2.4. Western Blotting
In addition to functional activity assays, expression of specific apoptotic pathway intermediates at the protein level was assessed in ECM-treated Jurkat cells through Western blotting with monoclonal antibodies against caspases-3, -6, -7, -8, -9, and -10, cleaved caspases -3, -7, -8, and -9, apoptotic pathway promoters Smac/DIABLO, APAF-1, FADD, and apoptotic pathway inhibitors FLIP, PARP, and XIAP.
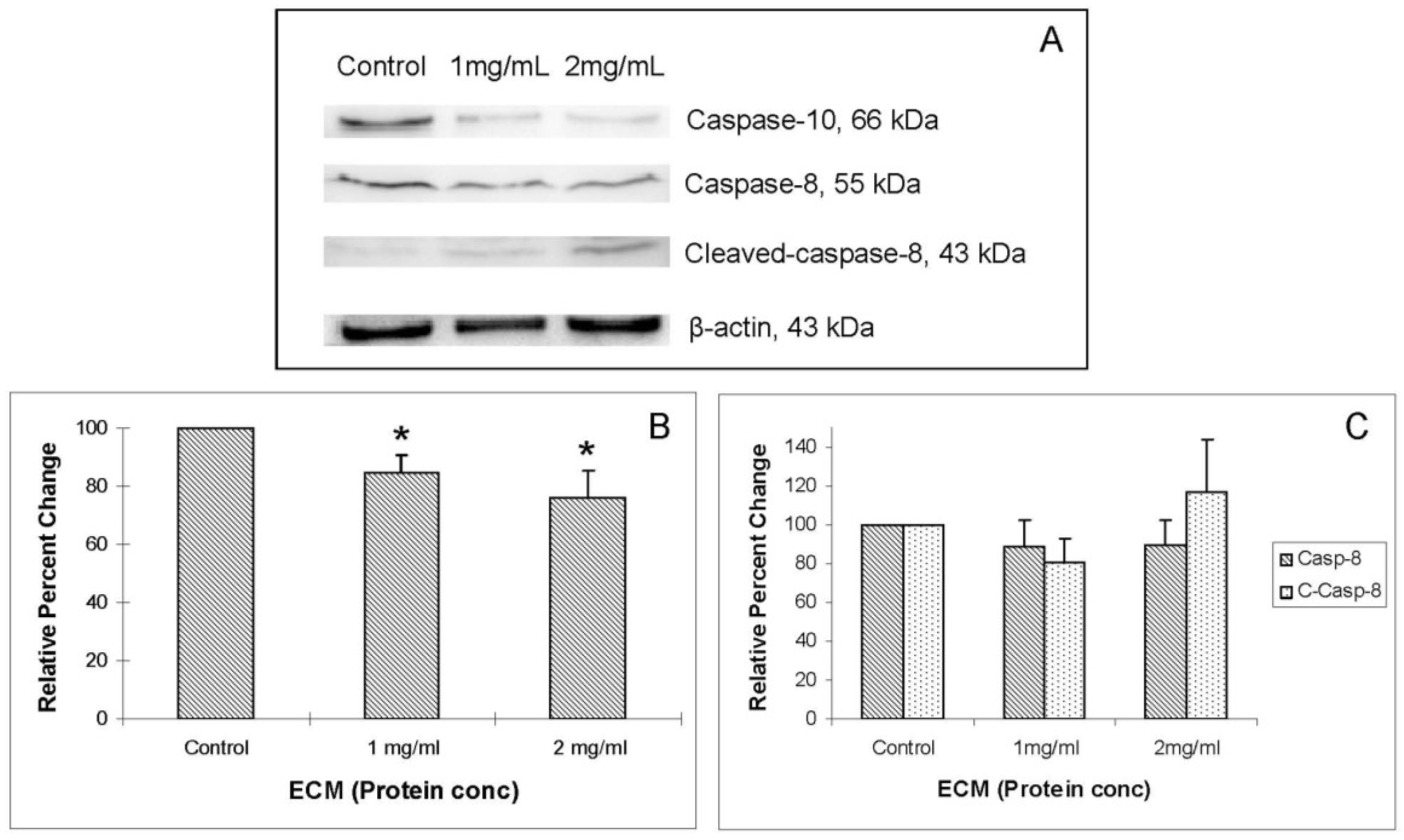
Western blot images demonstrating relative concentrations of initiator caspases -8 and -10, as well as cleaved caspase-8, are shown in
Figure 4A.
Figure 4B,C show relative band densities, normalized to β-actin as protein loading control, of each of these molecules in ECM-treated Jurkat cell lysates compared with control. A significant (
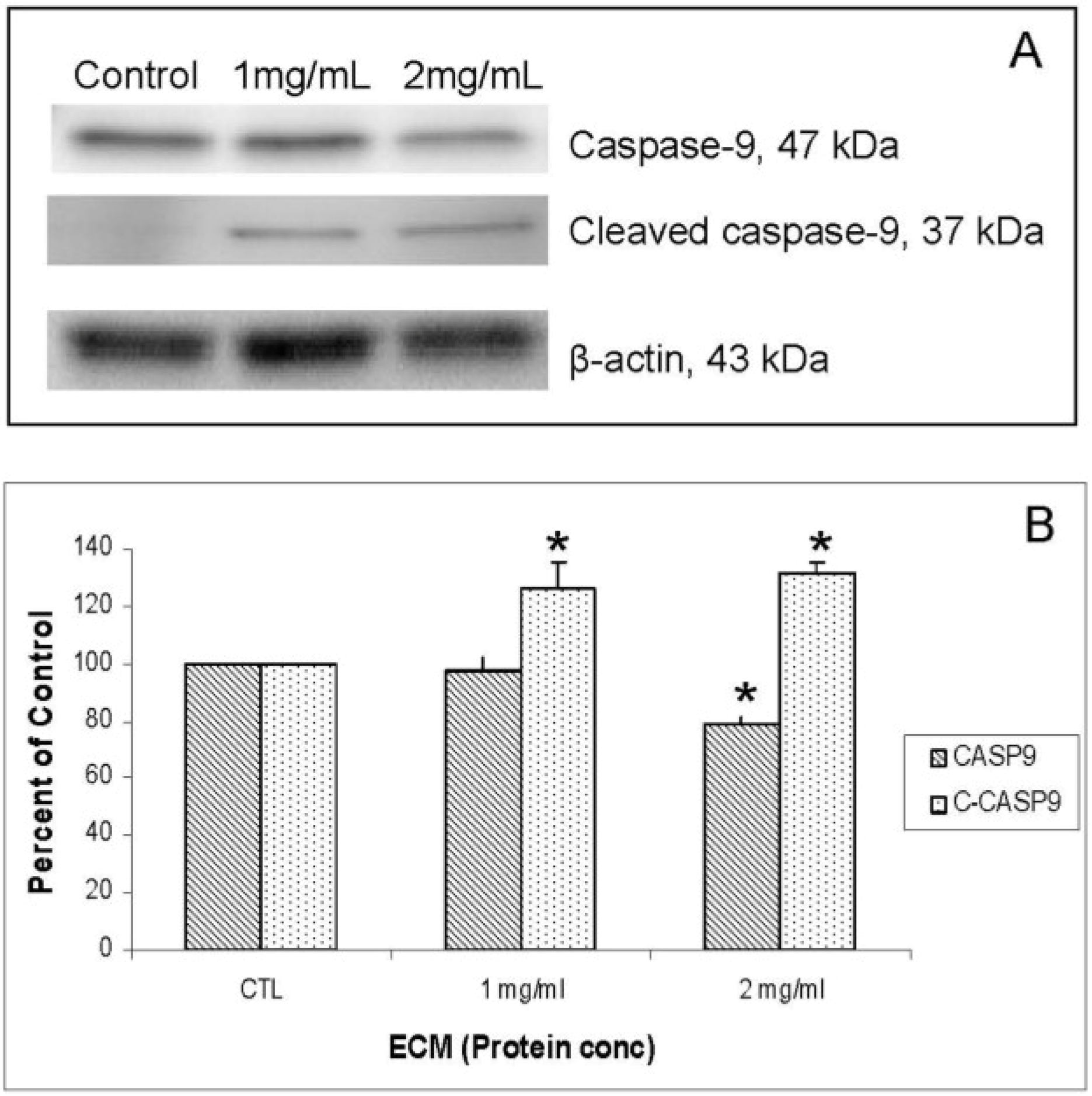
P < 0.05) decrease in the amount of full-length caspase-10 was observed after treatment with both 1 and 2 mg/mL ECM, which demonstrates activation of pro-caspase-10 to cleaved caspase-10. Differences in full-length and cleaved caspase-8 expression were not significant, although visually there appeared to be a difference in expression of these proteins in ECM-treated cells. These images demonstrate significant activation of caspase-10. A significant effect on protein expression of caspase-8 is not strongly observed through Western blotting. Once caspases-8 and -10 are activated, caspase-9 is cleaved, followed by activation of effector caspases-6 and -7, and finally by the terminal caspase, caspase-3. Western blot images demonstrating relative concentrations of caspase-9 and cleaved caspase-9 in Jurkat cells treated with ECM for 24 h are shown in
Figure 5A. Relative band densities, using β-actin as protein loading control, are shown in
Figure 5B. Full-length caspase-9 was significantly reduced in response to 2 mg ECM/mL (78.58% ± 2.36% of control;
P = 0.015; one-way ANOVA). Production of cleaved caspase-9 is apparent, with an increase in band density over untreated cells observed following 1 mg/mL ECM treatment (125.89% ± 9.74%,
N = 4,
P = 0.029,
t-test) and 2 mg/mL ECM treatment (131.20% ± 4.34%,
P = 0.029,
t-test). Activation of caspase-9 indicates progression of apoptosis through the mitochondrial (intrinsic) pathway.
