1. Introduction
Sleeping sickness (human African trypanosomiasis), a human disease caused by the single-celled protozoans
Trypanosoma brucei gambiense (in Western and Central Africa) and
T. b. rhodesiense (in Eastern and Southern Africa), is a devastating tropical disease and in 2012 the reported cases were over 7000 [
1]. After transmission into humans by bites of
Glossina flies, trypanosomes multiply in several tissues, including blood and lymph and, in a second stage, the immune response against the metabolites released causes the neurological symptoms, including behavioural changes, coma, and ultimately, if untreated, death. Disturbance of the sleep cycle, which gives the disease its name, is a characteristic feature of the cerebral stage of the disease.
The dramatic figures about spread and consequences of human African trypanosomiasis should be, at least partially, ascribed to the scarcity of efficacious, cheap and safe treatments. Eflornithine (in combination with nifurtimox) and the trivalent arsenic derivative melarsoprol are practically the only therapeutic options to treat the cerebral stage and their efficacy is reduced by the increasingly observed cases of cross-resistance [
2]. Thus, there is an urgent need to find new, effective and, above all, affordable alternatives to the existing options for treatment of sleeping sickness.
In the course of our ongoing research investigation aimed at the discovery of marine secondary metabolites with potential activity against malaria and other tropical diseases [
3,
4,
5], we have recently reported the isolation of manadoperoxide B (
1) and its analogues manadoperoxides C–K from the sponge
Plakortis cfr.
lita de Laubenfels [
6], a species widely distributed in the Indo-West Pacific, collected along the coasts of the Bunaken Marine Park of Manado (North Sulawesi, Indonesia). Some of these endoperoxyketal polyketides revealed a potent
in vitro activity against
T. b. rhodesiense and, remarkably, manadoperoxide B (
1,
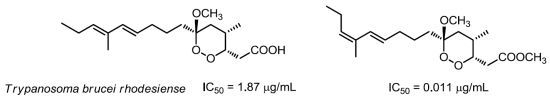
Figure 1) proved to be an ultrapotent trypanocidal agent with an IC
50 value of 3.0 ng/mL (8.8 nM), qualifying it as one of the most potent natural products, either marine or terrestrial, to possess such activity [
6].
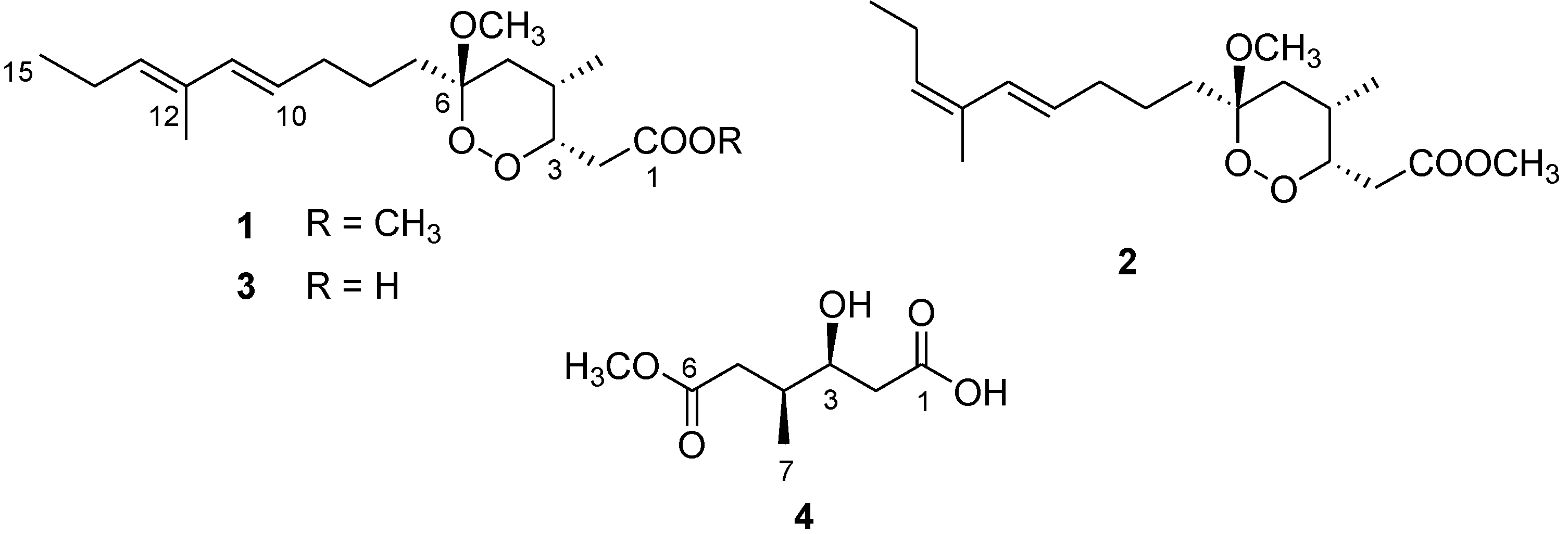
Figure 1.
