3.1. Synthesis of Marinopyrrole Derivatives
All chemicals were purchased from commercial suppliers and used without further purification. All solvents were dried and distilled before use. Tetrahydrofuran was distilled from sodium/benzophenone. Dichloromethane and acetonitrile were distilled over calcium hydride. Flash column chromatography was performed with silica gel (200–300 mesh). 1H NMR spectra were recorded at either 400 MHz or 600 MHz at ambient temperature. 13C NMR spectra were recorded at either 100 or 150 MHz at ambient temperature. Infrared spectra were recorded on a spectrophotometer (Perkin-Elmer Spectrum 100). Copies of NMR spectra of all the described compounds are provided in a Supporting Information Document. Melting points were determined with melting point apparatus (Fukai X-4). High resolution mass spectra were performed by electrospray ionization (ESI) on an Agilent ESI-TOF LC-MS 6200 system. Analytical HPLC was performed on an Agilent 1100 series with diode array detectors and auto samplers. All tested compounds possessed a purity of not less than 95%.
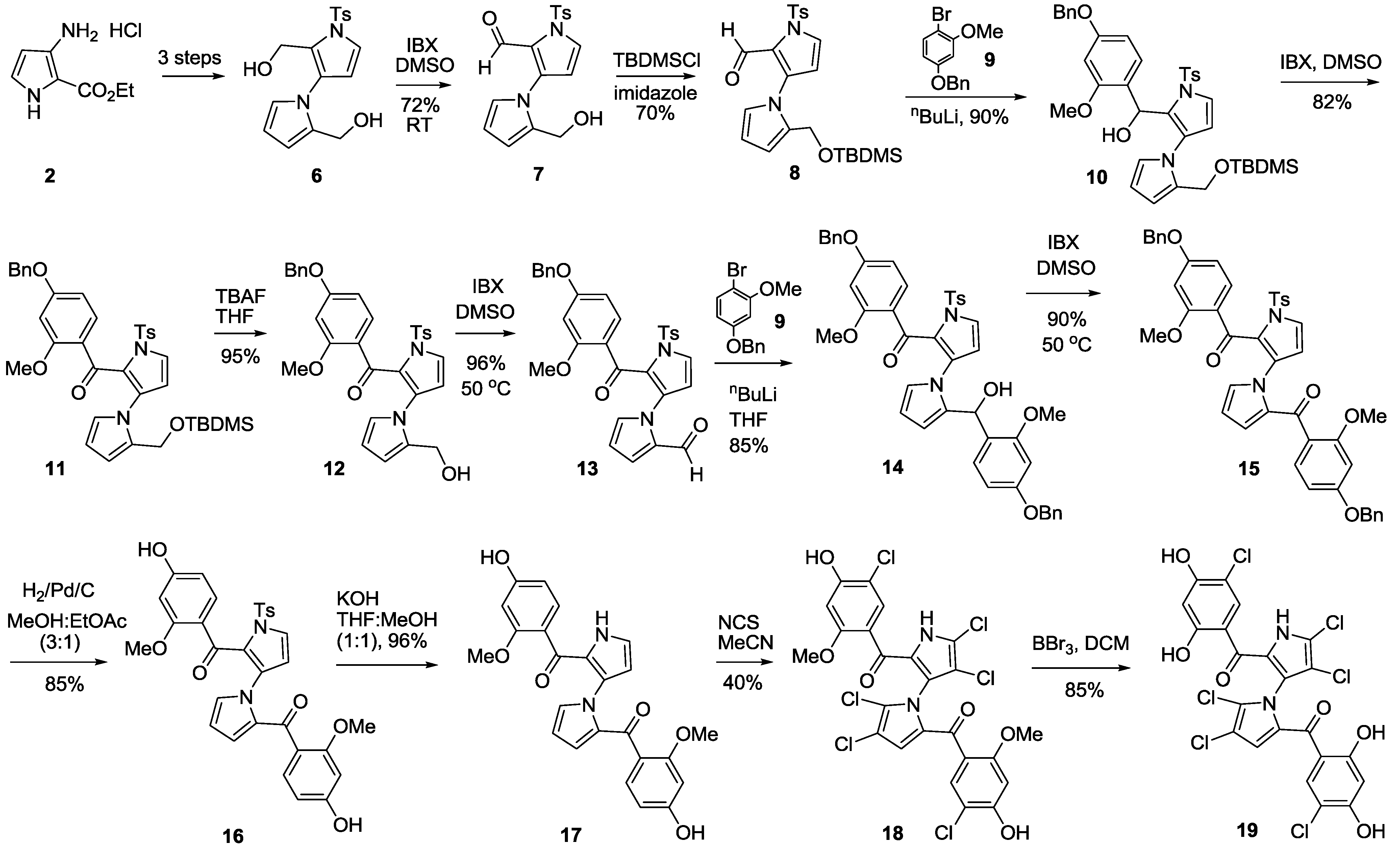
2-(Hydroxymethyl)-1′-tosyl-1′
H-1,3′-bipyrrole-2′-carbaldehyde (
7). To a solution of (1′-tosyl-1′
H-1,3′-bipyrrole-2,2′-diyl)dimethanol
6 obtained from ethyl 3-amino-1
H-pyrrole-2-carboxylate hydrochloride
2 in three steps [
3] (2.00 g, 5.8 mmol) in DMSO (30 mL) was added IBX (1.78 g, 6.4 mmol) at room temperature. After being stirred for 5 h, the mixture was quenched with water (50 mL). The suspension was filtered and the filtrate was extracted with EtOAc (25 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by flash column chromatography (25% EtOAc/petroleum ether,
Rf = 0.2) to give
7 (1.43 g, 72%) as a liquid.
1H NMR (400 MHz, CDCl
3) δ 2.43 (s, 3H), 2.87 (br, s, 1H), 4.44 (s, 2H), 6.33 (d,
J = 3.2 Hz, 1H), 6.38 (dd,
J = 4.0, 2.8 Hz, 1H), 7.05 (s, 1H), 7.09 (dd,
J = 4.4, 2.0 Hz, 1H), 7.31 (d,
J = 3.6 Hz, 1H), 7.35 (d,
J = 8.4 Hz, 2H), 7.82 (d,
J = 8.4 Hz, 2H), 9.46 (s, 1H) ppm;
13C NMR (CDCl
3, 100 MHz) δ 21.70, 53.00, 111.00, 111.16, 121.55, 123.57, 127.11, 127.11, 127.32, 128.70, 130.24, 130.24, 132.90, 132.99, 135.55, 145.77, 179.27 ppm; HRMS (M + Na
+) calcd. for C
17H
16N
2NaO
4S 367.0728, found 367.0733; IR (KBr) 3425, 3118, 2924, 2875, 2730, 1662, 1595, 1369, 1317, 1177, 1086, 1012, 774, 754 and 669 cm
−1.
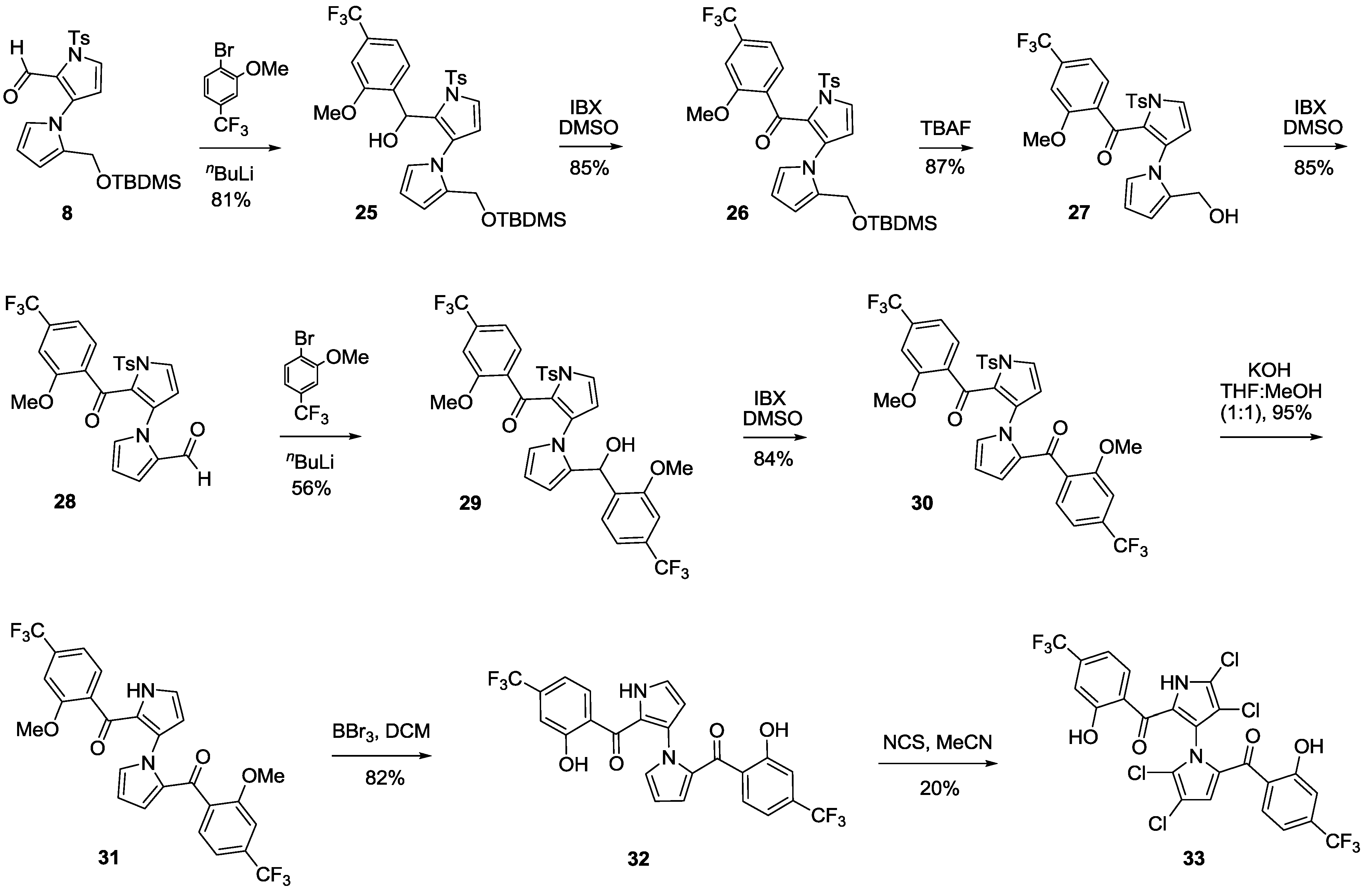
2-((tert-Butyldimethylsilyloxy)methyl)-1′-tosyl-1′H-1,3′-bipyrrole-2′-carbaldehyde (8). To a solution of 7 (6.00 g, 17.4 mmol) in dry CH2Cl2 (60 mL) was added imidazole (2.37 g, 34.8 mmol) at room temperature. After being stirred for 5 min, TBDMSCl (5.30 g, 34.8 mmol) was added. The mixture was stirred for about 2.5 h and quenched with water (50 mL) and extracted with CH2Cl2 (25 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by flash column chromatography (20% EtOAc/petroleum ether, Rf = 0.35) to give 8 (5.59 g, 70%) as a liquid. 1H NMR (400 MHz, CDCl3) δ −0.04 (s, 6H), 0.82 (s, 9H), 2.42 (s, 3H), 4.61 (s, 2H), 6.34–6.36 (m, 2H), 7.03 (t, J = 1.6 Hz, 1H), 7.11 (dd, J = 4.0, 1.6 Hz, 1H), 7.26 (d, J = 2.4 Hz, 1H), 7.29 (d, J = 8.4 Hz, 2H), 7.85 (d, J = 8.4 Hz, 2H), 9.46 (s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ −5.67, −5.67, 18.44, 21.65, 25.88, 25.88, 25.88, 53.34, 110.65, 111.12, 121.08, 121.73, 127.09, 127.09, 127.46, 128.65, 129.90, 129.90, 132.11, 133.25, 136.34, 145.19, 179.04 ppm; HRMS (M + Na+) calcd. for C23H30N2NaO4SSi 481.1593, found 481.1591; IR (KBr) 3444, 3130, 2952, 2928, 2857, 2787, 1665, 1595, 1463, 1336, 1252, 1182, 1009, 841, 668, 604 cm−1.