Figure 4.
Western blot analysis demonstrating relative concentrations of caspase-10, caspase-8, and cleaved caspase-8 in Jurkat cells treated with ECM for 24 h, using β-actin as protein loading control. (a) Western blot images and relative band densities for (b) caspase-10; and (c) caspase-8 and cleaved caspase-8. * Significantly different (P < 0.05) from control.
Figure 4.
Western blot analysis demonstrating relative concentrations of caspase-10, caspase-8, and cleaved caspase-8 in Jurkat cells treated with ECM for 24 h, using β-actin as protein loading control. (a) Western blot images and relative band densities for (b) caspase-10; and (c) caspase-8 and cleaved caspase-8. * Significantly different (P < 0.05) from control.
Figure 5.
Western blot analysis demonstrating relative concentrations of caspase-9 and cleaved caspase-9 in Jurkat cells treated with ECM for 24 h, using β-actin as protein loading control. (a) Western blot images; (b) relative band densities for caspase-9 and cleaved caspase-9. * Significantly different (P < 0.05) from control.
Figure 5.
Western blot analysis demonstrating relative concentrations of caspase-9 and cleaved caspase-9 in Jurkat cells treated with ECM for 24 h, using β-actin as protein loading control. (a) Western blot images; (b) relative band densities for caspase-9 and cleaved caspase-9. * Significantly different (P < 0.05) from control.
Western blot images demonstrating relative concentrations of caspase-6, -7, and -3 and cleaved caspases -7 and -3 in Jurkat cells treated with ECM for 24 h are shown in
Figure 6A. Corresponding relative band densities are shown in
Figure 6B,C. Caspase-6 is one of the major executioner caspases functioning in cellular apoptotic processes [
11]. With full-length caspase-6 (35 kDa), there was very little detectable change in protein expression with ECM treatment (
Figure 6A), although there was a slight (86.89% ± 6.52% of control), but statistically insignificant, decrease in expression at 2 mg/mL (
Figure 6B). No significant differences in expression of full length caspase-6 were detected. Likewise, with caspase-7, there were no detectable changes in expression of full-length caspase-7 following ECM treatment for 24 h (
Figure 6A,C). A slight increase (120% ± 15%) was seen in amount of cleaved caspase-7 in response to treatment with 2 mg/mL ECM, but this expression did not differ significantly from control. With caspase-3, there were slight decreases in amount of protein for full-length caspase protein (
Figure 6A,D). Amounts of cleaved caspase-3 were increased by 117.72% ± 17.07% in response to 1 mg/mL and 205.84% ± 20.64% in response to 2 mg/mL (
P = 0.037; one way ANOVA;
Figure 6D). These observations indicate cleavage, and consequently, activation of initiator caspase-9 and terminal caspase-3. No significant differences in expression of proteins for caspase-6 or -7 were observed. Molecular pathways in Jurkat cells treated with ECM at concentrations of 1 and 2 mg protein/mL for 24 h indicate activation of apoptotic pathways and progression of cell death through apoptotic mechanisms.
Figure 6.
Western blot analysis demonstrating relative concentrations of caspases -6, -7, and -3 and cleaved caspase -7 and -3 in Jurkat cells treated with ECM for 24 h, using β-actin as protein loading control. (a) Western blot images; and relative band densities for (b) caspase-6; (c) caspase-7 and cleaved caspase-7; and (d) caspase-3 and cleaved caspase-3. * Significantly different from untreated control.
Figure 6.
Western blot analysis demonstrating relative concentrations of caspases -6, -7, and -3 and cleaved caspase -7 and -3 in Jurkat cells treated with ECM for 24 h, using β-actin as protein loading control. (a) Western blot images; and relative band densities for (b) caspase-6; (c) caspase-7 and cleaved caspase-7; and (d) caspase-3 and cleaved caspase-3. * Significantly different from untreated control.