Chemical structure of manadoperoxide B (1) and of the new metabolites 2–4.
Figure 1.
Chemical structure of manadoperoxide B (1) and of the new metabolites 2–4.
Structure–activity relationships within the series of isolated compounds disclosed the crucial role of substituents around the six-membered ring and, in particular of the methyl group attached at C-4 [
6]. Surprisingly, when this methyl was linked at C-2 in place of C-4 (as found in peroxyplakoric ester B3) the activity was almost completely lost. As for the “western” side chain, the indications were not unambiguous. Also the non-dienic derivatives, such as manadoperoxides I and K, retained a very good activity with IC
50 values 62 and 87 ng/mL, corresponding to 170 and 240 nM, respectively [
6].
With this information in our hands, we have undertaken the chemical analysis of another specimen of
Plakortis cfr.
lita de Laubenfels, collected in the same area of the previous one, in order to obtain larger amounts of manadoperoxide B (
1). During the fractionation of the organic extract of this sponge, we isolated two new analogues of 1, namely 12-isomanadoperoxide B (
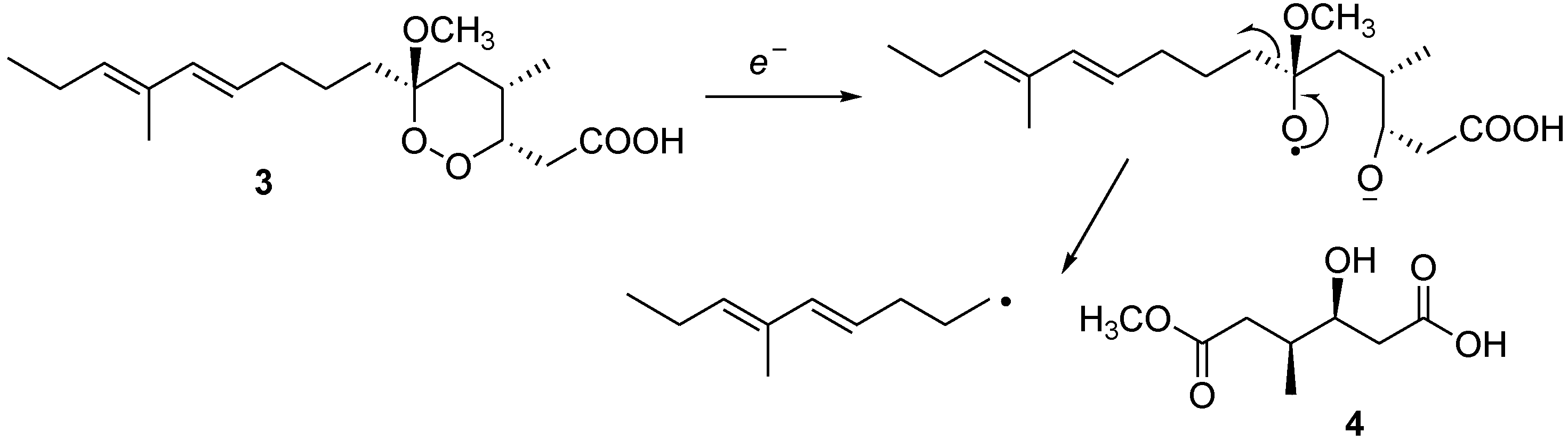
2) and manadoperoxidic acid B (
3), along with a new dicarboxylate monoester derivative 4, whose structural elucidation is herein described (
Figure 1). In this paper we also report on the preparation of three semisynthetic analogues of manadoperoxide B (
