) | HolF (70%) | Globin |
| ORF3483(HlmA) | Hom3 (38%) | HolE (45%) | N-acyltransferase |
| ORF3485(HlmB) | Hom4 (58%) | HolD (63%) | Acyl-CoA dehydrogenase |
| ORF3486(HlmC) | Hom5 (36%) | HolC (42%) | Thioesterase |
| ORF3487(HlmD) | Hom6 (47%) | HolB (59%) | FMN-dependent oxdioreductase |
| ORF3488(HlmE) | Hom7 (47%) | HolA (55%) | NRPS (Cy-A-T) |
| ORF3491(HlmH) | Hom8 (61%) | MFS efflux protein |
5.2.2. Characterization of Key Enzymes during the Holomycin Biosynthesis in S. clavuligerus


5.2.3. Regulation of the Biosynthesis of Holomycin in S. clavuligerus
5.2.4. Identification of the Holomycin Gene Cluster in the Fish Pathogen Yersinia ruckeri
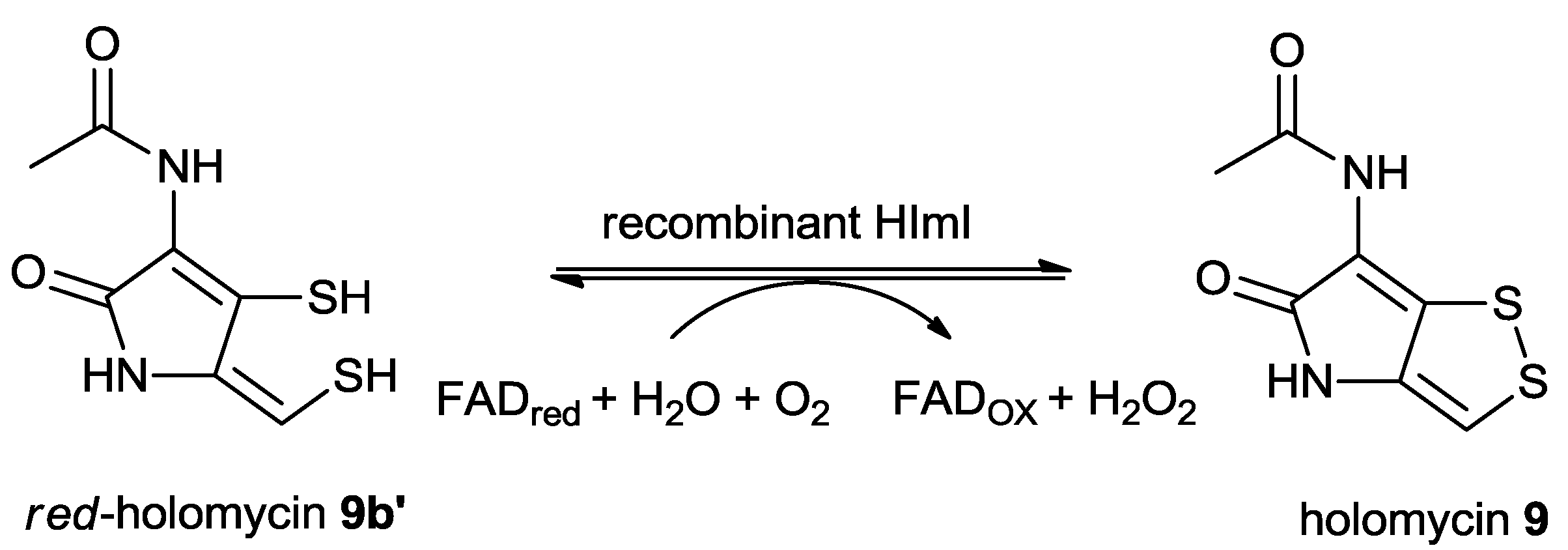
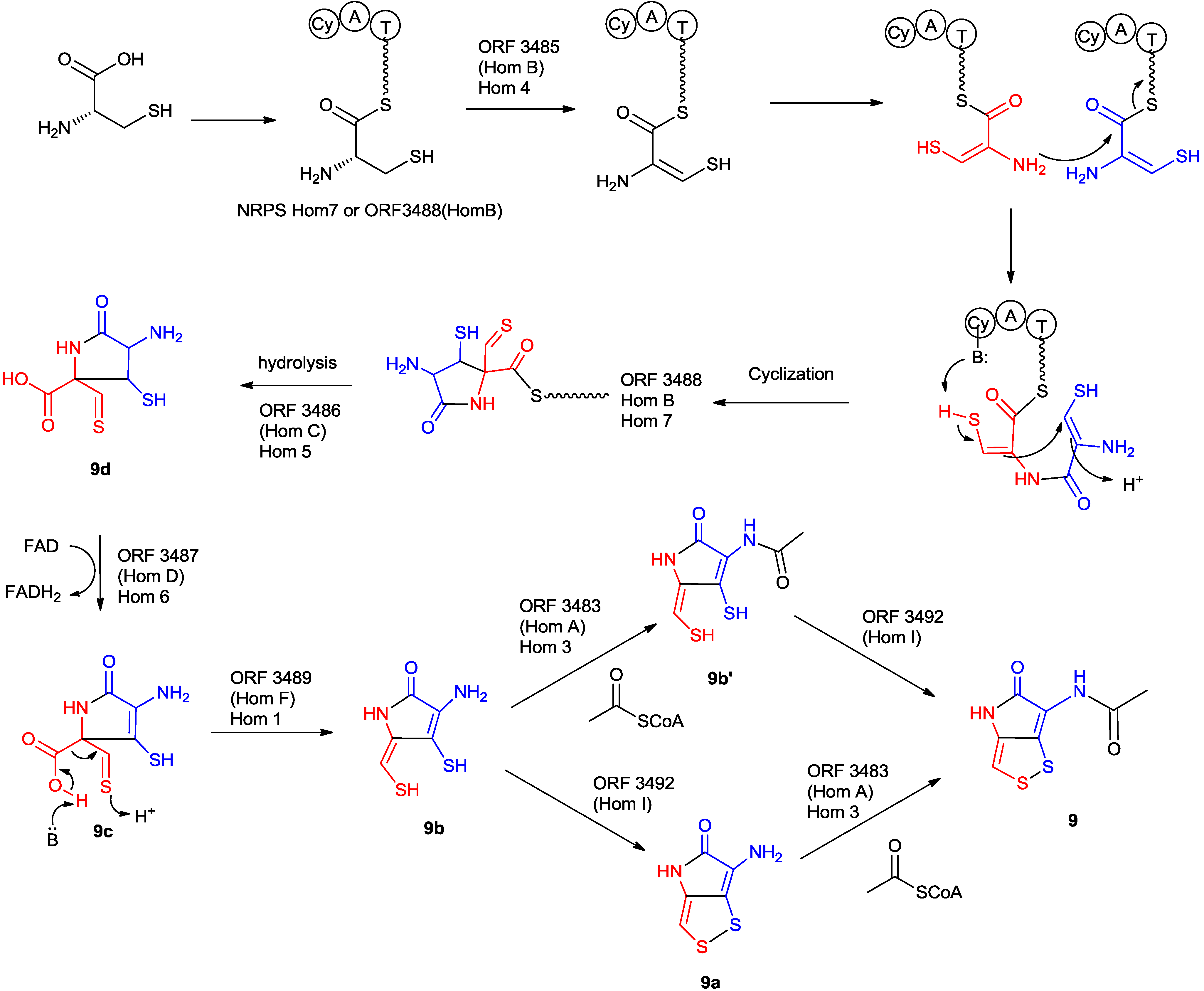
5.2.5. The Proposed Mechanism of the Formation of Holomycin

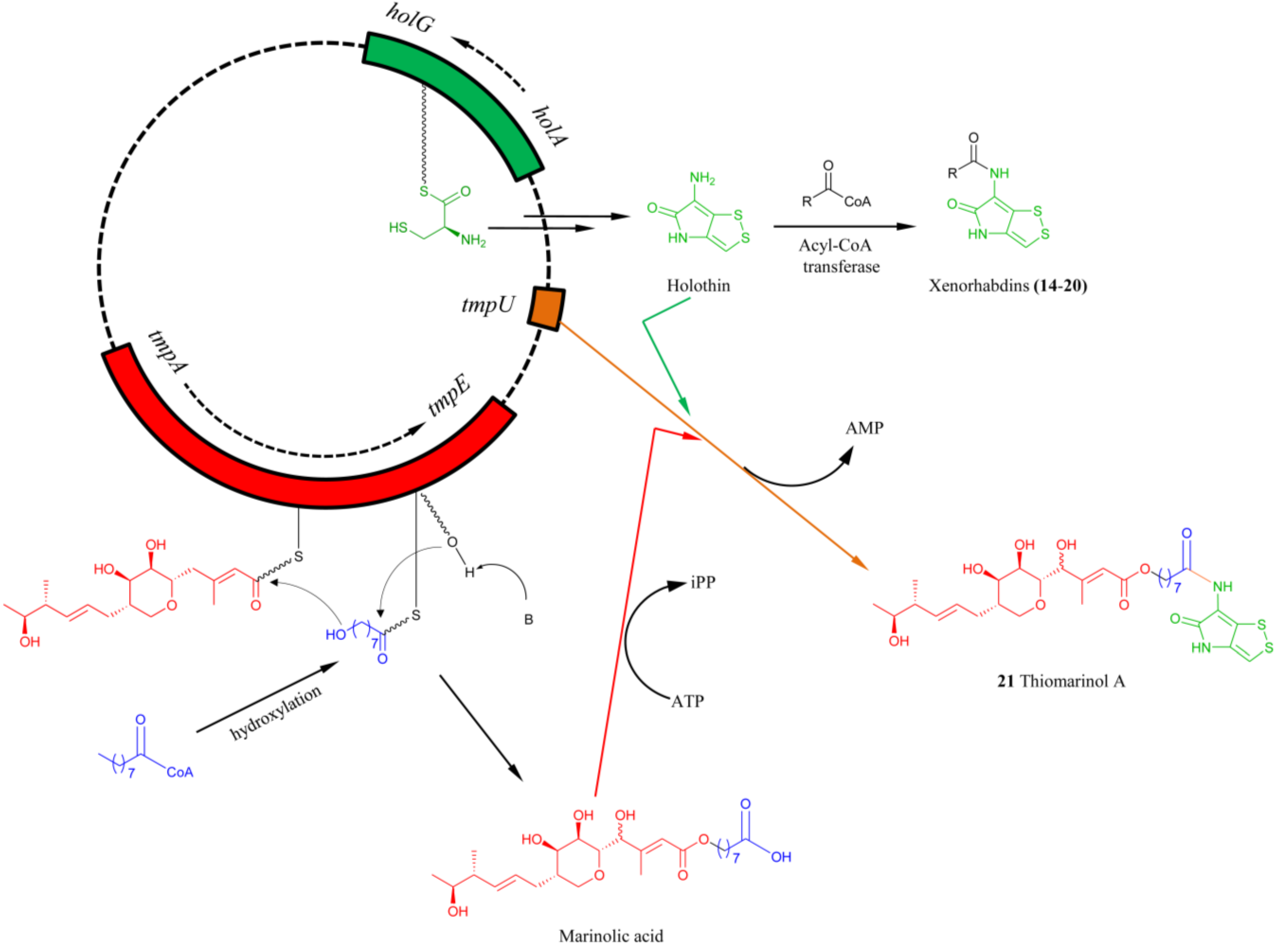
5.3. Biosynthesis of Thiomarinol Natural Products

6. Conclusions
Acknowledgements
Conflict of Interest
References
- Walsh, C.T.; Wright, G.D. Antimicrobials. Curr. Opin. Microbiol. 2009, 12, 473–475. [Google Scholar] [CrossRef]
- Fischbach, M.A.; Walsh, C.T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. [Google Scholar] [CrossRef]
- Li, J.W.H.; Vederas, J.C. Drug discovery and natural products: End of an era or an endless frontier? Science 2009, 325, 161–165. [Google Scholar] [CrossRef]
- Jiang, C.; Muller, W.E.G.; Schroder, H.C.; Guo, Y. Disulfide- and multisulfide-containing metabolites from marine organisms. Chem. Rev. 2012, 112, 2179–2207. [Google Scholar] [CrossRef]
- Umezawa, H.; Maeda, K.; Kosaka, H. Isolation of a new antibiotic substance, aureothricin from a strain of streptomyces. Jpn. Med. J. 1948, 1, 512–517. [Google Scholar]
- Tanner, F.W.; Means, J.A.; Davisson, J.W.; English, A.R. Thiolutin, an Antibiotic Produced by Certain Strains of Streptomyces albus. In Proceedings of the 118th Meeting of the American Chemical Society, Chicago, IL, USA, 1950.
- Chen, G.; Li, B.; Li, J.; Webster, J. Dithiolopyrrolone derivatives useful in the treatment of prolifeative disease. Patent WO03080624, 2 October 2003. [Google Scholar]
- Bhate, D.S.; Hulyalkar, R.K.; Menon, S.K. Isolation of iso-butyropyrrothine along with thiolutin and aureothricin from a Streptomyces sp. Experientia 1960, 16, 504–505. [Google Scholar]
- Lamari, L.; Zitouni, A.; Boudjelli, H.; Badji, H.; Sabaou, N.; Lebrihi, A.; Lefebvre, G.; Seguin, E.; Tillequin, F. New dithiolopyrrolone antibiotics from Saccharothrix sp. SA 233. I. Taxonomy, fermentation, isolation and biological activities. J. Antibiot. 2002, 55, 696–701. [Google Scholar] [CrossRef]
- Lamari, L.; Zitouni, A.; Dob, T.; Sabaou, N.; Lebrihi, A.; Germain, P; Seguin, E.; Tillequin, F. New dithiolopyrrolone antibiotics from Saccharothrix sp. SA 233. II. Physicochemical properties and structure elucidation. J. Antibiot. 2002, 55, 702–706. [Google Scholar] [CrossRef]