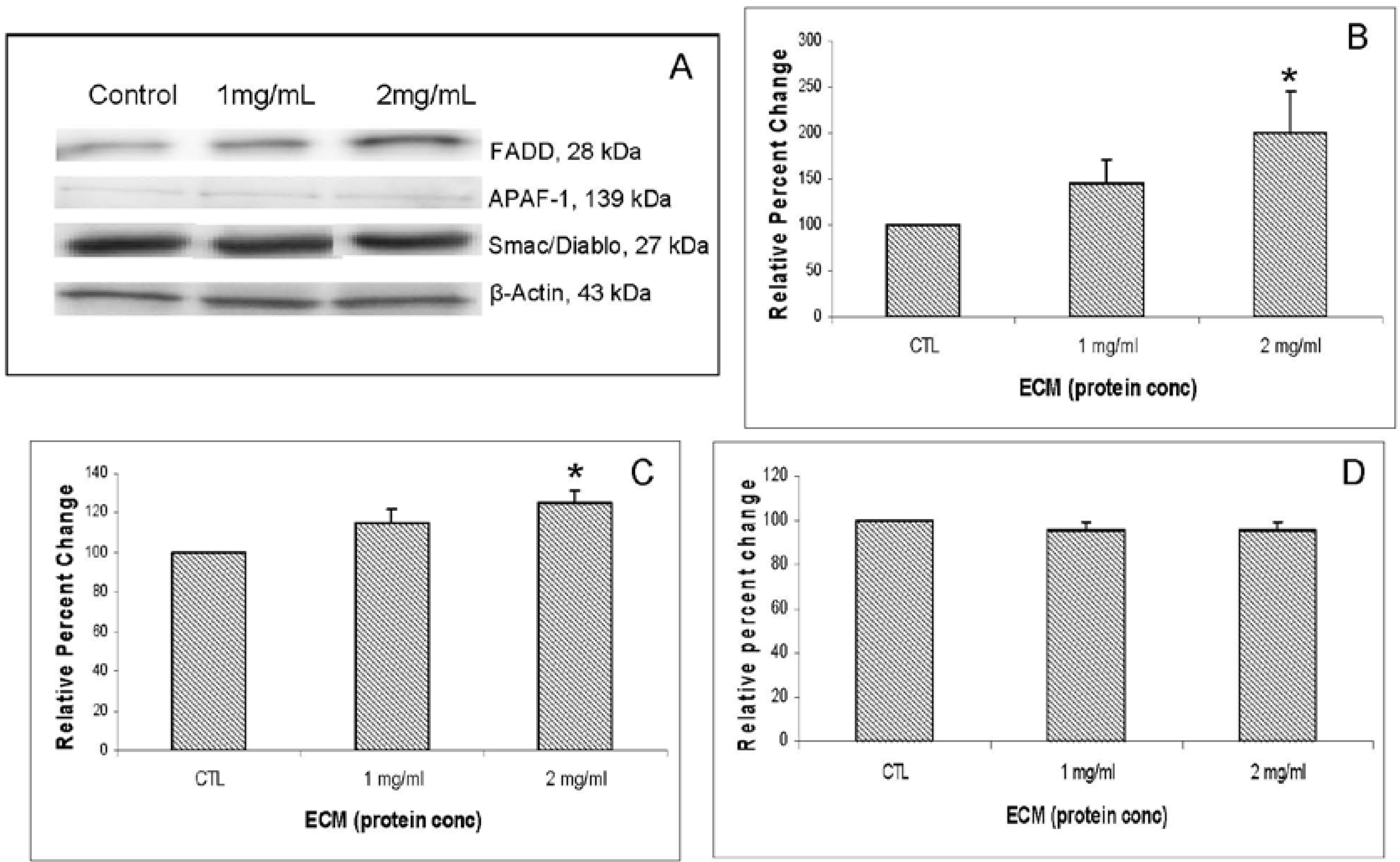
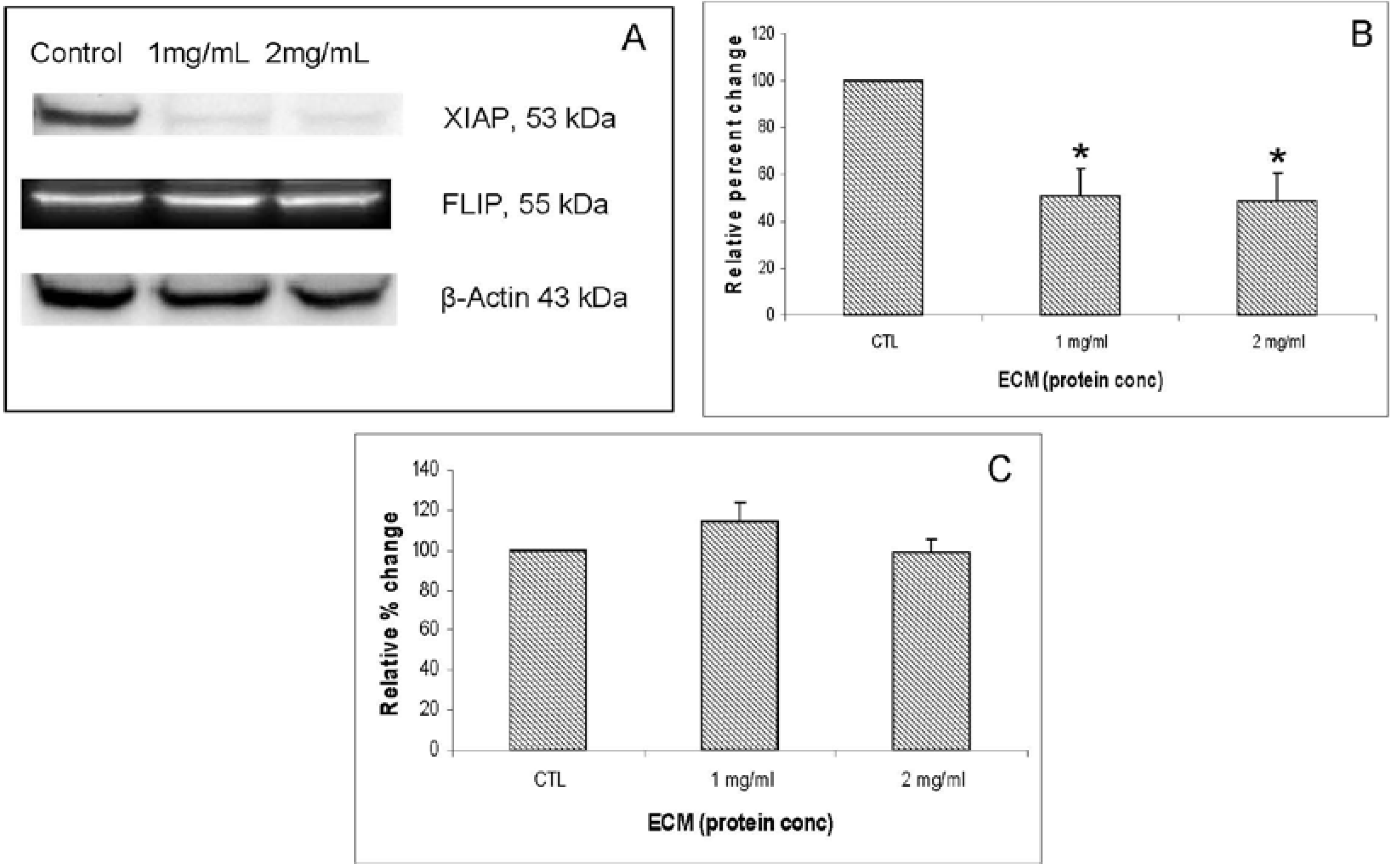
Other apoptosis pathway intermediates that were examined for their role in ECM-induced apoptosis in Jurkat cells include regulators involved in activation (FADD, APAF-1, Smac/DIABLO) as well as inhibitors of apoptosis (FLIP, XIAP, PARP). Western blotting results for FADD, APAF-1, and Smac/DIABLO are shown in
Figure 7. Western blotting results for inhibitors FLIP and XIAP are shown in
Figure 8. FADD (Fas-associated via death domain) functions as an apoptotic adaptor molecule that recruits caspase-8 or -10 to activated Fas (CD95) or TNFR-1 to form a death-inducing signaling complex (DISC) [
12]. Relative band densities demonstrating effects of 24 h ECM on expression of FADD through Western blotting are shown in
Figure 7B. With FADD, relative band density changed in response to 1 mg/mL (144.73% ± 25.45%,
N = 3) and in response to 2 mg/mL (199.32% ± 45.2%,
N = 3,
P <0.05, one-way ANOVA followed by Tukey’s). APAF-1 (apoptotic protease activating factor 1) is a cytoplasmic protein involved in apoptosis by forming part of the apoptosome that binds and activates caspase-9. Relative band densities for APAF-1 are shown in
Figure 7C. APAF-1 changed in relative band density by 114.79% ± 6.82% at 1 mg/mL ECM and 124.98% ± 6.07% at 2 mg/mL ECM (
P = 0.021, one way ANOVA followed by Tukey’s). Smac/DIABLO is a mitochondrial protein that functions as an apoptotic promoter by translocating to the cytosol and activating caspases in the cytochrome c/APAF-1/caspase-9 pathway and opposing inhibitory activity of inhibitor of apoptosis proteins (IAP), including XIAP [
13]. No detectable change in expression of Smac/DIABLO was observed among ECM treatments. In summary, results indicate an increase in FADD and APAF-1 expression, but no change in expression of Smac/DIABLO. An increase in FADD expression indicates that ECM is involved in recruitment of FADD to the receptor, possibly through binding to TRAIL receptor [
14], which would also be consistent with observed increased in caspase-8 activity. The increase in APAF-1 expression provides evidence that ECM initiates pathways that involve apoptosome formation and activation of caspase-9 [
15]. Logically, a translocation of Smac/DIABLO would be involved in this process as well, but a significant change in expression of this molecule was not observed. This is not unexpected; however, since the whole cell lysate method used in these experiments does not provide information about protein localization within the cell.