6–
8) and on the evaluation of the entire series of endoperoxyketal derivatives for
in vitro trypanocidal activity against
T. b. rhodesiense.
3. Experimental Section
3.1. General Experimental Procedures
Low and high resolution ESI-MS spectra were performed on a LTQ OrbitrapXL (Thermo Scientific) mass spectrometer. 1H (700 MHz) and 13C (175 MHz) NMR spectra were measured on Varian INOVA spectrometers. Chemical shifts were referenced to the residual solvent signal (CDCl3: δH 7.26, δC 77.0; CD3OD: δH 3.34). Homonuclear 1H connectivities were determined by the COSY experiment. Through-space 1H connectivities were evidenced using a ROESY experiment with a mixing time of 500 ms. One-bond heteronuclear 1H-13C connectivities was determined by the HSQC experiment; two- and three-bond 1H-13C connectivities by gradient-HMBC experiments optimized for a 2,3J of 8 Hz. Medium pressure liquid chromatography was performed on a Büchi apparatus using a reverse-phase (230–400 mesh) column. HPLC were achieved on a Knauer apparatus equipped with a refractive index detector and LUNA (Phenomenex) SI60 or Kinetex (2.6 µ, 100 × 4.60 mm Phenomenex) C18 columns.
3.3. 12-Isomanadoperoxide B (2)
Colorless amorphous solid; [α]25D −7.5 (c 0.1 in CHCl3); 1H NMR (CDCl3, 500 MHz) δH 6.41 (1H, d, J = 15.9 Hz, H-11), 5.60 (1H, dt, J = 15.9, 6.0 Hz, H-10), 5.26 (1H, t, J = 6.1 Hz, H-13), 4.43 (1H, m, H-3), 3.74 (3H, s, 1-OMe), 3.26 (3H, s, 6-OMe), 2.97 (1H, dd, J = 15.5, 9.5 Hz, H-2a), 2.57 (1H, m, H-4), 2.44 (1H, dd, J = 15.5, 4.5 Hz, H-2b), 2.14 (2H, overlapped, H-14), 2.10 (2H, overlapped, H-9), 1.79 (3H, s, 12-Me), 1.69 (1H, overlapped, H-5a), 1.66 (1H, overlapped, H-7a), 1.40 (2H, overlapped, H-8), 1.36 (1H, overlapped, H-7b), 1.28 (1H, m, H-5b), 0.98 (3H, t, J = 7.1 Hz, H-15), 0.84 (3H, d, J = 7.1 Hz, 4-Me); 13C NMR (CDCl3, 125 MHz) δC 172.5 (C, C-1), 138.5 (CH, C-11), 136.8 (CH, C-13), 133.9 (C, C-12), 126.4 (CH, C-10), 103.2 (C, C-6), 80.12 (CH, C-3), 52.0(CH3, 1-OMe), 48.8 (CH3, 6-OMe), 34.6 (CH2, C-5), 33.9 (CH2, C-9), 32.1 (CH2, C-7), 31.4 (CH2, C-2), 27.6 (CH, C-4), 23.8 (CH2, C-8), 22.7 (CH2, C-14), 17.0 (CH3, C-4-Me), 15.0 (CH3, C-12-Me), 13.4 (CH3, C-15); (+) ESI-MS m/z 341 [M + H]+, 363 [M + Na]+. HR-ESIMS m/z 341.2325 (calcd for C19H33O5 341.2328).