- McInerney, B.V.; Gregson, R.P.; Lacey, M.J.; Akhurst, R.J.; Lyons, G.R.; Rhodes, S.H.; Smith, D.R.; Engelhardt, L.M.; White, A.H. Biologically active metabolites from Xenorhabdus spp., part 1. Dithiolopyrrolone derivatives with antibiotic activity. J. Nat. Prod. 1991, 54, 774–784. [Google Scholar] [CrossRef]
- Gaeumann, E.; Prelog, V. Holothin and derivatives thereof. U.S. Patent 3,014,922 A, 16 December 1961. [Google Scholar]
- Okamura, K.; Soga, K.; Shimauchi, Y.; Ishikura, T. Holomycin and N-propionyl-holothin, antibiotics produced by a cephamycin C producer. J. Antibiot. 1977, 30, 334–336. [Google Scholar] [CrossRef]
- Von Daehne, W.; Godtfredsen, W.O.; Tybring, L.; Schaumburg, K. New antibiotics containing the 1,2-dithiolo[4,3-b] pyrrole ring system. J. Antibiot. 1969, 22, 233–236. [Google Scholar] [CrossRef]
- Murphy, A.C.; Fukuda, D.; Song, Z.; Hothersall, J.; Cox, R.J.; Willis, C.L.; Thomas, C.M.; Simpson, T.J. Engineered thiomarinol antibiotics active against MRSA are generated by mutagenesis and mutasynthesis of Pseudoalteromonas SANK73390. Angew. Chem. Int. Ed. 2011, 50, 3271–3274. [Google Scholar] [CrossRef]
- Shiozawa, H.; Kagasaki, T.; Haruyama, H.; Domon, H.; Utsui, Y.; Kodama, K.; Takahashi, S. Thiomarinol, a new hybrid antimicrobial antibiotic produced by a marine bacterium: Fermentation, isolation, structure, and antimicrobial activity. J. Antibiot. 1993, 46, 1834–1842. [Google Scholar] [CrossRef]
- Shiozawa, H.; Kagasaki, T.; Torikata, A.; Tanka, N.; Fujimoto, K.; Hata, T.; Furukawa, Y.; Takahashi, S. Thiomarinol B and C, new antimicrobial antibiotics produced by a marine bacterium. J. Antibiot. 1995, 48, 907–909. [Google Scholar] [CrossRef]
- Shiozawa, H.; Shimada, A.; Takahashi, S. Thiomarinol D, E, F and G, new hybrid antimicrobial antibiotic produced by a marine bacterium: fermentation, isolation, structure and antimicrobial activity. J. Antibiot. 1997, 50, 449–452. [Google Scholar] [CrossRef]
- Celmer, W.D.; Tanner, M.H., Jr.; Lees, T.M.; Solomons, I.A. Characterization of the antibiotic thiolutin and its relationship with aureothricin. J. Am. Chem. Soc. 1952, 74, 6304–6305. [Google Scholar]
- Seneca, H.; Kane, J.H.; Rockenbach, J. Bactericidal, protozoicidal and fungicidal properties of thiolutin. Antibiot. Chemother. 1952, 2, 357. [Google Scholar]
- Celmer, W.D.; Solomons, I.A. The structures of thiolutin and aureothricin, antibiotics containing a unique pyrrolinodithiole nucleus. J. Am. Chem. Soc. 1955, 77, 2861–2865. [Google Scholar] [CrossRef]
- Ninomiya, Y.T.; Yamada, Y.; Shirai, H.; Onistsuka, M.; Suhara, Y.; Maruyama, H.B. Biochemically Active Substances from Microorganisms. V. Pyrrothines, Potent Platelet Aggregation Inhibitors of Microbial Origin. Chem. Pharm. Bull. 1980, 28, 3157–3162. [Google Scholar] [CrossRef]
- Miyamoto, N.; Fukuoka, D.; Utimoto, K.; Nozaki, H. The reaction of styryl sulfoxides or sulfones with boranes. Bull. Chem. Soc. Jpn. 1974, 47, 503. [Google Scholar] [CrossRef]