(4-(Benzyloxy)-2-methoxyphenyl)(2-((tert-butyldimethylsilyloxy)methyl)-1′-tosyl-1′H-1,3′-bipyrrol-2′-yl)methanol (10). To a solution of 4-(benzyloxy)-1-bromo-2-methoxybenzene (9) (0.80 g, 2.7 mmol) in dry THF (5 mL) at −78 °C under N2 was slowly added N-BuLi (1.09 mL, 2.5 M in N-pentane, 2.7 mmol). After being stirred for 40 min, a solution of 8 (0.50 g, 1.1 mmol) in dry THF (1.5 mL) was added slowly via a syringe. The mixture was stirred for about 1 h and quenched by addition of a saturated aqueous NH4Cl (15 mL) solution and extracted with EtOAc (15 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by flash column chromatography (15% EtOAc/petroleum ether, Rf = 0.2) to give 10 (0.66 g, 90%) as a pale yellow solid. mp 39.3–41.7 °C; 1H NMR (400 MHz, CDCl3) δ −0.03 (s, 3H), −0.02 (s, 3H), 0.84 (s, 9H), 2.40 (s, 3H), 2.73 (d, J = 5.2 Hz, 1H), 3.68 (s, 3H), 4.38 (d, J = 12.0 Hz, 1H), 4.63 (d, J = 11.6 Hz, 1H), 5.04 (s, 2H), 5.74 (d, J = 5.2 Hz, 1H), 6.02 (t, J = 2.0 Hz, 1H), 6.12 (t, J = 3.2 Hz, 1H), 6.28 (d, J = 3.6 Hz, 1H), 6.47–6.49 (m, 2H), 6.66 (t, J = 2.4 Hz, 1H), 7.14–7.17 (m, 2H), 7.28 (d, J = 10.4 Hz, 2H), 7.33–7.44 (m, 5H), 7.85 (d, J = 8.4 Hz, 2H) ppm; 13C NMR (CDCl3, 100 MHz) δ −5.70, −5.70, 18.44, 21.49, 25.87, 25.87, 25.87, 53.07, 55.21, 63.61, 69.97, 99.11, 104.80, 107.70, 107.96, 111.32, 121.26, 123.50, 123.74, 126.97, 126.97, 127.46, 127.46, 127.87, 128.01, 128.31, 128.31, 128.46, 129.21, 129.67, 129.67, 136.36, 136.40, 136.79, 144.84, 157.16, 159.22 ppm; HRMS (M + Na+) calcd. for C37H44N2NaO6SSi 695.2587, found 695.2598; IR (KBr) 3450, 3032, 2953, 2885, 2855, 1707, 1611, 1591, 1502, 1467, 1376, 1333, 1255, 1180, 1021, 838, 700, 600 cm−1.
(4-(Benzyloxy)-2-methoxyphenyl)(2-((tert-butyldimethylsilyloxy)methyl)-1′-tosyl-1′H-1,3′-bipyrrol-2′-yl)methanone (11). To a solution of 10 (3.67 g, 5.5 mmol) in dry DMSO (40 mL) was added IBX (3.06 g, 10.9 mmol) at room temperature. The mixture was allowed to warm up to 30 °C and stir for about 2 h. The mixture was quenched with water (60 mL) and extracted with EtOAc (25 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by flash column chromatography (15% EtOAc/petroleum ether, Rf = 0.3) to give 11 (3.00 g, 82%) as a pale brown solid. mp 44.7–47.0 °C; 1H NMR (400 MHz, CDCl3) δ 0.004 (s, 3H), 0.01 (s, 3H), 0.86 (s, 9H), 2.40 (s, 3H), 3.73 (s, 3H), 4.67 (s, 2H), 5.10 (s, 2H), 6.21 (t, J = 3.6 Hz, 1H), 6.30 (d, J = 3.6 Hz, 1H), 6.52–6.54 (m, 2H), 6.69 (dd, J = 4.0, 1.6 Hz, 1H), 7.06 (s, 1H), 7.17 (d, J = 3.6 Hz, 1H), 7.26 (d, J = 7.2 Hz, 2H), 7.36–7.46 (m, 6H), 7.84 (d, J = 8.4 Hz, 2H) ppm; 13C NMR (CDCl3, 100 MHz) δ −5.53, −5.53, 18.50, 21.63, 25.96, 25.96, 25.96, 53.61, 55.65, 70.22, 99.68, 104.56, 108.92, 111.55, 121.34, 122.70, 127.03, 127.03, 127.17, 127.62, 127.62, 128.22, 128.69, 128.69, 129.79, 130.04, 130.04, 131.65, 131.65, 132.33, 132.90, 136.45, 136.80, 144.72, 159.28, 161.84, 183.51 ppm; HRMS (M + H+) calcd. for C37H43N2O6SSi 671.2611, found 671.2607; IR (KBr) 3737, 3144, 2953, 2929, 2855, 1639, 1603, 1499, 1413, 1375, 1274, 1179, 1158, 1026, 840 cm−1.
(4-(Benzyloxy)-2-methoxyphenyl)(2-(hydroxymethyl)-1′-tosyl-1′H-1,3′-bipyrrol-2′-yl)methanone (12). To a solution of 11 (2.92 g, 4.4 mmol) in dry THF (20 mL) was added TBAF (3.41 g, 13.1 mmol) at room temperature. After being stirred for about 5 h at room temperature, the mixture was quenched with water (25 mL) and extracted with EtOAc (15 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by flash column chromatography (25% EtOAc/petroleum ether, Rf = 0.2) to give 12 (2.30 g, 95%) as a white solid. mp 55.7–58.3 °C; 1H NMR (400 MHz, CDCl3) δ 2.43 (s, 3H), 3.76 (s, 3H), 4.55 (s, 1H), 5.10 (s, 2H), 5.30 (s, 2H), 6.24 (dd, J = 4.0, 2.4 Hz, 1H), 6.31 (d, J = 3.6 Hz, 1H), 6.52–6.56 (m, 2H), 6.65 (dd, J = 4.0, 1.6 Hz, 1H), 7.00 (t, J = 2.4 Hz, 1H), 7.27 (d, J = 3.2 Hz, 1H), 7.30–7.45 (m, 8H), 7.84 (d, J = 8.4 Hz, 2H) ppm; 13C NMR (CDCl3, 100 MHz) δ 21.64, 53.17, 55.65, 70.18, 99.70, 104.57, 109.59, 110.96, 121.11, 122.58, 123.35, 127.20, 127.20, 127.55, 127.55, 128.20, 128.21, 128.66, 128.66, 128.90, 130.09, 130.09, 131.47, 132.47, 133.12, 135.81, 136.33, 145.34, 159.15, 161.85, 184.48 ppm; HRMS (M + H+) calcd. for C31H29N2O6S 557.1746, found 557.1743; IR (KBr) 3436, 2929, 2878, 1603, 1498, 1459, 1367, 1276, 1175, 1115, 1022, 867, 671 cm−1.