Figure 7.
Western blot analysis demonstrating effects of 24 h ECM treatment on expression of apoptotic pathway proteins FADD, APAF-1, and Smac/DIABLO. Western blot images (a) and relative band densities for (b) FADD; (c) APAF-1; and (d) Smac/DIABLO, using β-actin as protein loading control. *Significantly different (P < 0.05) from untreated control.
Figure 7.
Western blot analysis demonstrating effects of 24 h ECM treatment on expression of apoptotic pathway proteins FADD, APAF-1, and Smac/DIABLO. Western blot images (a) and relative band densities for (b) FADD; (c) APAF-1; and (d) Smac/DIABLO, using β-actin as protein loading control. *Significantly different (P < 0.05) from untreated control.
XIAP (X-linked inhibitor of apoptosis) encodes a potent apoptotic suppressor protein that functions by suppressing activities of caspases -3 and 7 [
13]. FLIP (FLICE-like inhibitory protein), also known as CFLAR (CASP8 and FADD-like apoptosis regulator), is structurally similar to caspase-8 and regulates apoptosis by functioning as a crucial link between cell survival and cell death pathways by inhibiting TNFRSF6-mediated apoptosis [
16,
17]. As the major protein that prevents caspase-8 from activation by death receptors, c-FLIP protein can be recruited to the death-inducing signaling complex (DISC) to inhibit caspase-8 activation [
18,
19]. Western blot images demonstrating effects of 24 h ECM treatment on expression of apoptotic pathway inhibitor proteins, XIAP and FLIP, are shown in
Figure 8A. Graphs representing relative band densities are shown in
Figure 8B,C. XIAP was strongly affected by ECM treatment, with a relative band density 51.13% ± 11.29% of control at 1 mg/mL and 48.24% ± 12.14% of control at 2 mg/mL. These differences were significant at 1 mg/mL (
P = 0.014, One way ANOVA Tukey’s) and 2 mg/mL (
P = 0.01, Tukey’s). These results indicate a strong down-regulation of the key inhibitor protein, XIAP. Down-regulation of XIAP functions in facilitating progression of apoptotic pathways towards cell death by inhibiting caspase activity. Relative band density of FLIP did not change significantly in response to ECM treatment at either 1 or 2 mg/mL. Since FLIP is involved in inhibiting recruitment of caspase-8 [
17], the absence of significant effects on expression of this inhibitor protein indicates no inhibition of apoptosis proceeding through caspase-8 recruitment and activation.
Figure 8.
Western blot analysis demonstrating effects of 24 h ECM treatment on expression of apoptotic pathway proteins XIAP and FLIP. (a) Western blot images; and relative band densities for (b) XIAP; and (c) FLIP, using β-actin as protein loading control. * Significantly different (P < 0.05) from untreated control.
Figure 8.
Western blot analysis demonstrating effects of 24 h ECM treatment on expression of apoptotic pathway proteins XIAP and FLIP. (a) Western blot images; and relative band densities for (b) XIAP; and (c) FLIP, using β-actin as protein loading control. * Significantly different (P < 0.05) from untreated control.
Another protein involved in inhibition of apoptosis is PARP (poly ADP-ribose polymerase), a nuclear protein that plays an important role in DNA repair [
20] and is inactivated by enzymatic cleavage. Once the enzyme is inactivated by caspase-3, DNA repair is prevented, which allows DNA fragmentation, and thus apoptosis, to proceed. Effects of ECM treatment on the apoptotic inhibitor, PARP, were investigated using Western blotting, with results shown in
Figure 9A (blot) and 9B (relative band densities for PARP/cleaved PARP). Cleavage of PARP was strongly observed in ECM-treated Jurkat cells, as evidenced by significant (
P < 0.05) decreases in full-length PARP and significant increases in cleaved PARP at both 1 and 2 mg ECM protein/mL. These results indicate that the nuclear DNA repair protein, PARP, is inactivated in Jurkat cells treated with ECM.
Figure 9.
Western blot analysis demonstrating effects of 24 h ECM treatment on expression of apoptotic pathway protein PARP. (a) Western blot images; (b) relative band densities for PARP and cleaved PARP, using β-actin as protein loading control. * Significantly different from untreated control.
Figure 9.
Western blot analysis demonstrating effects of 24 h ECM treatment on expression of apoptotic pathway protein PARP. (a) Western blot images; (b) relative band densities for PARP and cleaved PARP, using β-actin as protein loading control. * Significantly different from untreated control.
2.5. Antibody Array
An antibody array that measures 43 different apoptotic pathway intermediates was utilized to assess effects of ECM on Jurkat cells. The change in expression of apoptotic pathway proteins in Jurkat cell lysates detected on the antibody array following 24 h treatment with ECM is shown in
Figure 10. Results are expressed as relative percent change compared to control (untreated) Jurkat cells using a graph representing relative band densities (
Figure 10A) and a heat map (
Figure 10B). A representative image of antibody arrays from control and ECM-treated (1 mg/mL) Jurkat cell lysates is also given (
Figure 10C). Responses to ECM varied with concentration; of the 43 proteins that were on the array, only 9 were significantly (
P < 0.05) changed from control in response to treatment with either 1 or 2 mg/mL ECM (ANOVA,
P < 0.05). No pathway intermediates changed significantly in response to both ECM concentrations. The nine proteins which changed significantly at either 1 or 2 mg/mL ECM included the mitochondrial proteins BAD and BAX, apoptosis inhibitor, survivin, initiator caspase-8, death receptors DR6 and Fas, insulin growth factor binding protein IGFBP-1, and TNF family members TNF-α and sTNF-R2.
Figure 10.
Relative percent change in expression of apoptotic pathway proteins in Jurkat cell lysates following 24 h treatment with ECM. Protein expression was determined using an antibody array (RayBio) containing 43 different antibodies for apoptotic pathway intermediates. Results are expressed as relative percent change compared to control (untreated) Jurkat cells using a heat map according to the color scale on the lower right. Only proteins that were significantly different (P < 0.05) compared to control at either 1 or 2 mg/mL ECM are shown. (a) Relative expression of intermediates significantly increased or decreased (d) from control at either 1 or 2 mg/mL. * Significantly different from control (P < 0.05); (b) heat map showing relative change in expression of proteins with significant increases or decreases; (c) representative array blot. N = 4 trials for the experiment.
Figure 10.
Relative percent change in expression of apoptotic pathway proteins in Jurkat cell lysates following 24 h treatment with ECM. Protein expression was determined using an antibody array (RayBio) containing 43 different antibodies for apoptotic pathway intermediates. Results are expressed as relative percent change compared to control (untreated) Jurkat cells using a heat map according to the color scale on the lower right. Only proteins that were significantly different (P < 0.05) compared to control at either 1 or 2 mg/mL ECM are shown. (a) Relative expression of intermediates significantly increased or decreased (d) from control at either 1 or 2 mg/mL. * Significantly different from control (P < 0.05); (b) heat map showing relative change in expression of proteins with significant increases or decreases; (c) representative array blot. N = 4 trials for the experiment.