- Bouras, N.; Mathieu, F.; Sabaou, N.; Lebrihi, A. Influence on dithiolopyrrolone antibiotic production by organic acids in Saccharothrix algeriensis NRRL B-24137. Process Biochem. 2007, 42, 925–933. [Google Scholar] [CrossRef]
- Paik, S.; Park, Y.H.; Suh, S.I.; Kim, H.S.; Lee, I.S.; Park, M.K.; Lee, C.S.; Park, S.H. Unusual cytotoxic phenethylamides from Xenorhabdus nematophilus. Bull. Korean Chem. Soc. 2001, 22, 372–374. [Google Scholar]
- Alexander, O.B. Isolation and identification of natural products and biosynthetic pathways from Photorhabdus and Xenorhabdus. Ph.D. Thesis, Saarland University, Saarbrücken, Germany, 18 December 2009. [Google Scholar]
- Akhurst, R.J. Taxonomic study of Xenorhabdus, a genus of bacteria symbiotically associated with insect pathogenic nematodes. Int. J. Syst. Bacteriol. 1983, 33, 38–45. [Google Scholar] [CrossRef]
- Thomas, G.M.; Poinar, G.O. Xenorhabdus gen. nov., a genus of entomopathogenic nematophilic bacteria of the family Entero-bacteriaceae. Int. J. Syst. Bacteriol. 1979, 29, 352–360. [Google Scholar] [CrossRef]
- Kenig, M.; Reading, C. Holomycin and an antibiotic (MM 19290) related to tunicamycin, metabolites of Streptomyces clavuligerus. J. Antibiot. 1979, 32, 549–554. [Google Scholar] [CrossRef]
- Trown, P.W.; Abraham, E.P.; Newton, G.G.F. Incorporation of acetate into cephalosporin C. Biochem. J. 1962, 84, 157–161. [Google Scholar]
- Trown, P.W.; Smith, B.; Abraham, E.P. Biosynthesis of cephalosporin C from amino acids. Biochem. J. 1963, 86, 284–291. [Google Scholar]
- Mamoru, A.; Yashuhiro, I.; Masaki, N.; Hisashi, K.; Shinichi, S. Process for the production of antibiotic substance cephemimycin. Patent US3865693, 11 February 1975. [Google Scholar]
- Miller, A.K.; Celozzi, E.; Kong, Y.; Pelak, B.A.; Kropp, H.; Stapley, E.O.; Hendlin, D. Cephamycins, a new family of β-lactam antibiotics. IV. In vivo studies. Antimicrob. Agents Chemother. 1972, 2, 287–290. [Google Scholar] [CrossRef]
- Neu, H.C.; Fu, K.P. Clavulanic acid, a novel inhibitor of β-lactamases. Antimicrob. Agents Chemother. 1978, 14, 650–655. [Google Scholar] [CrossRef]
- Kirby, R. An unstable genetic element affecting the production of the antibiotic holomycin by Streptomyces clavuligerus. FEMS Microbiol. Lett. 1978, 3, 283–286. [Google Scholar] [CrossRef]
- Hou, Y.H.; Li, F.C.; Wang, S.J.; Wang, Q.F. Intergeneric conjugation in holomycin-producing marine Streptomyces sp. strain M095. Microbiol. Res. 2008, 163, 96–104. [Google Scholar] [CrossRef]
- Wietz, M.; Mansson, M.; Gotfredsen, C.H.; Larsen, T.O.; Gram, L. Antibacterial compounds from marine Vibrionaceae isolated on a global expedition. Mar. Drugs 2010, 8, 2946–2960. [Google Scholar] [CrossRef]
- Qin, Z.; Baker, A.T.; Raab, A.; Huang, S.; Wang, T.H.; Yu, Y.; Jaspars, M.; Secombes, C.J.; Deng, H. The fish pathogen Yersinia ruckeri produces holomycin and uses an RNA methyltransferase for self-resistance. J. Biol. Chem. 2013, 288, 14688–14697. [Google Scholar]