2′-(4-(Benzyloxy)-2-methoxybenzoyl)-1′-tosyl-1′H-1,3′-bipyrrole-2-carbaldehyde (13). To a solution of 12 (2.31 g, 4.2 mmol) in dry DMSO (30 mL) was added IBX (2.33 g, 8.3 mmol) at room temperature. The mixture was allowed to warm up to 50 °C and stir for about 3 h. The mixture was quenched with water (40 mL) and extracted with EtOAc (15 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by flash column chromatography (20% EtOAc/petroleum ether, Rf = 0.3) to give 13 (2.20 g, 96%) as a pale yellow solid. mp 132.7–135.0 °C; 1H NMR (400 MHz, CDCl3) δ 2.41 (s, 3H), 3.81 (s, 3H), 4.99 (s, 2H), 6.27–6.30 (m, 2H), 6.45 (d, J = 2.0 Hz, 1H), 6.49 (dd, J = 8.0, 2.0 Hz, 1H), 6.73 (dd, J = 3.6, 1.6 Hz, 1H), 6.98 (dd, J = 2.4, 1.6 Hz, 1H), 7.17–7.20 (m, 2H), 7.25–7.26 (m, 3H), 7.34 (d, J = 8.4 Hz, 2H), 7.38 (d, J = 8.4 Hz, 1H), 7.63 (d, J = 3.2 Hz, 1H), 7.88 (d, J = 8.4 Hz, 2H), 9.56 (s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 21.70, 55.65, 70.17, 99.64, 104.78, 110.13, 111.57, 121.95, 122.69, 125.22, 127.62, 127.62, 127.69, 128.04, 128.04, 128.21, 128.68, 128.68, 130.03, 130.03, 131.59, 132.02, 134.07, 134.78, 136.41, 139.25, 145.95, 159.37, 162.13, 177.08, 183.45 ppm; HRMS (M + H+) calcd. for C31H27N2O6S 555.1590, found 555.1600; IR (KBr) 3454, 3124, 3083, 2872, 2792, 1685, 1633, 1566, 1440, 1347, 1169, 1112, 1026, 763 cm−1.
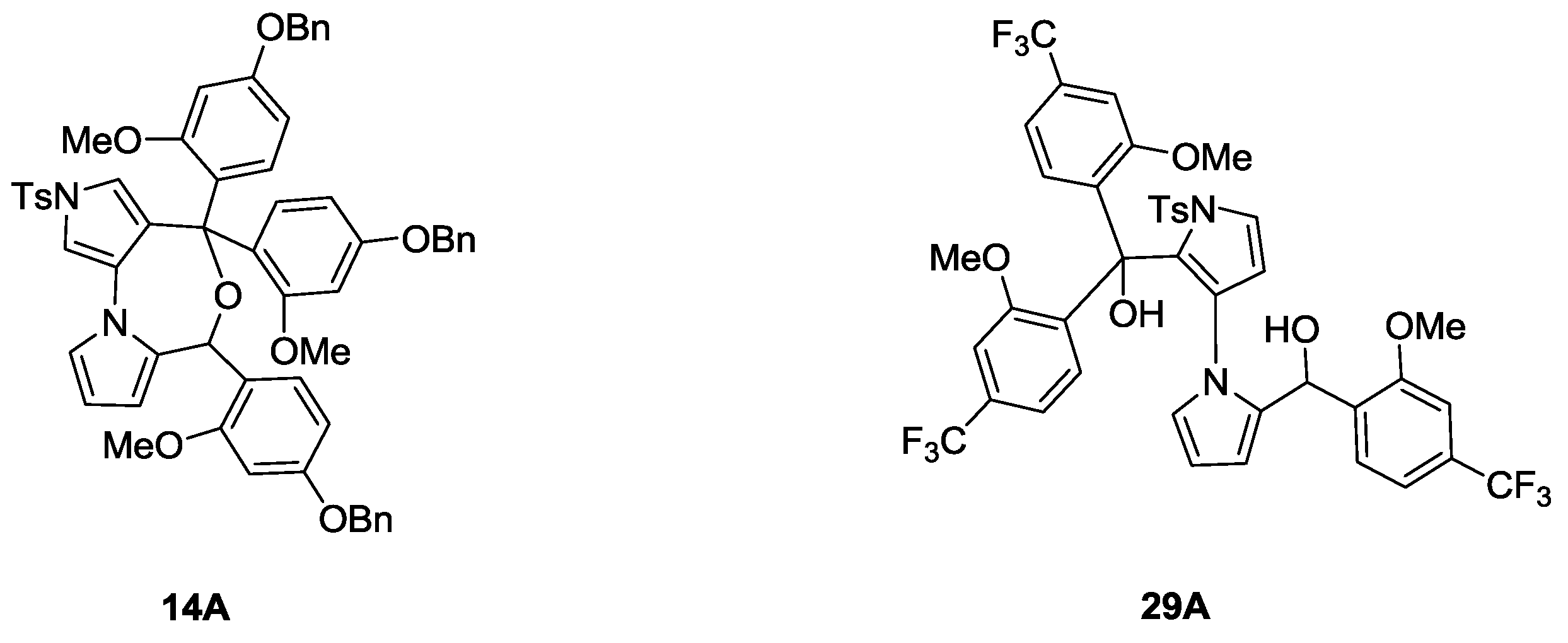
(4-(Benzyloxy)-2-methoxyphenyl)(2-((4-(benzyloxy)-2-methoxyphenyl)(hydroxy)methyl)-1′-tosyl-1′H-1,3′-bipyrrol-2′-yl)methanone (14). To a solution of 4-(benzyloxy)-1-bromo-2-methoxybenzene (9) (198 mg, 0.68 mmol) in dry THF (5 mL) at −78 °C under N2 was slowly added N-BuLi (0.27 mL, 2.5 M in N-pentane, 0.68 mmol). After being stirred for 30 min, a solution of 13 (150 mg, 0.27 mmol) in dry THF (1.5 mL) was added slowly via a syringe. After the mixture was stirred at −78 °C for 2 h, the reaction was quenched by addition of a saturated aqueous solution of NH4Cl (10 mL) and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified quickly by flash column chromatography (20% EtOAc/petroleum ether, Rf = 0.2) to give unstable 14 (177 mg, 85%) as a light red solid and 14A (10 mg, 5%) as a light yellow solid. mp for 14, 76.5–77.4 °C; 1H NMR (600 MHz, CDCl3) δ 2.39 (s, 3H), 3.38 (br s, 3H), 3.45 (s, 3H), 4.90 (d, J = 11.4 Hz, 1H), 4.94 (d, J = 11.4 Hz, 1H), 5.01 (br s, 2H), 5.78 (br s, 1H), 6.15 (d, J = 8.4 Hz, 1H), 6.18 (s, 1H), 6.30 (s, 1H), 6.35–6.46 (m, 2H), 6.52 (s, 1H), 7.04 (s, 1H), 7.18 (br s, 1H), 7.28–7.42 (m, 16H), 7.87 (d, J = 7.8 Hz, 2H) ppm; 13C NMR (CDCl3, 150 MHz) δ 21.59, 54.85, 55.56, 61.78, 69.69, 70.00, 98.22, 99.63, 100.55, 104.06, 104.23, 108.66, 111.62, 120.16, 122.53, 123.22, 126.26, 127.26, 127.26, 127.48, 127.48, 127.52, 127.52, 127.78, 127.87, 128.20, 128.41, 128.41, 128.57, 128.57, 129.75, 129.75, 130.03, 131.52, 132.41, 135.98, 136.32, 137.07, 144.86, 156.26, 158.52, 158.81, 159.07, 161.46, 171.10 ppm; HRMS (M + Na+) calcd. for C45H40N2NaO8S 791.2403, found 791.2410; IR (KBr) 3423, 1608, 1502, 1454, 1411, 1372, 1278, 1198, 1175, 1123, 1034, 945, 738, 697, 593 cm−1.
4,4,6-Tris(4-(benzyloxy)-2-methoxyphenyl)-2-tosyl-4,6-dihydro-2H-dipyrrolo[2,1-c:3′,4′-e][1,4]oxazepine (14A). mp 105.6–106.4 °C; 1H NMR (400 MHz, CDCl3) δ 2.25 (s, 3H), 3.08 (s, 3H), 3.55 (s, 3H), 3.83 (s, 3H), 4.95 (s, 2H), 5.00 (s, 4H), 5.59 (s, 1H), 6.09 (d, J = 2.4 Hz, 1H), 6.16–6.18 (m, 2H), 6.38–6.42 (m, 4H), 6.50–6.51 (m, 2H), 6.67 (td, J = 8.8, 2.4 Hz, 2H), 7.00–7.02 (m, 6H), 7.30–7.44 (m, 15H), 7.72 (dd, J = 8.8, 2.4 Hz, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 21.68, 54.24, 54.69, 55.14, 67.95, 68.37, 68.62, 78.68, 98.24, 98.55, 98.63, 98.86, 99.70, 100.29, 103.14, 103.63, 103.91, 103.97, 105.72, 107.05, 109.16, 117.71, 118.71, 119.37, 120.79, 122.51, 124.31, 125.42, 125.42, 125.52, 126.38, 126.38, 126.58, 126.58, 126.84, 126.99, 127.47, 127.47, 127.47, 127.47, 127.59, 127.59, 128.40, 128.40, 128.49, 128.59, 129.78, 131.74, 135.03, 135.96, 136.14, 136.84, 142.76, 155.71, 157.34, 157.80, 157.85, 158.09, 158.73 ppm; HRMS (M + Na+) calcd. for C59H52N2NaO9S 987.3291, found 987.3297; IR (KBr) 3429, 2925, 1609, 1585, 1502, 1453, 1416, 1262, 1193, 1139, 1037, 964, 806, 698, 590 cm−1.