BAD and BAX are mitochondrial proteins that belong to the bcl-2 family of apoptosis regulators [
21]. BAD, also known as BCL2-associated agonist of cell death, positively regulates apoptosis by forming heterodimers with BCL-XL and BCL-2 and reversing their repressor activity. BAX, also known as BCL2-associated X protein, activates apoptosis by opening mitochondrial voltage-dependent anion channel (VDAC) which leads to loss in membrane potential and subsequent release of cytochrome c from the mitochondria. BAX and BAD were both up-regulated greater than >150% in response to 1 mg/mL ECM (
P < 0.05), but not significantly up-regulated in response to 2 mg/mL. Although an antibody for cytochrome c was included on the array, expression of cytochrome c was not significantly changed in response to ECM treatment. An increase in cytochrome c release, however, was previously demonstrated using mitochondrial and cytosolic fractions from another cell line, a B-cell lymphoma (Daudi) exposed to ECM [
5,
6]. Similar to Smac/DIABLO, it is likely that no change was observed in these experiments with ECM-treated Jurkat cells because whole cell lysates rather than separated mitochondrial and cytosolic fractions were utilized.
Only two caspases (caspase-3 and caspase-8) were included on the array. Interestingly, caspase-3 was not significantly changed in response to ECM treatment on the array, although blot results with individual antibodies did show a significant change in both full-length procaspase enzyme forms as well as cleaved (active) forms of the enzyme. Levels of caspase-8 were up-regulated on the antibody array in response to 1 mg/mL ECM, a response different from the results obtained using Western blotting and individual antibodies, however, functional enzyme activity assays also indicated induction of caspase-8 activity with ECM treatment.
Several members of the TNF receptor superfamily were included on the array. DR6 and Fas are both cell surface receptors belonging to the TNF receptor superfamily that were significantly up-regulated in response to ECM. Expression of DR6 was significantly increased in response to 1 mg/mL ECM, but not 2 mg/mL. DR6 is TNF receptor superfamily member 21 (TNFRSF21) and is involved in inducing apoptosis. Through its death domain, DR6 interacts with TRADD (TNFRSF1A-associated via death domain), an adaptor molecule that mediates signal transduction occurring through TNF-receptors and triggers the caspase cascade [
12,
22]. DR6 also activates NF-κB and MAPK8/JNK. Fas, another member of the TNF-receptor superfamily (TNFRSF6), was also significantly up-regulated in response to 1 mg/mL ECM, but not 2 mg/mL. Fas, also known as CD95 or APO-1, plays a central role in regulation of apoptosis and contains a death domain [
12]. The interaction of Fas with FasL forms a death-inducing signaling complex that includes Fas-associated death domain protein (FADD), caspase-8 and caspase-10, and leads to apoptosis [
12]. Results indicate activation of Fas and DR6 by ECM, thus likely leading to induction of the caspase cascade through caspase-8. Other TNF family members on the array included soluble TNF receptors sTNF-R1 and sTNF-R2, with sTNF-R2 significantly down-regulated in response to 2 mg/mL ECM. TNF-R2 mediates most effects of TNF-α [
23]. TNF-α was significantly down-regulated in response to ECM treatment at 2 mg/mL. Although several TRAIL receptors, death receptors TRAIL-R1 (DR4) and TRAIL-R2 (DR5) and decoy receptors TRAIL-R3 (DcR3) and TRAIL-R4 (DcR2), were included on the array, none of these were significantly altered in response to ECM treatment. TRAIL-R1 (DR4) is activated by TRAIL and functions in transducing cell death signals and inducing apoptosis [
12]. TRAIL-R2 (DR5) is also activated by TRAIL binding and transduces apoptosis signals [
12]. The decoy receptor, TRAIL-R3 (DcR1), binds TRAIL but lacks a cytoplasmic death domain and therefore is not capable of inducing apoptosis. This receptor is believed to protect against TRAIL-mediated apoptosis by competing with TRAIL-R1 and R2 for ligand binding [
12]. Another decoy receptor, TRAIL-R4 (DcR2), also binds TRAIL but contains a truncated cytoplasmic death domain and thus does not transmit signals for apoptosis induction [
12]. Several heat shock proteins (HSP27, HSP60, HSP70), markers of cell stress, were included on the array, although none were significantly affected by ECM treatment.
Apoptosis is tightly regulated through careful balance of pro- and anti-apoptotic factors. Survivin and IGFBP-1 are apoptotic inhibitor proteins that were present on the array. IGFBP-1 has been reported to function as a negative regulator of BAK-dependent apoptosis, and thus functions to inhibit apoptosis [
24]. In response to ECM treatment, IGFBP-1 was significantly down-regulated at 2 mg/mL. Based on its role as an inhibitor of apoptosis, this decrease in IGFBP-1 protein levels indicates ECM facilitates apoptosis by decreasing inhibitor expression. Although a number of pro-apoptotic molecules were positively up-regulated, a significant increase in the anti-apoptotic molecule, survivin, was observed in response to 1 mg/mL ECM. Survivin, also known as BIRC5, is an inhibitor of apoptosis and functions in promoting cell survival by inhibiting activities of caspase-3 and caspase-7 [
25,
26]. Survivin is a member of the IAP (inhibitor of apoptosis proteins) family of proteins and acts downstream of mitochondria to prevent processing of caspase-9 from the apoptosome, which prevents activation of downstream effector caspases, and is thought to modulate both extrinsic and intrinsic apoptotic pathways [
25,
26]. In contrast to malignant tissues, survivin is not usually expressed in normal cells, thus making it an ideal target for cancer therapy [
27]. In experiments reported here, ECM treatment significantly increased survivin expression at 1 mg/mL, observations which are inconsistent with the large amount of data indicating activation of pro-apoptotic molecules. Despite up-regulation of survivin, however, ECM induces apoptosis in Jurkat cells. The explanation for this is unclear, although it appears that the pro-apoptotic signaling activated by ECM overcomes the anti-apoptotic signaling provided by survivin. Similar responses have been observed with another potential therapeutic agent, P2-341 (Bortezomib), in which survivin expression was increased in presence of apoptosis, an observation hypothesized to be a consequence of proteasome inhibition [
28]. In other systems, Fas-induced apoptosis was associated with release of cytochrome c as well as up-regulation of survivin in mitochondria, nucleus, and cytosol [
29]. Survivin is known to be a multifunctional protein with roles in cell division as well as apoptosis [
30]. Survivin biology includes a link to multiple pathways of cellular homeostasis [
31]. XIAP binds survivin that is released from mitochondria in response to cell death stimuli [
32]. Since XIAP was dramatically decreased, it is possible that the increase in survivin was a reflection of lower amounts of available XIAP. Alternatively, the observed increase in survivin expression may be related to one of the other cellular roles attributed to survivin. Overexpression of survivin has been reported to be more efficient at blocking mitochondrial but not death-receptor-induced apoptosis [
33]. Also of interest is that a complex between survivin and caspase-9 has been demonstrated [
34]; survivin is also believed to associated with Smac/DIABLO [
35].
2.7. PCR Array
The relative fold change in expression of apoptotic pathway genes in Jurkat cells following 24 h treatment with ECM is shown in
Figure 12. Gene expression was determined using a PCR array (Applied Biosystems) containing primers for 84 different genes coding for apoptotic pathway intermediates. Results are expressed as relative fold change compared to control (untreated) Jurkat cells using a heat map. Only genes with expression that was significantly changed (
P < 0.05) in response to ECM treatment at 1 or 2 mg/mL were included on the heat map. Genes that encode positive inducers of apoptosis are shown in
Figure 12A; inhibitors of apoptosis are shown in
Figure 12B. The first several genes listed in the array data in
Figure 12A encode mitochondrial proteins (BAD, BCL10 BCL2L11, BCL2L14, and BID). Expression of the genes encoding BAD, a mitochondrial protein that promotes apoptosis, was down-regulated in response to 1 and 2 mg/mL ECM treatment; an opposite effect to that observed at the protein level. Expression of mitochondrial BCL family members BCL10, BCL2L11, BCL2L14, all inducers of apoptosis, was up-regulated in response to 2 mg/mL ECM. BCL10 contains a caspase recruitment domain (CARD) and has been shown to induce apoptosis through activation of pro-caspase-9 and NF-κB. BCL2L11, also known as BIM, encodes a protein that acts as an apoptotic activator, as does BCL2L14. A gene coding for another mitochondrial protein and BCL2 family member, BID, was down-regulated in response to 1 and 2 mg/mL ECM. BID functions by mediating mitochondrial damage induced by caspase-8 and ultimately triggering cytochrome c release [
21].