- El-Sayed, A.K.; Hotherall, J.; Cooper, S.M.; Stephens, E.; Simpson, T.J.; Thomas, C.M. Characterization of the mupirocin biosynthesis gene cluster from Pseudomonas fluorescens NCIMB 10586. Chem. Biol. 2003, 10, 419–430. [Google Scholar] [CrossRef]
- Thomas, C.M.; Hotherall, J.; Willis, C.L.; Simpson, T.J. Resistance and synthesis of the antibiotic mupirocin. Nat. Rev. Microbiol. 2010, 8, 281–289. [Google Scholar] [CrossRef]
- Bowman, J.P. Bioactive compound synthetic capacity and ecological significance of marine bacterial genus Pseudoalteromonas. Mar. Drugs 2007, 5, 220–241. [Google Scholar] [CrossRef]
- Holmström, C.; Kjelleberg, S. Marine Pseudoalteromonas species are associated with higher organisms and produce biologically active extracellular agents. FEMS Microbiol. Ecol. 1999, 30, 285–293. [Google Scholar] [CrossRef]
- Shiozawa, H.; Takahashi, S. Configurational studies on thiomarinol studies. J. Antibiot. 1994, 47, 851–853. [Google Scholar] [CrossRef]
- Stierle, D.B.; Stierle, A.A. Pseudomonic acid derivatives from a marine bacterium. Experientia 1992, 48, 1165–1169. [Google Scholar] [CrossRef]
- Jimenez, A.; Tipper, D.J.; Davies, J. Mode of Action of Thiolutin, an inhibitor of macromolecular synthesis in Saccharomyces cerevisia. Antimicrob. Agents Chemother. 1973, 3, 729–738. [Google Scholar] [CrossRef]
- Tipper, D.J. Inhibition of Yeast Ribonucleic-Acid Polymerases by Thiolutin. J. Bacteriol. 1973, 116, 245–256. [Google Scholar]
- Jia, Y.F.; Wu, S.L.; Isenberg, J.S.; Dai, S.J.; Sipes, J.M.; Field, L.; Zeng, B.X.; Bandle, R.W.; Ridnour, L.A.; Wink, D.A.; Ramchandran, R.; Karger, B.L.; Roberts, D.D. Thiolutin inhibits endothelial cell adhesion by perturbing Hsp27 interactions with components of the actin and intermediate filament cytoskeleton. Cell Stress Chaperon 2010, 15, 165–181. [Google Scholar] [CrossRef]
- Deb, P.R.; Dutta, B.K. Activity of thiolutin against certain soil borne plant-pathogens. Curr. Sci. India 1984, 53, 659–660. [Google Scholar]
- Dai, S.; Jia, Y.; Wu, S.-L.; Isenberg, J.S.; Ridnour, L.A.; Bandle, R.W.; Wink, D.A.; Roberts, D.D.; Karger, B.L. Comprehensive characterization of heat shock protein 27 phosphorylation in human endothelial cells stimulated by the microbial dithiole thiolutin. J. Proteome Res. 2008, 7, 4384–4395. [Google Scholar] [CrossRef]
- Oliva, B.; O’Neill, A.; Wilson, J.M.; O’Hanlon, P.J.; Chopra, I. Antimicrobial properties and mode of action of the pyrrothine holomycin. Antimicrob. Agents Chemother. 2001, 45, 532–5329. [Google Scholar] [CrossRef]
- Shiozawa, H.; Fukuoka, T.; Fujimoto, K.; Kodama, K. Thiomarinols: Discovery from a marine bacterium, structure-activity relationship, and efficacy as topical antibacterial agents. Annu. Rep. Sankyo. Res. Lab. 1999, 51, 45–72. [Google Scholar]
- Khachatourians, G.G.; Tipper, D.J. Inhibition of messenger ribonucleic acid synthesis in Escherichia coli by thiolutin. J. Bacteriol. 1974, 119, 795–804. [Google Scholar]
- Khachatourians, G.G.; Tipper, D.J. In vivo effect of thiolutin on cell growth and macromolecular synthesis in Escherichia coli. Antimicrob. Agents Chemother. 1974, 6, 304–310. [Google Scholar] [CrossRef]