(1′-Tosyl-1′H-1,3′-bipyrrole-2,2′-diyl)bis((4-(benzyloxy)-2-methoxyphenyl)methanone) (15). To a solution of 14 (127 mg, 0.17 mmol) in dry DMSO (4 mL) was stepwise added IBX (116 mg, 0.41 mmol) at room temperature. The mixture was allowed to warm up to 50 °C and stir for about 3.5 h. The mixture was quenched with water (10 mL) and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuum. The residue was purified by flash column chromatography (20% EtOAc/petroleum ether, Rf = 0.2) to give 15 (115 mg, 90%) as a pale brown solid. mp 147.9–152.7 °C; 1H NMR (400 MHz, CDCl3) δ 2.42 (s, 3H), 3.64 (s, 3H), 3.73 (s, 3H), 4.98 (s, 2H), 5.03 (s, 2H), 5.87 (t, J = 3.2 Hz, 1H), 6.28–6.33 (m, 3H), 6.42–6.45 (m, 2H), 6.54 (d, J = 2.0 Hz, 1H), 6.72 (t, J = 2.0 Hz, 1H), 7.09 (d, J = 8.4 Hz, 1H), 7.31–7.46 (m, 15H), 7.84 (d, J = 8.4 Hz, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 21.55, 40.76, 55.49, 55.56, 69.75, 69.95, 98.89, 99.58, 103.93, 104.75, 108.56, 111.99, 120.90, 122.10, 122.65, 122.94, 127.18, 127.18, 127.38, 127.38, 128.03, 128.03, 128.08, 128.08, 128.48, 128.48, 128.50, 128.57, 129.47, 129.47, 131.54, 131.78, 132.10, 132.32, 134.01, 135.79, 136.01, 136.22, 144.86, 159.22, 160.38, 161.52, 163.17, 182.48, 183.62 ppm; HRMS (M + H+) calcd. for C45H39N2O8S 767.2427, found 767.2414; IR (KBr) 3433, 3113, 2937, 2874, 1634, 1599, 1499, 1450, 1265, 1170, 1029, 746, 667, 585 cm−1.
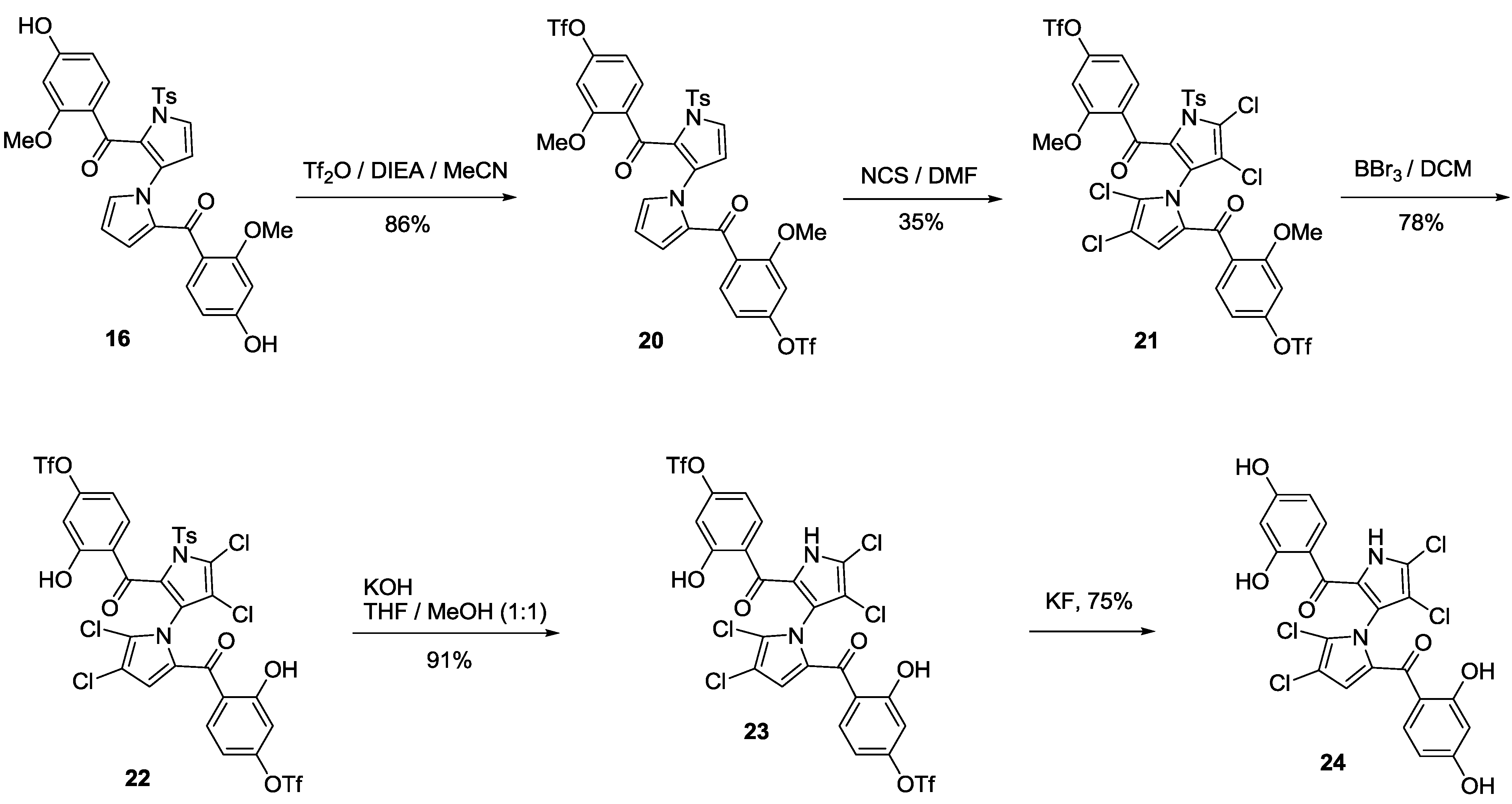
(1′-Tosyl-1′H-1,3′-bipyrrole-2,2′-diyl)bis((4-hydroxy-2-methoxyphenyl)methanone) (16). To a solution of 15 (1.00 g, 1.30 mmol) in a 3:1 mixture of MeOH/EtOAc (10 mL) was added Pd/C (0.54 g, 0.13 mmol, purity: 5%) under 1 atm H2. The mixture was stirred for about 12 h at room temperature. The suspension was filtered and the filtrate was washed with acetone (100 mL). The combined organic layers were concentrated in vacuum and the residue was purified by flash column chromatography (20% acetone/petroleum ether, Rf = 0.3) to give 16 (0.65 g, 85%) as a gray solid. mp 157.3–160.3 °C; 1H NMR (400 MHz, CDCl3) δ 2.31 (s, 3H), 3.35 (s, 3H), 3.38 (s, 3H), 5.90 (s, 1H), 5.99 (d, J = 8.4 Hz, 1H), 6.05 (s, 1H), 6.20 (d, J = 8.0 Hz, 1H), 6.24 (s, 1H), 6.39 (d, J = 2.4 Hz, 1H), 6.42 (d, J = 2.8 Hz, 1H), 6.74 (s, 1H), 6.94 (d, J = 8.0 Hz, 1H), 7.20–7.24 (m, 3H), 7.49 (d, J = 3.2 Hz, 1H), 7.85 (d, J = 7.6 Hz, 2H), 8.29 (br s, 1H), 8.88 (br s, 1H) ppm; 13C NMR (acetone-d6, 100 MHz) δ 21.52, 55.70, 55.79, 99.45, 100.04, 106.99, 107.75, 109.35, 112.76, 120.50, 122.35, 122.46, 123.34, 128.81, 128.81, 130.01, 130.46, 130.46, 131.23, 132.07, 132.20, 134.03, 134.68, 137.20, 146.06, 160.09, 161.31, 162.12, 163.86, 183.31, 183.35 ppm; HRMS (M + Na+) calcd. for C31H26N2NaO8S 609.1308, found 609.1313; IR (KBr) 3422, 2934, 2853, 1606, 1466, 1436, 1313, 1268, 1174, 936, 671 cm−1.
1′H-1,3′-Bipyrrole-2,2′-diylbis((4-hydroxy-2-methoxyphenyl)methanone) (17). To a solution of 16 (100 mg, 0.17 mmol) in a 1:1 mixture of MeOH/THF (5 mL) was added KOH (39 mg, 0.69 mmol) at room temperature. After being stirred for 2 h, the mixture was adjusted to pH 7.0 with 0.5 N HCl and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by flash column chromatography (50% EtOAc/petroleum ether, Rf = 0.3) to give 17 (66 mg, 90%) as a brown solid. mp 83.0–85.7 °C; 1H NMR (400 MHz, CD3OD) δ 3.55 (s, 3H), 3.66 (s, 3H), 6.00 (t, J = 2.4 Hz, 1H), 6.09 (dd, J = 8.4, 2.0 Hz, 1H), 6.17 (d, J = 2.0 Hz, 1H), 6.22 (d, J = 2.0 Hz, 1H), 6.33 (dd, J = 8.4, 2.0 Hz, 1H), 6.36 (dd, J = 4.0, 2.0 Hz, 1H), 6.41 (d, J = 2.0 Hz, 1H), 6.83 (s, 1H), 6.99 (d, J = 8.4 Hz, 1H), 7.02 (t, J = 1.6 Hz, 1H), 7.04 (d, J = 8.4 Hz, 1H) ppm; 13C NMR (CD3OD, 100 MHz) δ 55.69, 55.85, 99.75, 100.10, 106.98, 107.78, 109.96, 110.64, 120.78, 121.46, 124.03, 124.43, 127.17, 132.42, 132.57, 133.67, 133.84, 133.96, 160.55, 161.11, 162.41, 162.56, 185.53, 185.61 ppm; HRMS (M + H+) calcd. for C24H21N2O6 433.1400, found 433.1379; IR (KBr) 3293, 2938, 1697, 1610, 1465, 1407, 1308, 1269, 1201, 1163, 1121, 1031, 959, 868, 748 cm−1.