Figure 12.
Relative fold change in expression of apoptotic pathway genes in Jurkat cells following 24 h treatment with ECM. Gene expression was determined using a PCR array (Applied Biosystems) containing primers for 84 different genes for apoptotic pathway intermediates. Results are expressed as relative fold change compared to control (untreated) Jurkat cells using a heat map according to the color scale. (a) relative fold change of pro-apoptotic intermediates; (b) relative fold change of anti-apoptotic intermediates. N = 3 trails for the experiment. Only genes that were significantly different among treatments (P < 0.05, N =3) are included in the heat map.
Figure 12.
Relative fold change in expression of apoptotic pathway genes in Jurkat cells following 24 h treatment with ECM. Gene expression was determined using a PCR array (Applied Biosystems) containing primers for 84 different genes for apoptotic pathway intermediates. Results are expressed as relative fold change compared to control (untreated) Jurkat cells using a heat map according to the color scale. (a) relative fold change of pro-apoptotic intermediates; (b) relative fold change of anti-apoptotic intermediates. N = 3 trails for the experiment. Only genes that were significantly different among treatments (P < 0.05, N =3) are included in the heat map.
The genes BNIP3, BNIP3L, and NOD1 were all down-regulated. BNIP3 and BNIP3L are apoptosis-inducing proteins that can overcome BCL2 and BCL-XL suppression [
36]. Nods, a growing family of proteins containing a nucleotide-binding oligomerization domain (NOD), are involved in regulation of programmed cell death and immune responses. APAF-1, ced-4, and Nod1 are members of this family. The NOD module is homologous to ATP-binding cassette (ABC) found in a large number of proteins with diverse biological function, and result in activation of diverse signaling pathways involved in elimination of cells via programmed cell death. NOD1 encodes a cytosolic APAF-1 like molecule that contains a caspase recruitment domain (CARD) and promotes apoptosis by enhancing caspase-9-mediated apoptosis [
37]. The most strongly up-regulated molecule on the array was CARD6, with greater than 8-fold increase in expression at both 1 and 2 mg/mL ECM. The protein encoded by this gene contains a CARD with a domain structure not shared by other CARD proteins and is a microtubule-associated protein that interacts with receptor-interacting protein kinases (RIPK) and positively modulates signal transduction pathways activating NF-κB [
38,
39]. Most proteins containing a CARD are involved in pathways regulating apoptosis [
38,
39]. Examples of prominent CARD proteins are caspase-9 and APAF-1, which are involved in the intrinsic death pathway; BCL10 and CARD11, which mediate antigen receptor-induced NF-κB activation, and receptor-interacting protein (RIP)-like interacting caspase-like apoptosis regulatory protein kinase (RICK) and the nucleotide-binding oligomerization domain (NOD) proteins, which induce NF-κB activation [
38].
With regard to caspase genes on the array, the genes coding for CASP4 and CASP6 were up-regulated, whereas the genes coding for CASP5 and CASP8 were down-regulated. Caspase-8 was the only caspase that appeared on both the protein and gene arrays. CASP8 was down-regulated in the gene array and up-regulated on the protein array. CASP4, CASP5, CASP6, and CASP8 all encode caspase enzymes which are initially present in the cell as inactive precursor forms that become activated with cellular processing; overexpression of these genes promotes cell death. HIP1, also known as huntingtin interacting protein, was up-regulated. The protein encoded by this gene is important in cell filament networks and promotes apoptosis through the intrinsic apoptosis pathway [
40]. The gene coding for LRDD, also known as PIDD (p53-induced death domain protein), was down-regulated. LRDD is involved in apoptosis through interaction with death domain proteins such as FADD and MADD and may function as an adaptor protein in cell death-related signaling processes and play a role as an effector of p53-dependent apoptosis by promoting apoptosis as a component of the DNA damage/stress response pathway that connects p53 to apoptosis [
41]. PIDD is implicated in activation of pro-caspase-2 and may mediate apoptosis induction by tumor suppressor p53 [
42]. LTA encodes a protein, lymphotoxin alpha—a member of the tumor necrosis factor family, that functions as a death receptor ligand and plays a role in promoting apoptosis by binding to TNFRs [
43]. LTA was up-regulated in response to both 1 and 2 mg/mL ECM. NLRP1 is a gene that encodes a member of the ced-4 family of apoptosis proteins; ced-family members contain a CARD and are known to be key mediators of programmed cell death by enhancing APAF-1 and cytochrome c-dependent activation of pro-caspase 9. NLRP1 was up-regulated in response to 1 mg/mL ECM, but unchanged in response to 2 mg/mL ECM. The higher expression of genes for certain caspase enzymes, genes coding for death domain proteins, and genes coding for proteins that promote apoptosis through death receptors in response to ECM treatment indicates apoptosis induction in target cells by ECM, proceeding through death receptor-initiated mechanisms.
The transcription factor NFκB is activated in response to death receptor signaling [
44]. The pattern of NFkB pathway activation in Jurkat cells treated with ECM was complex. In the NFκB family, some mediators were up-regulated and some were down-regulated in response to ECM treatment. NFKB2, NFKBIE, and NFKBIZ were up-regulated. NFKB2, a subunit of the NFκB transcription factor complex, was up-regulated. REL (also designated c-Rel) was up-regulated, which functions in promoting cell death [
44]; RELB was also up-regulated. Certain NFkB pathway regulators, NFKBIE and NFKBIZ, were also up-regulated. PMAIP, also known as NOXA, was up-regulated. This molecule is known to promote activation of caspases, and thus induction of apoptosis. PMAIP1, REL, and RELB were up-regulated. PMAIP1 promotes activation of caspases and mitochondrial membrane changes that result in efflux of apoptogenic proteins from the mitochondria, while REL and RELB form part of NFKB complex.
RIPK1, also known as receptor (TNFRSF)-interacting serine-threonine kinase, was down-regulated. The protein encoded by this gene induces apoptosis following death receptor ligation and forms a necroptosis inducing complex [
45]. TNFRSF10A, also known as DR4 or TRAIL-R1, is activated by TRAIL and transduces cell death signals and induces apoptosis [
43]. TNFRSF10A was down-regulated on the gene array in response to both 1 and 2 mg/mL ECM. TNFRSF25 was up-regulated in response to 1 mg/mL ECM and TRADD was up-regulated in response to both 1 and 2 mg/mL ECM. TNFRSF25, also known as DR3, regulates apoptosis by interacting directly with TRADD and activating NFκB [
46]. Overexpression of TRADD (TNFR1-associated death domain protein) is involved in Fas-induced cell death pathway and activation of NFκB [
46].
BCL2A1, an inhibitor of apoptosis, was up-regulated in response to both 1 and 2 mg/mL. BCL2A1 is a member of the BCL2 family and encodes a protein that decreases mitochondrial release of cytochrome c and blocks caspase activation [
47]. Another apoptosis inhibitor, BCL2L1, was down-regulated. BCL2L1 also works by preventing release of caspase activators from the mitochondrial membrane and thus preventing activation of caspases [
21]. Other inhibitors, BCL3, NAIP, and BIRC3, were up-regulated. NAIP (NLR family, apoptosis inhibitory protein) is involved in suppressing apoptosis by inhibiting activities of caspases-3, -7, and -9 [
48]. BIRC3 encodes a member of the IAP family of proteins that inhibit apoptosis by binding to tumor necrosis receptor-associated factors TRAF1 and TRAF2 [
49]. BIRC5, also known as survivin, was down-regulated on the PCR array in response to 1 mg/mL ECM, but was up-regulated at the protein level on the antibody array in response to treatment with the same concentration of ECM. ICEBERG was strongly (>8-fold) up-regulated in response to 2 mg/mL ECM. ICEBERG is a CARD family member that inhibits caspase-1 [
50]. In other studies, ICEBERG was up-regulated in response to TNF [
50]. TBK1 (TANK-binding kinase 1), which is also known as NF-kappa-B-activating kinase, was up-regulated; this molecule plays a role in mediate NFKB activation in response to certain growth factors.