- Sivasubramanian, N.; Jayaraman, R. Thiolutin resistant mutants of Escherichia coli are the RNA chain initiation mutants? Mol. Gen. Genet. 1976, 145, 89–96. [Google Scholar] [CrossRef]
- O’Neill, A.; Oliva, B.; Storey, C.; Hoyle, A.; Fishwick, C.; Chopra, I. RNA polymerase inhibitors with activity against rifampin-resistant mutants of Staphylococcus aureus. Antimicrob. Agents Chemother. 2000, 44, 3163–3166. [Google Scholar] [CrossRef]
- Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.H.; Nishiyama, N.; Nakajima, I.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; Horinouchi, S. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002, 62, 4916–4921. [Google Scholar]
- Yakushiji, F.; Miyamoto, Y.; Kunoh, Y.; Okamoto, R.; Nakaminami, H.; Yamazaki, Y.; Noguchi, N.; Hayashi, Y. Novel hybrid-type antimicrobial agents targeting the switch region of bacterial RNA polymerase. ACS Med. Chem. Lett. 2013, 4, 220–224. [Google Scholar] [CrossRef]
- Li, B.; Forseth, R.R.; Bowers, A.A.; Schroeder, F.C.; Walsh, C.T. A backup plan for self-protection: S-methylation of holomycin biosynthetic intermediates in Streptomyces clavuligerus. ChemBioChem 2012, 13, 2521–2526. [Google Scholar] [CrossRef]
- Juhl, M.J.; Clark, D.P. Thiophene-degrading Escherichia coli mutants possess sulfone oxidase activity and show altered resistance to sulfur-containing antibiotics. Appl. Environ. Microbiol. 1990, 56, 3179–3185. [Google Scholar]
- Mariani, R.; Maffioli, S.I. Bacterial RNA Polymerase Inhibitors: An Organized Overview of their Structure, Derivatives, Biological Activity and Current Clinical Development Status. Curr. Med. Chem. 2009, 16, 430–454. [Google Scholar] [CrossRef]
- Floss, H.G.; Yu, T.W. Rifamycin-mode of action, resistance, and biosynthesis. Chem. Rev. 2005, 105, 621–632. [Google Scholar] [CrossRef]
- Schmidt, U.; Geiger, F. Total Synthesis of the Antibiotics Thiolutin, Aureothricin, and Holomycin. Angew. Chem. Int. Ed. 1962, 1, 265. [Google Scholar]
- Buchi, G.; Lukas, G. A total synthesis of holomycin. J. Am. Chem. Soc. 1964, 86, 5654–5658. [Google Scholar] [CrossRef]
- Hagio, K.; Yoneda, N. Total synthesis of holomycin, thiolutin, and aureothricin. Bull. Chem. Soc. Jpn. 1974, 47, 1484. [Google Scholar] [CrossRef]
- Ellis, J.E.; Fried, J.H.; Harrison, I.T.; Rapp, E.; Ross, C.H. Synthesis of holomycin and derivatives. J. Org. Chem. 1977, 42, 2891–2893. [Google Scholar] [CrossRef]
- Kishi, Y.; Fukuyama, T.; Nakatsuka, S. A new method for the synthesis of epidithiodiketopiperazines. J. Am. Chem. Soc. 1973, 95, 6490–6492. [Google Scholar] [CrossRef]
- Hjelmgaard, T.; Givskov, M.; Nielsen, J. Expedient total synthesis of pyrrothine natural products and analogs. Org. Biomol. Chem. 2007, 5, 344–348. [Google Scholar] [CrossRef]
- Stachel, H.D.; Nienaber, J.; Zoukas, T. Ring-fused 1,2-dithioles, I. Synthesis of thiolutine and related compounds. Ann. Chem. 1992, 5, 473–480. [Google Scholar]