3.4. Manadoperoxidic Acid B (3)
Colorless solid; [α]25D −5.0 (c 0.2 in CHCl3); 1H NMR (CDCl3, 500 MHz) δH 6.03 (1H, d, J = 15.9 Hz, H-11), 5.47 (1H, dt, J = 15.9, 6.0 Hz, H-10), 5.40 (1H, t, J = 6.1 Hz, H-13), 4.46 (1H, m, H-3), 3.26 (3H, s, 6-OMe), 2.95 (1H, dd, J = 15.5, 9.5 Hz, H-2a), 2.57 (1H, m, H-4), 2.47 (1H, dd, J = 15.5, 4.5 Hz, H-2b), 2.09 (2H, overlapped, H-14), 2.07 (2H, overlapped, H-9), 1.70 (3H, s, 12-Me), 1.69 (1H, overlapped, H-5a), 1.66 (1H, overlapped, H-7a), 1.40 (2H, overlapped, H-8), 1.36 (1H, overlapped, H-7b), 1.28 (1H, m, H-5b), 1.00 (3H, t, J = 7.1 Hz, H-15), 0.84 (3H, d, J = 7.1 Hz, 4-Me); 13C NMR (CDCl3, 125 MHz) δC 177.3 (C, C-1), 136.0 (CH, C-11), 133.8 (CH, C-13), 133.3 (C, C-13), 125.8 (CH, C-10), 103.4 (C, C-6), 80.2 (CH, C-3), 52.2 (CH3, 1-OMe), 48.8 (CH3, 6-OMe), 34.6 (CH2, C-5), 33.6 (CH2, C-9), 32.1 (CH2, C-7), 31.4 (CH2, C-2), 27.6 (CH, C-4), 23.5 (CH2, C-8), 21.9 (CH2, C-14), 17.0 (CH3, C-4-Me), 15.0 (CH3, C-12-Me), 13.0 (CH3, C-15); (−) ESI-MS m/z 325 [M − H]−. HR-ESIMS m/z 325.2013 (calcd for C18H29O5 325.2015).
3.5. Diazomethane Reaction of Manadoperoxidic Acid B (3)
Manadoperoxidic acid B (3, 1.0 mg) was dissolved in ethyl ether and the resulting solution was added dropwise to an ethereal solution of CH2N2 (ca. 15 equiv) at 0 °C. The mixture was stirred for 10 min and then concentrated under reduce pressure to give semisynthetic manadoperoxide B (1) identified by means of NMR and [α]25D.
3.6. Compound 4
Colorless solid; [α]25D −23.0 (c 0.1 in CHCl3); 1H NMR (CDCl3, 500 MHz) δH 4.04, (1H, m, H-3), 3.66 (3H, s, 6-OMe), 2.97 (1H, bs, 3-OH), 2.52 (1H, dd, J = 12.2, 6.0 Hz, H-5a), 2.48 (1H, overlapped, H-2a), 2.43 (1H, overlapped, H-2b), 2.23 (1H, dd, J = 12.2, 4.5 Hz, H-5b), 2.11 (1H, m, H-4), 0.95 (3H, d, J = 7.1 Hz, 4-Me); (13C NMR (CDCl3, 125 MHz) δC 177.8 (C, C-1), 173.6 (C, C-6), 70.2 (CH, C-3), 52.7 (CH3, 6-OMe), 37.7 (CH2, C-5) 35.0 (CH, C-4), 31.8 (CH2, C-2), 17.4 (CH3, C-4-Me); (−) ESI-MS m/z 189 [M − H]−. HR-ESIMS m/z 189.0770 (calcd for C8H13O5 189.0763).