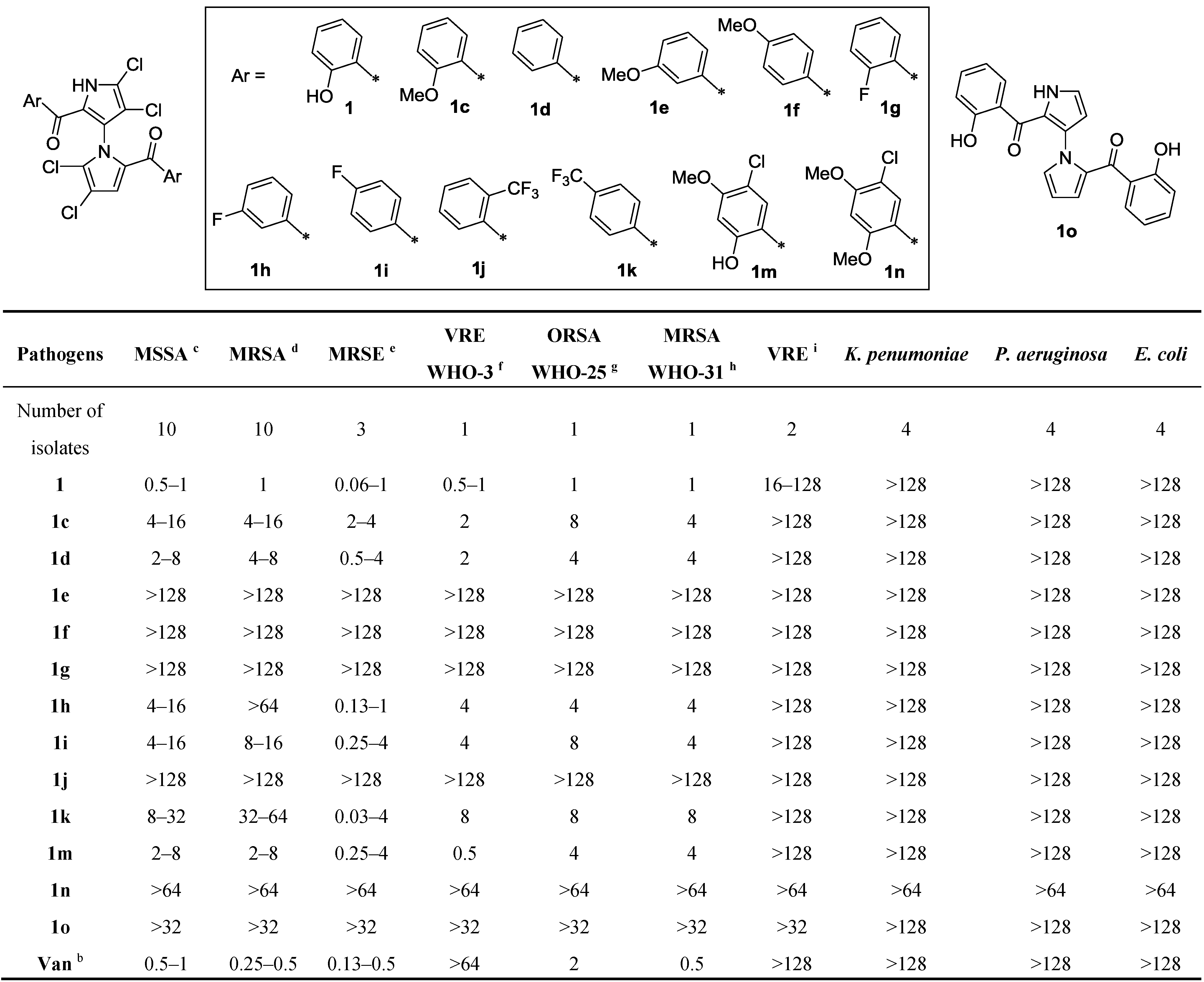
(4,4′,5,5′-Tetrachloro-1′H-1,3′-bipyrrole-2,2′-diyl)bis((5-chloro-4-hydroxy-2-methoxyphenyl) methanone) (18). To a solution of 17 (20 mg, 0.05 mmol) in dry MeCN (2 mL) at room temperature was gradually added NCS (37 mg, 0.28 mmol). After being stirred for about 6 h at room temperature, the mixture was quenched with water (15 mL) and extracted with EtOAc (15 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuum. The residue was purified by flash column chromatography (33% EtOAc/petroleum ether, Rf = 0.2) to give 18 (11 mg, 40%) as a yellow solid. mp 102.7–104.7 °C; 1H NMR (400 MHz, acetone-d6) δ 3.65 (s, 3H), 3.71 (s, 3H), 6.52 (s, 1H), 6.55 (s, 1H), 6.71 (s, 1H), 7.10 (s, 1H), 7.23 (s, 1H) ppm; 13C NMR (CD3OD + CDCl3, 100 MHz) δ 55.99, 56.03, 100.22, 100.86, 110.21, 111.77, 111.83, 112.37, 119.31, 120.11, 120.80, 120.86, 124.20, 124.960, 125.98, 130.88, 131.42, 132.46, 157.01, 157.51, 158.39, 159.10, 181.27, 181.86 ppm; HRMS (M + H+) calcd. for C24H15Cl6N2O6 636.9061, found 636.9073; IR (KBr) 3441, 3230, 3130, 2936, 2855, 1723, 1628, 1602, 1403, 1298, 1271, 1215, 1037, 994, 745, 666 cm−1.
(4,4′,5,5′-Tetrachloro-1′H-1,3′-bipyrrole-2,2′-diyl)bis((5-chloro-2,4-dihydroxyphenyl)methanone) (19). To a solution of 18 (14 mg, 0.02 mmol) in dry CH2Cl2 (4 mL) was slowly added a solution of BBr3 (19 mg, 0.08 mmol) in dry CH2Cl2 (1 mL) via a syringe under N2 at −78 °C. After being stirred for 0.5 h, the mixture was quenched by addition of water (10 mL) and extracted with CH2Cl2 (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuum. The residue was purified by flash column chromatography (25% EtOAc/petroleum ether, Rf = 0.3) to give 19 (11 mg, 85%) as a pale brown solid. mp 145.7–147.7 °C; 1H NMR (400 MHz, CD3OD) δ 6.27 (s, 1H), 6.32 (s, 1H), 7.31 (s, 1H), 7.41 (s, 1H), 7.96 (s, 1H) ppm; 13C NMR (CD3OD, 100 MHz) δ 101.49, 105.13, 105.25, 110.00, 112.90, 113.27, 114.67, 119.42, 120.24, 123.00, 123.87, 125.98, 130.91, 132.64, 132.66, 133.99, 134.07, 163.85, 164.48, 165.02, 184.46, 185.64 ppm; HRMS (M + Na+) calcd. for C22H10Cl6N2NaO6 630.8568, found 630.8581; IR (KBr) 3425, 2961, 2924, 2854, 1654, 1622, 1414, 1384, 1358, 1258, 1024, 800 cm−1. HPLC purity, 95.4% (Flow rate: 1.0 mL/min; Column: Phenomenex C6-phenyl, 5 μm, 150 × 4.6 mm; Wavelength: UV 254 nm; Temperature: 25 °C; Mobile phase: MeOH:H2O = 80:20; tR = 5.1 min).
(1′-Tosyl-1′H-1,3′-bipyrrole-2,2′-diyl)bis(((2-methoxy-4-hydroxytrifluoromethanesulfonate)phenyl) methanone) (20). To a solution of 16 (0.50 g, 0.85 mmol) in dry MeCN (10 mL) at −30 °C under N2 was slowly added DIEA (0.44 g, 3.4 mmol). After being stirred for 5 min, Tf2O (0.72 g, 2.60 mmol) was added slowly via a syringe. The mixture was stirred for about 3 h at room temperature and quenched with water (10 mL) and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by flash column chromatography (15% EtOAc/petroleum ether, Rf = 0.3) to give 20 (0.62 g, 86%) as a pale brown solid. mp 116.3–120.0 °C; 1H NMR (400 MHz, CDCl3) δ 2.46 (s, 3H), 3.71 (s, 3H), 3.79 (s, 3H), 5.90 (t, J = 2.8 Hz, 1H), 6.31 (d, J = 2.8 Hz, 1H), 6.38 (d, J = 3.2 Hz, 1H), 6.53 (d, J = 1.6 Hz, 1H), 6.63 (dd, J = 8.4, 1.6 Hz, 1H), 6.70 (s, 1H), 6.80 (s, 1H), 6.83 (d, J = 8.4 Hz, 1H), 7.22 (d, J = 8.4 Hz, 1H), 7.39 (d, J = 8.4 Hz, 3H), 7.67 (d, J = 3.6 Hz, 1H), 7.99 (d, J = 8.0 Hz, 2H) ppm; 13C NMR (CDCl3, 100 MHz) δ 21.59, 56.01, 56.10, 104.81, 105.22, 109.65, 111.29, 112.18, 112.23, 117.08, 120.28, 123.86, 126.14, 127.57, 128.07, 128.46, 128.46, 128.97, 129.68, 129.68, 130.45, 131.78, 132.18, 132.70, 134.11, 135.64, 145.44, 150.99, 151.83, 158.38, 159.05, 181.57, 181.98 ppm; HRMS (M + H+) calcd. for C33H25F6N2O12S3 851.0474, found 851.0480; IR (KBr) 3444, 3121, 2950, 2871, 1642, 1600, 1493, 1426, 1269, 1243, 1141, 948, 827, 581 cm−1.