- Stachel, H.D.; Eckl, E.; Immerz-Winkler, E.; Kreiner, C.; Weigand, W.; Robl, C.; Wunsch, R.; Dick, S.; Drescher, N. Synthesis and reactions of new dithiolopyrrolones. Helvetica Chimica Acta 2002, 85, 4453–4467. [Google Scholar] [CrossRef]
- Chen, G.; Guo, Y.; Li, B. Dithiolopyrrolones compounds and their therapeutic applications. Patent WO2008038175 A3, 12 June 2008. [Google Scholar]
- Li, B.; Lyle, M.P.A.; Chen, G.; Li, J.; Hu, K.; Tang, L.; Alaoui-Jamali, A.; Webster, J. Substituted 6-amino-4H-[1,2]dithiolo[4,3-b]pyrrol-5-ones: Synthesis, structure-activity relationships, and cytotoxic activity on selected human cancer cell lines. Bioorg. Med. Chem. 2007, 15, 4601–4608. [Google Scholar] [CrossRef]
- Gao, X.; Hall, D.G. Catalytic asymmetric synthesis of a potent thiomarinol antibiotic. J. Am. Chem. Soc. 2005, 127, 1628–1629. [Google Scholar] [CrossRef]
- Goss, R.J.M.; Shankar, S.; Fayad, A.A. The generation of “unNatural” products: Synthetic biology meets synthetic chemistry. Nat. Prod. Rep. 2012, 29, 870–889. [Google Scholar] [CrossRef]
- Cane, D.E.; Kudo, F.; Kinoshita, K.; Khosla, C. Precursor-directed biosynthesis: Biochemical basis of the remarkable selectivity of the erythromycin polyketide synthase towards unsaturated triketides. Chem. Biol. 2002, 9, 131–142. [Google Scholar] [CrossRef]
- Merrouche, R.; Bouras, N.; Coppel, Y.; Mathieu, F.; Monje, M.C.; Sabaou, N.; Lebrihi, A. Dithiolopyrrolone antibiotic formation induced by adding valeric acid to the culture broth of Saccarothrix algeriensis. J. Nat. Prod. 2010, 73, 1164–1166. [Google Scholar] [CrossRef]
- Merrouche, R.; Bouras, N.; Coppel, Y.; Mathieu, F.; Sabaou, N.; Lebrihi, A. New dithiolopyrrolone antibiotics induced by adding sorbic acid to the culture medium of Saccharothrix algeriensis NRRL B-24137. FEMS Microbiol. Lett. 2011, 318, 41–46. [Google Scholar] [CrossRef]
- Pacholec, M.; Freel Meyers, C.L.; Oberthur, M.; Kahne, D.; Walsh, C.T. Characterization of the aminocoumarin ligase SimL from the simocyclinone pathway and tandem incubation with NovM,P,N from the novobiocin pathway. Biochemistry 2005, 44, 4949–4956. [Google Scholar] [CrossRef]
- Okanishi, M.; Umezawa, H. Plasmids involved in antibiotic production in Streptomyces. In Genetics of the Actinomyetales; Freerksen, E, Tarnok, I, Thumin, J.H., Eds.; Gustav Fischer Verlag: Stuttgard, NY, USA, 1978; pp. 19–38. [Google Scholar]
- Furumai, T.; Takeda, K.; Okanishi, M. Function of plasmid in the production of aureothricin 1. Elimination of plasmids and alteration of phenotypes caused by protoplast regeneration in Streptomyces kasugaensis. J Antibiot. 1982, 35, 1367–1373. [Google Scholar] [CrossRef]
- Fuente, A.; Lorenzana, L.M.; Martin, J.F.; Liras, P. Mutants of Streptomyces clavuligerus with disruptions in different genes for clavulanic acid biosynthesis produce large amounts of holomycin: Possible cross-regulation of two unrelated secondary metabolic pathways. J. Bacteriol. 2002, 184, 6559–6565. [Google Scholar] [CrossRef]