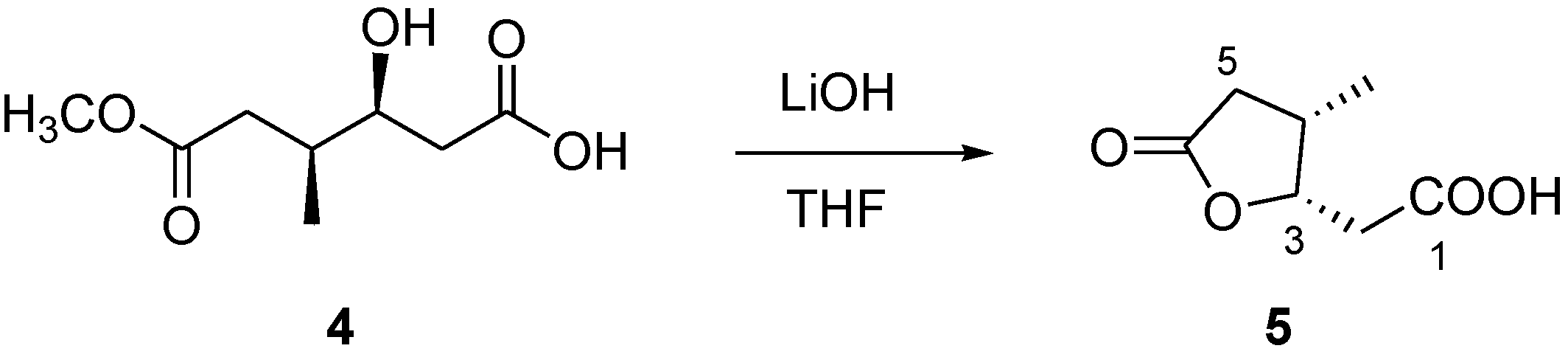
3.7. Conversion of Compound 4 into Lactone 5
Compound 4 (1.1 mg) was dissolved in a THF/H2O 3:1 solution (2.0 mL) and 2 mg of LiOH were added. The solution was stirred at 0 °C overnight. Then, the reaction mixture was partitioned between EtOAc and water. The organic phase, evaporated to dryness, contained compound pure compound 5 (0.6 mg).
3.8. Compound 5
Colorless solid; [α]25D −11.0 (c 0.1 in CHCl3); 1H NMR (CD3OD, 500 MHz) δH 4.97 (1H, m, H-3), 2.80 (1H, dd, J = 16.7, 7.5 Hz, H-5a), 2.77 (1H, m, H-4), 2.59 (1H, dd, J = 12.2, 4.5 Hz, H-2a), 2.43 (1H, dd, J = 12.2, 6.0 Hz, H-2b), 2.20 (1H, dd, J = 16.7, 3.3 Hz, H-5b), 1.07 (3H, d, J = 7.1 Hz, 4-Me);13C NMR (CDCl3, 125 MHz) δC 176.6 (C, C-1), 176.0 (C, C-6), 82.2 (CH, C-3), 37.1 (CH, C-4), 36.8 (CH2, C-2), 32.9 (CH2, C-5), 14.0 (CH3, C-4-Me); (−) ESI-MS m/z 157 [M − H]−. HR-ESIMS m/z 157.0507 (calcd for C7H9O4 157.0501).
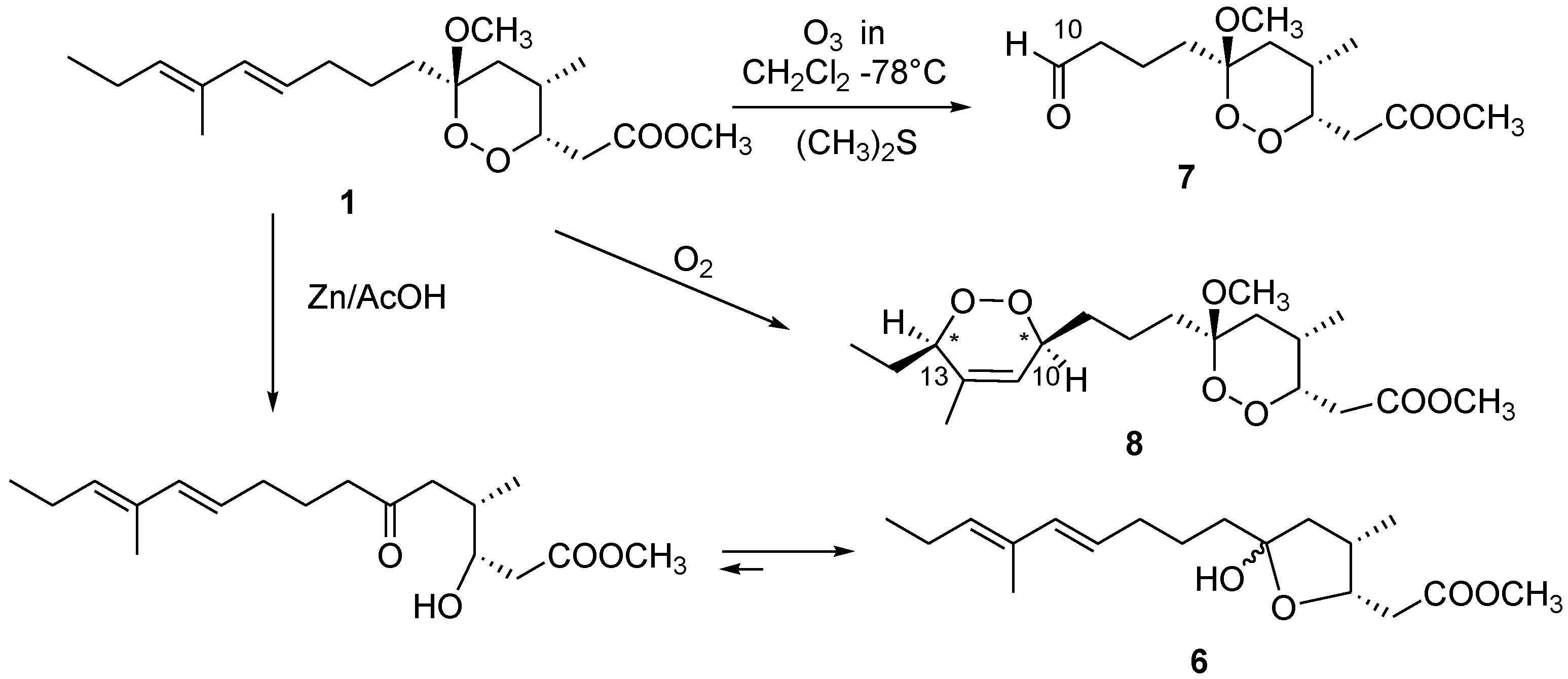
3.9. Reductive Cleavage of Manadoperoxide B
Semi-synthetic procedures and spectral data of
6 are reported in ref. [
7].
3.10. Reductive Ozonolysis of Manadoperoxide B
A stream of O3 was bubbled into a solution of manadoperoxide B (1, 4.1 mg, 0.012 mm) in CH2Cl2 (2 mL) kept at −78 °C until a blue-colored solution resulted. After stirring for 1 min, excess of O3 was removed upon bubbling N2 and dry Me2S (1 mL) was added to the colorless solution. The reaction mixture was left at room temperature overnight, then concentrated in vacuo, purified by reversed-phase HPLC (eluent MeOH/H2O 55:45) to yield compound 7 (1.1 mg) in the pure state.
3.11. Aldehyde 7
Colorless amorphous solid. [α]25D −3.5 (c 0.1 in CHCl3); 1H NMR (CDCl3): δH 9.78 (1H, bs, H-10), 4.43 (1H, ddd, J = 9.5, 4.3, 3.0 Hz, H-3), 3.72 (3H, s, 1-OMe), 3.26 (3H, s, 6-OMe), 2.97 (1H, dd, J = 15.5, 9.5 Hz, H-2a), 2.57 (1H, m, H-4), 2.44 (1H, overlapped, H-2b), 2.41 (2H, overlapped, H2-9), 1.69 (1H, overlapped, H-5a), 1.71 (1H, overlapped, H-7a), 1.67 (2H, overlapped, H2-8), 1.62 (1H, overlapped, H-5b), 1.34 (1H, m, H-7b), 0.86 (3H, d, J = 7.1 Hz, 4-Me); ESIMS: m/z 297 [M + Na]+, HRESIMS: m/z 297.1307, calcd. for C13H22O6Na m/z 297.1314.
3.12. Photo-Oxygenation Reaction
A solution of manadoperoxide B (1, 9.0 mg) in CHCl3/MeOH 95:5 (2 mL) was photolysed with a 500 W halogen lamp in the presence of methylene blue (0.01 mg) as photosensitizer, through which was bubbled a constant stream of oxygen at a flow rate of 50 mL/min for 24 h. The reaction was performed in a Pyrex flask fitted with an external cooling jacket. The reaction mixture was then concentrated in vacuo and the resulting residue was purified by HPLC chromatography (n-hexane/EtOAc mixtures 85:15) to yield pure compound 8 (3.0 mg).