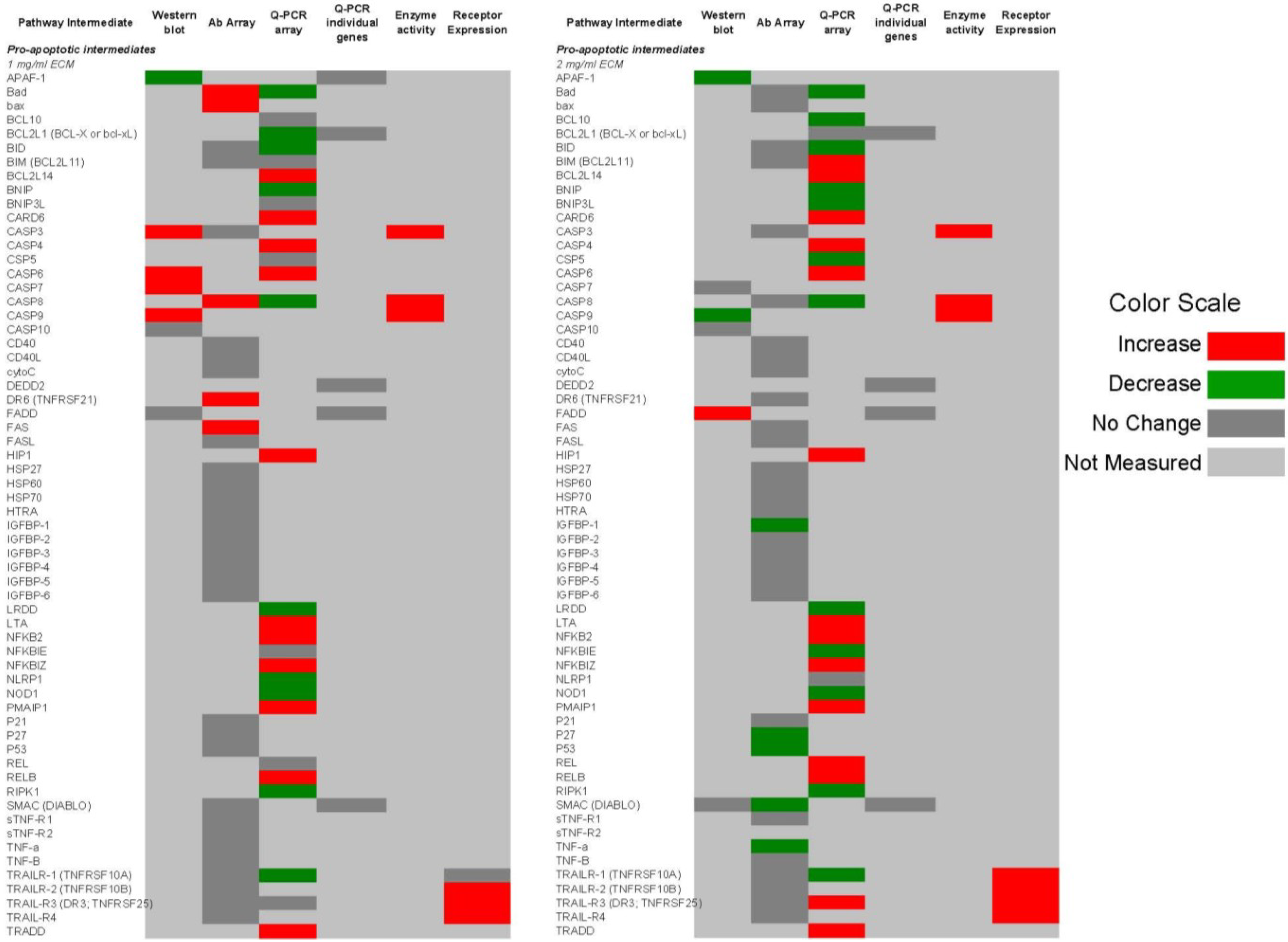
2.9. Comparison of Apoptosis Pathway Intermediates Using Different Means of Detection
Figure 14 and
Figure 15 show qualitative comparisons of effects of 1 and 2 mg/mL ECM on apoptosis mediators in Jurkat cells as measured by different methods. Observed increases or decreases of pro-apoptosis regulators (
Figure 14) and anti-apoptosis regulators (
Figure 15) among Western blotting with individual antibodies and antibody arrays, gene expression of pathway intermediates using Q-PCR of individual genes and PCR arrays, functional enzyme activity assays, and surface expression of receptors using flow cytometric detection are shown.
Figure 14.
Qualitative comparison of changes in 62 pro-apoptotic regulators in Jurkat T cell leukemia cells exposed to ECM at 1 and 2 mg/mL. Observed increases or decreases are based on antibody recognition of pathway intermediates using Western blotting with individual antibodies and antibody arrays, gene expression of pathway intermediates using Q-PCR of individual genes and PCR arrays, functional enzyme activity assays, and surface expression of receptors using flow cytometric detection.
Figure 14.
Qualitative comparison of changes in 62 pro-apoptotic regulators in Jurkat T cell leukemia cells exposed to ECM at 1 and 2 mg/mL. Observed increases or decreases are based on antibody recognition of pathway intermediates using Western blotting with individual antibodies and antibody arrays, gene expression of pathway intermediates using Q-PCR of individual genes and PCR arrays, functional enzyme activity assays, and surface expression of receptors using flow cytometric detection.
Figure 15.
Qualitative comparison of changes in 17 anti-apoptotic regulators in Jurkat T cell leukemia cells exposed to ECM at 1 and 2 mg/mL. Observed increases or decreases are based on antibody recognition of pathway intermediates using Western blotting with individual antibodies and antibody arrays, and gene expression of pathway intermediates using Q-PCR of individual genes and PCR array.
Figure 15.
Qualitative comparison of changes in 17 anti-apoptotic regulators in Jurkat T cell leukemia cells exposed to ECM at 1 and 2 mg/mL. Observed increases or decreases are based on antibody recognition of pathway intermediates using Western blotting with individual antibodies and antibody arrays, and gene expression of pathway intermediates using Q-PCR of individual genes and PCR array.
In these comparisons, inconsistencies among results obtained through different methods of analysis were observed. Some variations may be a result of the temporal nature of apoptotic signaling, with gene expression changes occurring generally earlier than changes reflected at the protein level.
With regard to mitochondrial proteins, BAD was up-regulated in response to 1 mg/mL at the protein level but down-regulated at the gene level in response to both 1 and 2 mg/mL treatments. BID showed no change in response to 1 mg/mL at the protein level, but was decreased in expression at the gene level. In response to 2 mg/mL, BIM was decreased at the protein level but increased at the gene level. BAX was up-regulated at the gene level. BAX and BAD are important pro-apoptotic in the mitochondria [
59].
With caspase enzymes, expression of caspase-8 was increased at the protein level and at the level of functional enzyme activity. Genetic expression of caspase-8, however, was decreased in response to 1 mg/mL. With 2 mg/mL ECM treatment, caspase-8 expression was decreased at both protein and gene levels. Caspase-3 expression was not significantly changed at the protein level as determined using the antibody array, but functional enzyme activity and amount of cleaved caspase-3 at the protein level were increased in response to 2 mg/mL ECM.