- Chorin, A.C.; Bijeire, L.; Monje, M.C.; Baziard, G.; Lebrihi, A.; Mathieu, F. Expression of pyrrothine N-acyltransferase activities in Saccharothrix algeriensis NRRL B-24137: New insights into dithiolopyrrolone antibiotic biosynthetic pathway. J Appl. Microbiol. 2009, 107, 1751–1762. [Google Scholar] [CrossRef]
- Medema, M.H.; Trefzer, A.; Kovalchuk, A.; van den Berg, M.; Müller, U.; Heijne, W.; Wu, L.; Alam, M.T.; Ronning, C.M.; Nierman, W.C.; et al. The sequence of a 1.8-Mb bacterial linear plasmid reveals a rich evolutionary reservoir of secondary metabolic pathways. Genome Biol. Evol. 2010, 2, 212–224. [Google Scholar] [CrossRef]
- Li, B.; Walsh, C.T. Identification of the gene cluster for the dithiolopyrrolone antibiotic holomycin in Streptomyces clavuligerus. Proc. Natl. Acad. Sci. USA 2010, 107, 19731–19735. [Google Scholar] [CrossRef]
- Huang, S.; Zhao, Y.; Qin, Z.; Wang, X.; Onega, M.; Chen, L.; He, J.; Yu, Y.; Deng, H. Identification and heterologous expression of the biosynthetic gene cluster for holomycin produced by Streptomyces clavuligerus. Process Biochem. 2011, 46, 811–816. [Google Scholar] [CrossRef]
- Robles-Reglero, V.; Santamarta, I.; Alvarez-Álvarez, R.; Martín, J.F.; Liras, P. Transcriptional analysis and proteomics of the holomycin gene cluster in overproducer mutants of Streptomyces clavuligerus. J. Biotechnol. 2013, 163, 69–76. [Google Scholar] [CrossRef]
- Wang, C.; Wesener, S.R.; Zhang, H.; Cheng, Y.Q. An FAD-dependent pyridine nucleotide-disulfide oxidoreductase is involved in disulfide bond formation in FK228 anticancer depsipeptide. Chem. Biol. 2009, 16, 585–593. [Google Scholar] [CrossRef]
- Fukuda, D.; Haines, A.S.; Song, Z.; Murphy, A.C.; Hothersall, J.; Stephens, E.R.; Gurney, R.; Cox, R.J.; Crosby, J.; Willis, C.L.; Simpson, J.T.; Thomas, C.M. A natural plasmid uniquely encodes two biosynthetic pathways creating a potent anti-MRSA antibiotic. PLoS One 2011, 6, e18031. [Google Scholar] [CrossRef]
- Scharf, D.H.; Remme, N.; Heinekamp, T.; Hortschansky, P.; Brakhage, A.A.; Hertweck, C. Transannular disulfide formation in gliotoxin biosynthesis and its role in self-resistance of the human pathogen Aspergillus fumigatus. J. Am. Chem. Soc. 2010, 132, 10136–10141. [Google Scholar] [CrossRef]
- Li, B.; Walsh, C.T. Streptomyces clavuligerus HlmI is an intramolecular disulfide-forming dithiol oxidase in holomycin biosynthesis. Biochemistry 2011, 50, 4615–4622. [Google Scholar] [CrossRef]
- Nárdiz, N.; Santamarta, I.; Lorenzana, L.M.; Martín, J.F.; Liras, P. A rhodanese-like protein is highly overrepresented in the mutant S. clavuligerus oppA2::aph: effect on holomycin and other secondary metabolites production. Microb. Biotechnol. 2011, 4, 216–225. [Google Scholar] [CrossRef]
- de la Fuente, A.; Martín, J.F.; Rodríguez-García, A.; Liras, P. Two proteins with ornithine acetyltransferase activity show different functions in Streptomyces clavuligerus: Oat2 modulates clavulanic acid biosynthesis in response to arginine. J. Bacteriol. 2004, 186, 6501–6507. [Google Scholar] [CrossRef]