(4,4′,5,5′-Tetrachloro-1′-tosyl-1′H-1,3′-bipyrrole-2,2′-diyl)bis(((2-methoxy-4-hydroxytrifluoromethanesulfonate)phenyl)methanone) (21). To a solution of 20 (0.50 g, 0.59 mmol) in dry DMF (10 mL) at room temperature was gradually added NCS (0.51 g, 3.8 mmol). After being stirred for about 3 h at room temperature, the mixture was quenched with water (15 mL), and extracted with EtOAc (15 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by flash column chromatography (12% EtOAc/petroleum ether, Rf = 0.2) to give 21 (0.20 g, 35%) as a yellow solid. mp 81.7–83.3 °C; 1H NMR (400 MHz, CD3OD) δ 2.49 (s, 3H), 3.50 (s, 3H), 3.61 (s, 3H), 6.41 (s, 1H), 6.57 (d, J = 8.0 Hz, 1H), 6.82 (d, J = 2.0 Hz, 1H), 6.90 (d, J = 2.0 Hz, 1H), 6.95 (dd, J = 8.8, 2.4 Hz, 1H), 7.32 (d, J = 8.4 Hz, 1H), 7.51 (d, J = 12.0 Hz, 2H), 7.64 (d, J = 8.4 Hz, 1H), 7.92 (d, J = 8.4 Hz, 2H) ppm; 13C NMR (CDCl3, 100 MHz) δ 21.61, 55.90, 56.13, 104.61, 105.00, 112.01, 112.54, 113.03, 113.61, 116.93, 119.59, 120.12, 120.80, 125.09, 127.11, 128.01, 128.40, 128.40, 129.32, 129.94, 129.94, 131.06, 131.30, 132.63, 132.86, 133.68, 146.57, 151.94, 152.94, 158.53, 159.68, 180.21, 181.56 ppm; HRMS (M + H+) calcd. for C33H21Cl4F6N2O12S3 986.8915, found 986.8926; IR (KBr) 3446, 2923, 2853, 1653, 1603, 1491, 1428, 1270, 1216, 1140, 996, 950, 831, 585 cm−1.
(4,4′,5,5′-Tetrachloro-1′-tosyl-1′H-1,3′-bipyrrole-2,2′-diyl)bis((2-hydroxy-4-hydroxytrifluoromethanesulfonate)phenyl)methanone) (22). To a solution of 21 (37 mg, 0.03 mmol) in dry CH2Cl2 (5 mL) was slowly added a solution of BBr3 (56 mg, 0.22 mmol) in dry CH2Cl2 (1 mL) via a syringe under N2 at −78 °C. After being stirred for 0.5 h, the mixture was quenched by addition of water (10 mL), and extracted with CH2Cl2 (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by flash column chromatography (10% EtOAc/petroleum ether, Rf = 0.3) to give 22 (28 mg, 78%) as a yellow solid. mp 71.3–73.0 °C; 1H NMR (400 MHz, CD3OD) δ 2.48 (s, 3H), 6.41 (s, 1H), 6.72–6.95 (m, 4H), 7.35 (d, J = 8.0 Hz, 2H), 7.50 (d, J = 8.8 Hz, 2H), 7.79 (d, J = 8.0 Hz, 2H), 11.28 (br s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 21.76, 110.86, 111.12, 111.12, 111.12, 112.21, 112.40, 112.87, 113.93, 116.93, 116.94, 118.33, 118.35, 120.15, 122.32, 125.44, 128.30, 128.30, 130.18, 130.18, 132.90, 134.18, 135.40, 135.42, 135.43, 147.32, 154.40, 163.51, 163.78, 186.18, 188.56 ppm; HRMS (M + H+) calcd. for C31H17Cl4F6N2O12S3 958.8602, found 958.8610; IR (KBr) 3445, 3134, 2920, 2851, 1742, 1631, 1598, 1430, 1385, 1430, 1216, 1140, 970, 842, 583 cm−1.
(4,4′,5,5′-Tetrachloro-1′H-1,3′-bipyrrole-2,2′-diyl)bis(((2-hydroxy-4-hydroxytrifluoromethanesulfonate)phenyl)methanone) (23). To a solution of 22 (165 mg, 0.17 mmol) in a 1:1 mixture of MeOH/THF (3 mL) was added KOH (39 mg, 0.69 mmol) at room temperature. After being stirred for 15 min, the mixture was adjusted to pH 7.0 with 0.5 N HCl and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuum. The residue was purified by flash column chromatography (33% EtOAc/petroleum ether, Rf = 0.3) to give 23 (125 mg, 90.6%) as a light yellow solid. mp 65.7–67.7 °C; 1H NMR (400 MHz, CDCl3) δ 6.17 (s, 1H), 6.53 (dd, J = 7.6, 2.0 Hz, 1H), 6.81 (dd, J = 8.8, 2.0 Hz, 1H), 6.87 (d, J = 2.4 Hz, 1H), 6.93 (d, J = 2.4 Hz, 1H), 7.40 (br s, 1H), 7.57 (d, J = 8.8 Hz, 1H), 9.61 (br s, 1H), 10.67 (s, 1H), 11.49 (s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 109.34, 111.21, 111.33, 111.61, 112.44, 115.90, 117.01, 118.50, 118.60, 118.89, 122.60, 123.61, 124.41, 124.92, 132.11, 135.49, 151.32, 153.72, 154.72, 161.95, 162.77, 164.22, 184.70, 186.90 ppm; HRMS (M + H+) calcd. for C24H11Cl4F6N2O10S2 804.8513, found 804.8529; IR (KBr) 3380, 3264, 1627, 1597, 1497, 1429, 1217, 1139, 1107, 970, 942, 605 cm−1. HPLC purity, 96.0% (Flow rate: 1.0 mL/min; Column: Phenomenex C6-phenyl, 5 μm, 150 × 4.6 mm; Wavelength: UV 254 nm; Temperature: 25 °C; Mobile phase: MeOH:H2O = 75:25; tR = 17.9 min).
(4,4′,5,5′-Tetrachloro-1′H-1,3′-bipyrrole-2,2′-diyl)bis((2,4-dihydroxyphenyl)methanone) (24). To a solution of 23 (5.0 mg, 0.006 mmol) in DMSO (1 mL) was added a solution of KF (1.1 mg, 0.018 mmol) in water (0.1 mL) at room temperature. After being stirred for about 3 h, the mixture was added with water (5 mL) and extracted with EtOAc (5 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuum. The residue was purified by flash column chromatography (40% EtOAc/petroleum ether, Rf = 0.3) to give 24 (2.6 mg, 75%) as a pale yellow solid. mp 103.3–105.3 °C; 1H NMR (400 MHz, acetone-d6) δ 6.10 (s, 1H), 6.19 (dd, J = 8.8, 2.4 Hz, 1H), 6.22–6.23 (m, 2H), 6.28 (d, J = 8.0 Hz, 1H), 7.62 (d, J = 8.8 Hz, 1H), 8.09 (br s, 1H), 12.03 (br s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 103.19, 103.49, 107.73, 108.14, 108.30, 108.35, 109.60, 111.44, 113.20, 113.77, 114.26, 115.07, 120.82, 123.89, 126.11, 134.16, 137.03, 162.89, 165.67, 166.61, 167.02, 185.94 ppm; HRMS (M + H+) calcd. for C22H13Cl4N2O6 540.9528, found 540.9536; IR (KBr) 3400, 3282, 2958, 2922, 2851, 1626, 1596, 1447, 1333, 1266, 1177, 978, 796 cm−1. HPLC purity, 98.2% (Flow rate: 1.0 mL/min; Column: Waters C8, 5 μm, 150 × 4.6 mm; Wavelength: UV 254 nm; Temperature: 25 °C; Mobile phase: MeOH:H2O = 65:35; tR = 6.2 min).