Expression of TRAIL-R1 was not changed at the protein level, but was decreased at the gene level. Surface expression was also not significantly up-regulated in response to 1 mg/mL ECM. Jurkat cells express higher levels of TRAIL-R2 than TRAIL-R1 [
56,
57]. Surface expression of TRAIL-R1 and TRAIL-R2, however, was significantly increased with 2 mg/mL ECM. No changed was observed at the protein level in expression of the decoy receptor TRAIL-R3, but an increase in expression was observed at the gene level in response to 2 mg/mL. Surprisingly, surface expression of the decoy receptors TRAIL-R3 and TRAIL-R4 was significantly increased. The observed effects with decoy and death receptors are difficult to interpret. In other studies, decoy receptors DcR1 and DcR2 were expressed on Jurkat cells and were increased in expression with stimulation with TRAIL [
60]. Generally, it has been believed that transient overexpression of DcR1 or DcR2 in TRAIL-sensitive tumor cells prevents cell death trigger by TRAIL [
61,
62] and recent evidence indicates that tumor and normal cells can acquire resistance to TRAIL-induced killing by up-regulating TRAIL antagonistic receptors [
55,
63,
64]. It has been proposed that decoy receptors actually are likely to play more of a regulatory rather than inhibitory role [
55].
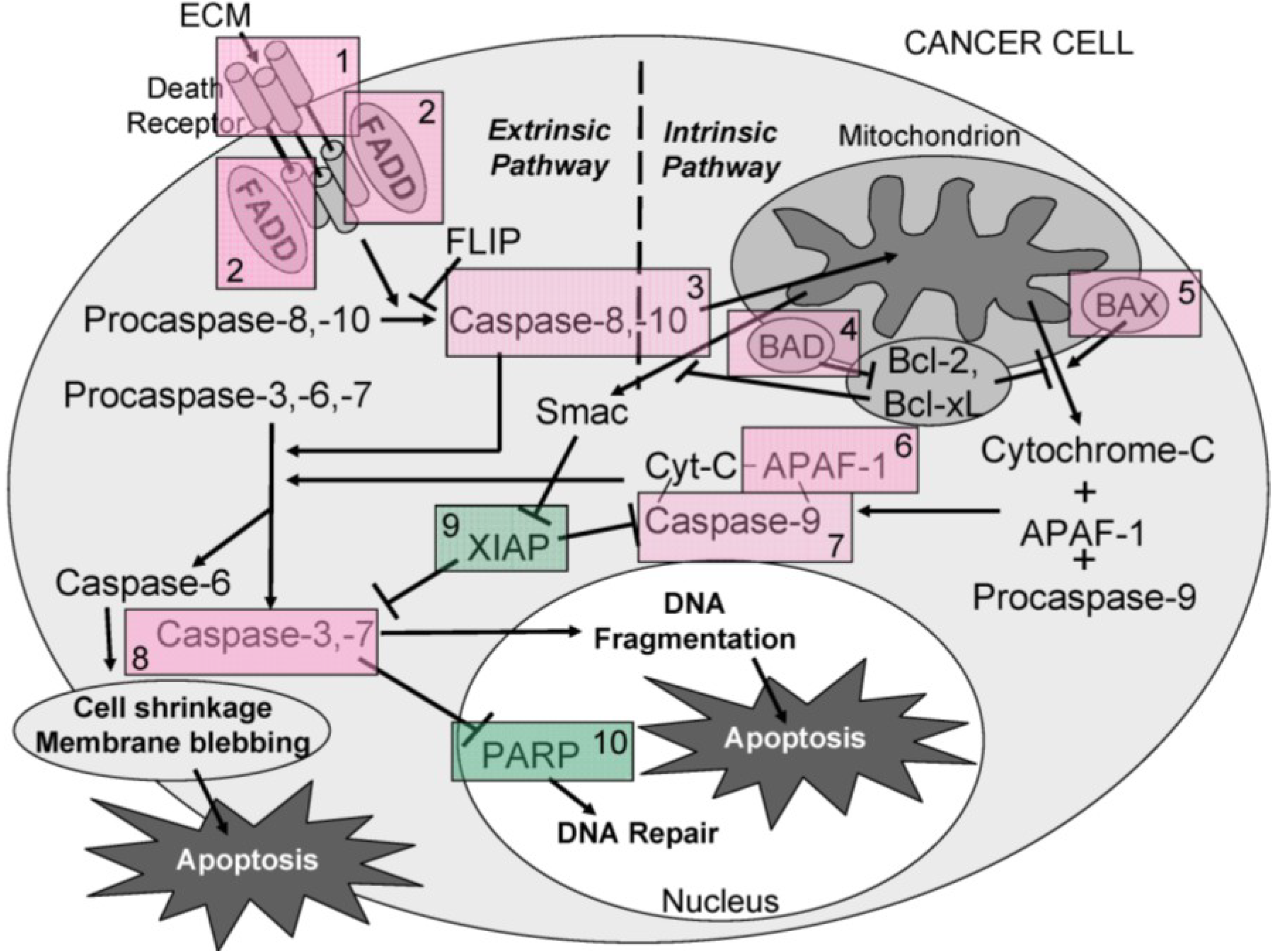
Overall, the results obtained indicate that ECM is cytotoxic (MTT) towards cultured Jurkat cells through triggering apoptosis (annexin V). The diagram in
Figure 16 can be used to illustrate proposed ECM-induced regulation of pathway intermediates resulting in apoptosis in ECM-treated Jurkat cells. Briefly, results presented here following ECM treatment are consistent with apoptotic activation pathways that would be observed with binding to death receptors (DR4 and DR5) on the cell surface (
Figure 16, Box 1). Based on the pattern of adaptor molecule (FADD; Box 2) and initiator caspase activation (caspase 8 and caspase 10; Box 3), activation of mitochondrial proteins BAD (Box 4) and BAX (Box 5), post-mitochondrial apoptosis pathway intermediates APAF-1 (Box 6) and caspase-9 (Box 7), and activation of the terminal caspase, caspase-3 (Box 9), these data are consistent with progression of apoptosis through the intrinsic (mitochondrial) pathway. Down-regulation of an important inhibitor of apoptosis (XIAP, Box 8) and cleavage of the nuclear DNA repair enzyme, PARP (Box 10) also facilitate progression of apoptosis.
Figure 16.
A schematic diagram showing the proposed ECM-induced regulation of pathway intermediates resulting in apoptosis in ECM-treated Jurkat cells. In this proposed schematic, ECM binds to death receptors DR4 and DR5 on the cell surface (Box 1) which stimulates the adapter molecule FADD (Box 2) to activate initiator caspases -8 and -10 (Box 3), mitochondrial proteins BAD (Box 4) and BAX (Box 5), post-mitochondrial apoptosis pathway intermediate APAF-1 (Box 6) and initiator caspase-9 (Box 7), resulting in the activation of terminal effector caspase-3 (Box 8). These data are consistent with progression of apoptosis through the intrinsic (mitochondrial) pathway. Down-regulation of an apoptotic inhibitor XIAP (Box 9) and cleavage of the nuclear DNA repair enzyme PARP (Box 10) also facilitate progression of apoptosis.
Figure 16.
A schematic diagram showing the proposed ECM-induced regulation of pathway intermediates resulting in apoptosis in ECM-treated Jurkat cells. In this proposed schematic, ECM binds to death receptors DR4 and DR5 on the cell surface (Box 1) which stimulates the adapter molecule FADD (Box 2) to activate initiator caspases -8 and -10 (Box 3), mitochondrial proteins BAD (Box 4) and BAX (Box 5), post-mitochondrial apoptosis pathway intermediate APAF-1 (Box 6) and initiator caspase-9 (Box 7), resulting in the activation of terminal effector caspase-3 (Box 8). These data are consistent with progression of apoptosis through the intrinsic (mitochondrial) pathway. Down-regulation of an apoptotic inhibitor XIAP (Box 9) and cleavage of the nuclear DNA repair enzyme PARP (Box 10) also facilitate progression of apoptosis.


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}