- Liras, P.; Gomez-Escribano, J.P.; Santamarta, I. Regulatory mechanisms controlling antibiotic production in Streptomyces clavuligerus. J. Ind. Microbiol. Biotechnol. 2008, 35, 667–676. [Google Scholar] [CrossRef]
- Yin, H.; Xiang, S.; Zheng, J.; Fan, K.; Yu, T.; Yang, X.; Peng, Y.; Wang, H.; Feng, D.; Luo, Y.; Bai, H.; Yang, K. Induction of holomycin production and complex metabolic changes by the argR mutation in Streptomyces clavuligerus NP1. Appl. Environ. Microbiol. 2012, 78, 3431–3441. [Google Scholar] [CrossRef]
- Chen, L.; Wang, Y.; Guo, H.; Xu, M.; Deng, Z.; Tao, M. High-throughput screening for Streptomyces antibiotic biosynthesis activators. Appl. Environ. Microbiol. 2012, 78, 4526–4528. [Google Scholar] [CrossRef]
- Oh, D.C.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Induced production of emericellamides A and B from the marine-derived fungus Emericella sp. in competing co-culture. J. Nat. Prod. 2007, 70, 515–520. [Google Scholar] [CrossRef]
- Slattery, M.; Rajbhandari, I.; Wesson, K. Competition-mediated antibiotic induction in the marine bacterium Streptomyces tenjimariensis. Microb. Ecol. 2001, 41, 90–96. [Google Scholar]
- Charusanti, P.; Fong, N.L.; Nagarajan, H.; Pereira, A.R.; Li, H.J.; Abate, E.A.; Su, Y.; Gerwick, W.H.; Palsson, B.O. Exploiting adaptive laboratory evolution of Streptomyces clavuligerus for antibiotic discovery and overproduction. PLoS One 2012, 7, e33727. [Google Scholar]
- Condurso, H.L.; Bruner, S.D. Structure and noncanonical chemistry of nonribosomal peptide biosynthetic machinery. Nat. Prod. Rep. 2012, 29, 1099–1110. [Google Scholar] [CrossRef]
- Luft, T.; Li, S.M.; Scheible, H.; Kammerer, B.; Heide, L. Overexpression, purification and characterization of SimL, an amide synthetase involved in simocyclinone biosynthesis. Arch. Microbiol. 2005, 183, 277–285. [Google Scholar] [CrossRef]
- Steffensky, M.; Li, S.M.; Heide, L. Cloning, overexpression, and purification of novobiocic acid synthetase from Streptomyces spheroides NCIMB 11891. J. Biol. Chem. 2000, 275, 21754–21760. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Qin, Z.; Huang, S.; Yu, Y.; Deng, H. Dithiolopyrrolone Natural Products: Isolation, Synthesis and Biosynthesis. Mar. Drugs 2013, 11, 3970-3997. https://doi.org/10.3390/md11103970
Qin Z, Huang S, Yu Y, Deng H. Dithiolopyrrolone Natural Products: Isolation, Synthesis and Biosynthesis. Marine Drugs. 2013; 11(10):3970-3997. https://doi.org/10.3390/md11103970
Chicago/Turabian StyleQin, Zhiwei, Sheng Huang, Yi Yu, and Hai Deng. 2013. "Dithiolopyrrolone Natural Products: Isolation, Synthesis and Biosynthesis" Marine Drugs 11, no. 10: 3970-3997. https://doi.org/10.3390/md11103970
APA StyleQin, Z., Huang, S., Yu, Y., & Deng, H. (2013). Dithiolopyrrolone Natural Products: Isolation, Synthesis and Biosynthesis. Marine Drugs, 11(10), 3970-3997. https://doi.org/10.3390/md11103970