(2-((tert-Butyldimethylsilyloxy)methyl)-1′-tosyl-1′H-1,3′-bipyrrol-2′-yl)(2-methoxy-4-(trifluoromethyl)phenyl)methanol (25). To a solution of 1-bromo-2-methoxy-4-(trifluoromethyl)benzene (69.0 mg, 0.27 mmol) in dry THF (4 mL) at −78 °C under N2 was slowly added N-BuLi (0.11 mL, 2.5 M in N-pentane, 0.27 mmol). After being stirred for 30 min, a solution of 8 (50 mg, 0.11 mmol) in dry THF (1 mL) was added slowly via a syringe. The mixture was stirred for about 2 h and quenched by addition of a saturated aqueous NH4Cl (15 mL) solution, and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by flash column chromatography (15% EtOAc/petroleum ether, Rf = 0.2) to give 25 (56 mg, 81%) as a pale brown solid. mp 34.7–36.7 °C; 1H NMR (400 MHz, CDCl3) δ 0.001 (s, 3H), 0.07 (s, 3H), 0.83 (s, 9H), 2.41 (s, 3H), 2.94 (d, J = 4.0 Hz, 1H), 3.73 (s, 3H), 4.43 (d, J = 12.0 Hz, 1H), 4.75 (d, J = 12.0 Hz, 1H), 5.81–5.84 (m, 2H), 6.10 (t, J = 3.2 Hz, 1H), 6.34 (d, J = 3.6 Hz, 1H), 6.66 (dd, J = 3.6, 2.0 Hz, 1H), 6.99 (s, 1H), 7.18 (d, J = 8.0 Hz, 1H), 7.24 (d, J = 3.6 Hz, 1H), 7.30 (d, J = 8.0 Hz, 2H), 7.55 (d, J = 8.0 Hz, 1H), 7.85 (d, J = 8.4 Hz, 2H) ppm; 13C NMR (CDCl3, 100 MHz) δ −5.71, −5.67, 18.53, 21.56, 25.92, 25.92, 25.92, 53.27, 55.53, 62.79, 106.87, 106.91, 108.05, 108.43, 111.37, 117.21, 117.25, 121.56, 123.91, 126.96, 127.01, 127.35, 128.40, 129.44, 129.87, 134.75, 134.76, 135.81, 136.29, 145.17, 156.02 ppm; HRMS (M + Na+) calcd. for C31H37F3N2NaO5SSi 657.2042, found 657.2040; IR (KBr) 3383, 3146, 2956, 2929, 2857, 1734, 1594, 1465, 1415, 1377, 1329, 1241, 1175, 1123, 1032, 841, 778, 670 cm−1.
(2-((tert-Butyldimethylsilyloxy)methyl)-1′-tosyl-1′H-1,3′-bipyrrol-2′-yl)(2-methoxy-4-(trifluoromethyl)phenyl)methanone (26). To a solution of 25 (463 mg, 0.73 mmol) in dry DMSO (10 mL) was added IBX (408 mg, 1.46 mmol) at room temperature. The mixture was allowed to warm up to 50 °C and stir additionally for about 3.5 h. The mixture was quenched with water (15 mL) and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuum. The residue was purified by flash column chromatography (15% EtOAc/petroleum ether, Rf = 0.2) to give 26 (403 mg, 85%) as a pale brown solid. mp 38.0–40.3 °C; 1H NMR (400 MHz, CDCl3) δ −0.006 (s, 6H), 0.85 (s, 9H), 2.40 (s, 3H), 3.82 (s, 3H), 4.68 (s, 3H), 6.23 (dd, J = 4.0, 2.4 Hz, 1H), 6.33 (d, J = 3.6 Hz, 1H), 6.64 (dd, J = 4.0, 1.6 Hz, 1H), 7.09 (t, J = 2.0 Hz, 1H), 7.21–7.23 (m, 2H), 7.28 (d, J = 8.0 Hz, 2H), 7.40 (d, J = 8.0 Hz, 1H), 7.85 (d, J = 8.4 Hz, 2H) ppm; 13C NMR (CDCl3, 100 MHz) δ −5.65, −5.65, 18.42, 21.55, 25.65, 25.86, 25.86, 53.48, 55.86, 108.06, 108.09, 109.50, 111.33, 116.83, 116.87, 121.33, 123.91, 127.00, 127.66, 129.27, 129.38, 129.77, 131.85, 132.60, 132.93, 132.99, 133.48, 136.64, 144.82, 157.04, 182.61 ppm; HRMS (M + Na+) calcd. for C31H35F3N2NaO5SSi 655.1886, found 655.1893; IR (KBr) 3145, 2955, 2929, 2856, 1737, 1647, 1598, 1499, 1463, 1411, 1376, 1328, 1245, 1176, 1130, 1077, 894, 838, 670 cm−1.
(2-(Hydroxymethyl)-1′-tosyl-1′H-1,3′-bipyrrol-2′-yl)(2-methoxy-4-(trifluoromethyl)phenyl)methanone (27). To a solution of 26 (400 mg, 0.63 mmol) in dry THF (10 mL) was added TBAF (495 mg, 1.90 mmol) at room temperature. After being stirred for about 5 h at room temperature, the mixture was quenched with water (10 mL) and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuum. The residue was purified by flash column chromatography (20% EtOAc/petroleum ether, Rf = 0.3) to give 27 (290 mg, 87%) as a light yellow solid. mp 53.3–56.0 °C; 1H NMR (400 MHz, CDCl3) δ 2.35 (s, 3H), 2.98 (t, J = 6.4 Hz, 1H), 3.76 (s, 3H), 4.54 (d, J = 6.8 Hz, 2H), 6.24 (t, J = 3.6 Hz, 1H), 6.34 (d, J = 6.8 Hz, 1H), 6.61 (dd, J = 3.6, 1.2 Hz, 1H), 7.04 (s, 1H), 7.14 (s, 1H), 7.22 (d, J = 3.6 Hz, 1H), 7.26–7.30 (m, 3H), 7.38 (d, J = 8.0 Hz, 1H), 7.84 (d, J = 8.4 Hz, 2H) ppm; 13C NMR (CDCl3, 100 MHz) δ 21.56, 53.09, 55.92, 108.14, 108.17, 110.27, 110.92, 116.94, 116.97, 122.31, 124.59, 127.16, 128.46, 128.55, 129.15, 130.11, 132.03, 132.68, 132.89, 133.00, 133.74, 135.81, 145.48, 157.03, 183.56 ppm; HRMS (M + Na+) calcd. for C25H21F3N2NaO5S 541.1021, found 541.1027; IR (KBr) 3425, 3119, 2956, 2925, 1642, 1596, 1500, 1460, 1411, 1328, 1175, 1133, 1078, 893, 673, 602 cm−1.
2′-(2-Methoxy-4-(trifluoromethyl)benzoyl)-1′-tosyl-1′H-1,3′-bipyrrole-2-carbaldehyde (28). To a solution of 27 (286 mg, 0.55 mmol) in dry DMSO (10 mL) was added IBX (309 mg, 1.10 mmol) at room temperature. After being stirred for about 3.5 h, the mixture was quenched with water (15 mL) and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuum. The residue was purified by flash column chromatography (20% EtOAc/petroleum ether, Rf = 0.3) to give 28 (242 mg, 85%) as a light brown solid. mp 114.3–117.0 °C; 1H NMR (400 MHz, CDCl3) δ 2.43 (s, 3H), 3.80 (s, 3H), 6.29 (t, J = 3.2 Hz, 1H), 6.48 (d, J = 3.6 Hz, 1H), 6.63 (d, J = 2.4 Hz, 1H), 7.03 (s, 1H), 7.13 (s, 1H), 7.24 (d, J = 7.6 Hz, 1H), 7.35 (d, J = 8.0 Hz, 2H), 7.46 (d, J = 7.6 Hz, 1H), 7.69 (d, J = 3.2 Hz, 1H), 7.89 (d, J = 8.4 Hz, 2H), 9.70 (s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 21.63, 55.89, 108.12, 108.15, 110.55, 111.54, 116.88, 116.92, 123.99, 125.49, 127.53, 127.90, 127.90, 129.62, 130.08, 130.08, 132.34, 132.63, 132.81, 133.15, 134.75, 138.21, 146.08, 177.23, 182.75 ppm; HRMS (M + Na+) calcd. for C25H19F3N2NaO5S 539.0864, found 539.0856; IR (KBr) 3433, 3141, 3089, 2927, 2855, 1679, 1641, 1562, 1408, 1327, 1173, 1128, 1023, 901, 812, 670 cm−1.
(2-(Hydroxy(2-methoxy-4-(trifluoromethyl)phenyl)methyl)-1′-tosyl-1′H-1,3′-bipyrrol-2′-yl)(2-methoxy-4-(trifluoromethyl)phenyl)methanone (29). To a solution of 1-bromo-2-methoxy-4-(trifluoromethyl)benzene (200 mg, 0.78 mmol) in dry THF (8 mL) at −78 °C under N2 was slowly added N-BuLi (0.31 mL, 2.5 M in N-pentane, 0.78 mmol). After being stirred for 30 min, a solution of 28 (200 mg, 0.39 mmol) in dry THF (2 mL) was added slowly via a syringe. After the mixture was stirred at −78 °C for 2 h, the reaction was quenched by addition of a saturated aqueous solution of NH4Cl (15 mL) and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered and concentrated in vacuum. The residue was purified by flash column chromatography (15% EtOAc/petroleum ether, Rf = 0.2) to give 29 (150 mg, 56%) as a light yellow solid, 29A (42 mg, 15%) as a light yellow solid and recovered 28 (48 mg, 24%). mp for 29, 85.1–86.3 °C; 1H NMR (400 MHz, CDCl3) δ 2.44 (s, 3H), 3.59 (br s, 3H), 3.84 (s, 3H), 4.12 (br s, 1H), 5.77 (br s, 1H), 6.17–6.32 (m, 3H), 6.49 (br s, 1H), 6.70 (br s, 1H), 6.83 (d, J = 7.6 Hz, 1H), 7.13 (br s, 1H), 7.21 (br s, 1H), 7.35–7.37 (m, 4H), 7.42 (d, J = 2.8 Hz, 1H), 7.92 (d, J = 8.0 Hz, 2H) ppm; 13C NMR (CDCl3, 100 MHz) δ 21.67, 55.75, 55.88, 67.96, 105.60, 105.64, 108.13, 108.03, 111.28, 116.78, 116.78, 116.78, 120.85, 120.85, 124.97, 126.20, 127.10, 127.30, 127.63, 127.63, 129.14, 129.14, 129.98, 129.98, 132.50, 132.56, 133.21, 133.45, 135.92, 145.38, 155.48, 157.01, 180.50 ppm; HRMS (M + Na+) calcd. for C33H26F6N2NaO6S 715.1313, found 715.1287; IR (KBr) 3444, 3137, 2923, 1668, 1562, 1447, 1361, 1180, 1014, 752, 668 cm−1.