3.13. Compound 8
Colorless oil. [α]25D −2.5 (c 0.1 in CHCl3); 1H NMR (CDCl3): δ 5.65 (1H, bs, H-11), 4.45 (1H, m, H-3), 4.30 (1H, overlapped, H-13), 4.29 (1H, overlapped, H-10), 3.72 (3H, s, 1-OCH3), 3.27 (3H, s, 6-OCH3); 2.92 (1H, dd, J =15.5, 9.4 Hz, H-2a), 2.56 (1H, m, H-4), 2.44 (1H, dd, J =15.5, 3.6 Hz, H-2b), 1.75 (1H, overlapped, H-9a), 1.72 (3H, s, 12-Me), 1.70 (1H, overlapped, H-5a), 1.69 (1H, overlapped, H-7a), 1.58 (2H, overlapped, H2-14), 1.55 (1H, overlapped, H-5b); 1.32 (1H, overlapped, H-7b), 1.25 (2H, overlapped, H2-8), 1.00 (3H, t, J =7 Hz, H3-15), 0.85 (3H, d, J =7 Hz, 4-Me). ESI-MS: m/z 395 [M + Na]+, HR-ESIMS: m/z 395.2051, calcd. for C19H32O7Na m/z 395.2046.
3.14. Activity against Trypanosoma brucei rhodesiense
Minimum Essential Medium (50 µL) supplemented with 25 mM HEPES, 1 g/L additional glucose, 1% MEM non-essential amino acids (100×), 0.2 mM 2-mercaptoethanol, 1mM Na-pyruvate and 15% heat inactivated horse serum was added to each well of a 96-well microtiter plate. Serial drug dilutions of eleven 3-fold dilution steps covering a range from 100 to 0.002 μg/mL were prepared. Then 4 × 10
4 bloodstream forms of STIB 900 strain (the stock was isolated in 1982 from a human patient in Tanzania and after several mouse passages was cloned and adapted to axenic culture conditions) [
11] of
T. b. rhodesiense in 50 μL was added to each well and the plate incubated at 37 °C under a 5% CO
2 atmosphere for 72 h. 10 µL of a resazurin solution (prepared dissolving 12.5 mg resazurin in 100 mL double distilled water) [
12] was then added to each well and incubation continued for a further 2–4 h. Then the plates were read in a Spectramax Gemini XS microplate fluorometer (Molecular Devices Cooperation, Sunnyvale, CA, USA) using an excitation wavelength of 536 nm and an emission wavelength of 588 nm. The IC
50 values were calculated by linear regression [
13] from the sigmoidal dose inhibition curves using SoftmaxPro software (Molecular Devices Cooperation, Sunnyvale, CA, USA). Melarsoprol was the standard drug.
3.15. Cytotoxicity against L6-Cells
Assays were performed in 96-well microtiter plates, each well containing 100 μL of RPMI 1640 medium supplemented with 1% L-glutamine (200 mM) and 10% fetal bovine serum, and 4 × 10
4 L-6 cells. Serial drug dilutions of eleven 3-fold dilution steps covering a range from 100 to 0.002 μg/mL were prepared. After 72 h of incubation the plates were inspected under an inverted microscope to assure growth of the controls and sterile conditions. 10 μL of a resazurin solution (prepared dissolving 12.5 mg resazurin in 100 mL double distilled water) was then added to each well and the plates incubated for another 2 h. The plates were read with a Spectramax Gemini XS microplate fluorometer using an excitation wavelength of 536 nm and an emission wavelength of 588 nm. The IC
50 values were calculated by linear regression [
13] from the sigmoidal dose inhibition curves using SoftmaxPro software (Molecular Devices Cooperation, Sunnyvale, CA, USA). The reported IC
50 values are the means of at least two separate experiments. Podophyllotoxin was used as control drug.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}