(2-(Hydroxy(2-methoxy-4-(trifluoromethyl)phenyl)methyl)-1′-tosyl-1′H-1,3′-bipyrrol-2′-yl)bis(2-methoxy-4-(trifluoromethyl)phenyl)methanol (29A). mp 103.7–104.5 °C; 1H NMR (600 MHz, CDCl3) δ 2.46 (s, 3H), 3.30 (s, 3H), 3.64 (s, 3H), 3.74 (br s, 3H), 5.30–5.42 (m, 5H), 5.78 (d, J = 8.4 Hz, 1H), 6.47 (d, J = 8.4 Hz, 2H), 6.75 (s, 1H), 6.81 (d, J = 7.2 Hz, 1H), 7.03–7.15 (m, 8H), 7.29 (d, J = 7.2 Hz, 1H), 7.34 (d, J = 7.2 Hz, 2H), 7.87 (d, J = 7.2 Hz, 2H) ppm; 13C NMR (CDCl3, 150 MHz) δ 21.66, 55.08, 55.37, 56.00, 61.74, 105.41, 106.67, 108.30, 109.09, 111.52, 112.15, 116.80, 116.89, 117.42, 119.68, 122.76, 122.90, 123.30, 124.57, 124.71, 125.11, 125.28, 125.78, 127.82, 128.68, 129.17, 129.38, 129.68, 129.95, 131.08, 131.36, 131.56, 132.38, 134.02, 136.38, 136.67, 144.93, 155.51, 155.51, 156.10, 157.72 ppm; HRMS (M + Na+) calcd. for C41H33F9N2NaO7S 891.1762, found 891.1716; IR (KBr) 3408, 1587, 1503, 1461, 1415, 1378, 1331, 1239, 1175, 1123, 1082, 1027, 922, 894, 860, 718, 672, 595 cm−1.
(1′-Tosyl-1′H-1,3′-bipyrrole-2,2′-diyl)bis((2-methoxy-4-(trifluoromethyl)phenyl)methanone) (30). To a solution of 29 (800 mg, 1.16 mmol) in dry DMSO (20 mL) was gradually added IBX (810 mg, 2.89 mmol) at room temperature. After being stirred for about 1 h, the mixture was quenched with water (30 mL) and extracted with EtOAc (15 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuum. The residue was purified by flash column chromatography (20% EtOAc/petroleum ether, Rf = 0.2) to give 30 (670 mg, 84%) as a light yellow solid. mp 81.3–83.3 °C; 1H NMR (400 MHz, CDCl3) δ 2.45 (s, 3H), 3.78 (s, 3H), 3.80 (s, 3H), 5.89 (t, J = 2.8 Hz, 1H), 6.26 (d, J = 2.8 Hz, 1H), 6.41 (d, J = 3.6 Hz, 1H), 6.74 (s, 1H), 6.85 (s, 1H), 6.98 (d, J = 8.0 Hz, 1H), 7.12–7.19 (m, 3H), 7.37 (d, J = 7.6 Hz, 3H), 7.65 (d, J = 3.2 Hz, 1H), 7.96 (d, J = 8.0 Hz, 2H) ppm; 13C NMR (CDCl3, 100 MHz) δ 21.68, 55.81, 55.85, 107.74, 108.17, 109.43, 111.60, 116.43, 116.63, 123.90, 125.74, 128.13, 128.36, 128.36, 129.29, 129.66, 129.66, 130.63, 131.01, 131.61, 132.13, 132.77, 133.06, 133.83, 134.05, 134.15, 135.56, 145.37, 145.37, 157.06, 157.69, 182.04, 183.06 ppm; HRMS (M + H+) calcd. for C33H25F6N2O6S 691.1338, found 691.1336; IR (KBr) 3633, 3433, 3148, 2940, 1650, 1586, 1461, 1413, 1330, 1175, 1129, 1028, 671 cm−1.
1′H-1,3′-Bipyrrole-2,2′-diylbis((2-methoxy-4-(trifluoromethyl)phenyl)methanone) (31). To a solution of 30 (670 mg, 0.97 mmol) in a 1:1 mixture of MeOH/THF (10 mL) was added KOH (218 mg, 3.88 mmol) at room temperature. After being stirred for 15 min, the mixture was adjusted to pH 7.0 with 0.5 N HCl and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuum. The residue was purified by flash column chromatography (33% EtOAc/petroleum ether, Rf = 0.3) to give 31 (494 mg, 95%) as a light yellow solid. mp 75.0–77.0 °C; 1H NMR (400 MHz, CDCl3) δ 3.76 (s, 3H), 3.84 (s, 3H), 5.84 (t, J = 4.0 Hz, 1H), 6.29–6.32 (m, 2H), 6.65 (s, 1H), 6.87 (s, 1H), 6.95 (d, J = 7.6 Hz, 1H), 7.11 (t, J = 3.2 Hz, 1H), 7.14 (s, 1H), 7.19–7.21 (m, 2H), 7.25–7.26 (m, 1H), 9.51 (br s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 55.61, 55.85, 107.31, 108.26, 109.08, 110.68, 116.64, 116.87, 116.91, 123.92, 124.21, 125.88, 128.88, 129.38, 131.32, 131.63, 131.75, 132.26, 132.50, 132.58, 132.65, 132.92, 156.39, 157.11, 182.18, 182.49 ppm; HRMS (M + H+) calcd. for C26H19F6N2O4 537.1249, found 537.1238; IR (KBr) 3295, 2943, 1636, 1462, 1413, 1331, 1126, 1076, 928, 829, 742 cm−1.
1′H-1,3′-Bipyrrole-2,2′-diylbis((2-hydroxy-4-(trifluoromethyl)phenyl)methanone) (32). To a solution of 31 (100 mg, 0.19 mmol) in dry CH2Cl2 (5 mL) was slowly added a solution of BBr3 (233 mg, 0.93 mmol) in dry CH2Cl2 (1 mL) via a syringe under N2 at −78 °C. After being stirred for 0.5 h, the mixture was quenched by addition of water (10 mL) and extracted with CH2Cl2 (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuum. The residue was purified by flash column chromatography (30% EtOAc/petroleum ether, Rf = 0.3) to give 32 (78 mg, 82%) as a yellow solid. mp 148.0–149.0 °C; 1H NMR (400 MHz, CDCl3) δ 6.26 (dd, J = 4.0, 2.8 Hz, 1H), 6.39 (s, 1H), 6.69 (d, J = 8.0 Hz, 1H), 6.75 (dd, J = 4.0, 1.2 Hz, 1H), 6.98 (t, J = 2.0 Hz, 1H), 7.01 (d, J = 8.4 Hz, 1H), 7.03 (s, 1H), 7.21 (d, J = 5.6 Hz, 2H), 7.37 (d, J = 8.0 Hz, 1H), 8.40 (d, J = 8.0 Hz, 1H), 9.46 (br s, 1H), 10.85 (br s, 1H), 11.42 (br s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ 109.89, 111.00, 114.80, 114.83, 114.98, 115.02, 115.42, 115.45, 121.36, 121.80, 123.28, 124.15, 124.47, 129.99, 130.53, 130.92, 132.19, 132.41, 136.17, 136.59, 161.01, 162.11, 186.42, 187.09 ppm; HRMS (M + H+) calcd. for C24H15F6N2O4 509.0936, found 509.0934; IR (KBr) 3334, 3148, 3080, 1636, 1591, 1562, 1412, 1337, 1231, 1130, 1068, 944, 875, 786, 748, 605 cm−1.
(4,4′,5,5′-Tetrachloro-1′H-1,3′-bipyrrole-2,2′-diyl)bis((2-hydroxy-4-(trifluoromethyl)phenyl)methanone) (33). To a solution of 32 (50 mg, 0.10 mmol) in dry MeCN (10 mL) at room temperature was stepwise added NCS (72 mg, 0.54 mmol). After being stirred for about 3.5 h at room temperature, the mixture was quenched with water (10 mL) and extracted with EtOAc (10 mL × 3). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuum. The residue was purified by flash column chromatography (12% EtOAc/petroleum ether, Rf = 0.2) to give 33 (12 mg, 20%) as a yellow solid. mp 70.7–72.3 °C; 1H NMR (400 MHz, CDCl3) δ 6.74 (d, J = 8.3 Hz, 1H), 6.80 (s, 1H), 7.17 (d, J = 8.3 Hz, 1H), 7.20 (s, 1H), 7.31 (s, 1H), 7.58 (d, J = 8.2 Hz, 1H), 7.77 (d, J = 8.3 Hz, 1H), 10.06 (s, 1H), 10.22 (s, 1H), 11.17 (s, 1H) ppm; 13C NMR (CDCl3, 100 MHz) δ ppm 110.94, 113.57, 114.93, 115.14, 115.54, 115.94, 120.82, 121.07, 121.21, 121.49, 123.41, 123.45, 125.00, 128.25, 130.59, 132.36, 136.70, 137.03, 137.20, 137.53, 160.53, 162.37, 184.81, 186.10; HRMS (M + H+) calcd. for C24H11Cl4F6N2O4 644.9372, found 644.9372; IR (KBr) 3789, 3661, 3577, 2917, 2847, 1726, 1636, 1601, 1489, 1407, 1332, 1218, 1133, 946, 832 cm−1. HPLC purity, 97.2% (Flow rate: 1.0 mL/min; Column: Waters C8, 5 μm, 150 × 4.6 mm; Wavelength: UV 254 nm; Temperature: 25 °C; Mobile phase: MeOH:H2O = 68:32; tR = 9.2 min).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






