Abstract
Four new jacaranone analogs, marinoids F–I (1–4), were isolated from the fruits of a Beibu Gulf mangrove Avicennia marina. The structures were elucidated based on analysis of spectroscopic data. Marinoids F and G are shown to be diastereoisomers of chlorocornoside, a new halogen containing marine secondary metabolite. The antioxidant activity of the isolates was evaluated using a cellular antioxidant assay, and 4 showed good antioxidant activity (EC50 = 26 μM).
1. Introduction
Avicennia marina (Forsk.) Vierh. is commonly known as the grey or white mangrove plant resident in the tropical and subtropical regions, it is extremely widespread along the coasts of eastern Africa, islands of the Indian Ocean, tropical Asia, Australia, New Zealand, and islands of the Pacific Ocean to Fiji [1]. The crude extracts are reported to possess antimalarial and cytotoxic activities [1]. Different parts of the plant are used in Egypt as a folk medicine cure for skin diseases [2]. Previous chemical investigation of plants of the genus Avicennina have exhibited the presence of iridoid glucosides, marinoids A–E [1,2,3,4,5], naphthoquinone derivatives [6,7], flavonoids [4,8], and diterpenoids [9]. However, these previous studies did not report any chemical and biological data from the fruits of A. marina. Searching for bioactive secondary metabolites from this specimen afforded four new jacaranone analogs, marinoids F–I (1–4 respectively) (Figure 1). In this paper, we describe the isolation, structural elucidation, and antioxidant activity of the four new secondary metabolites.
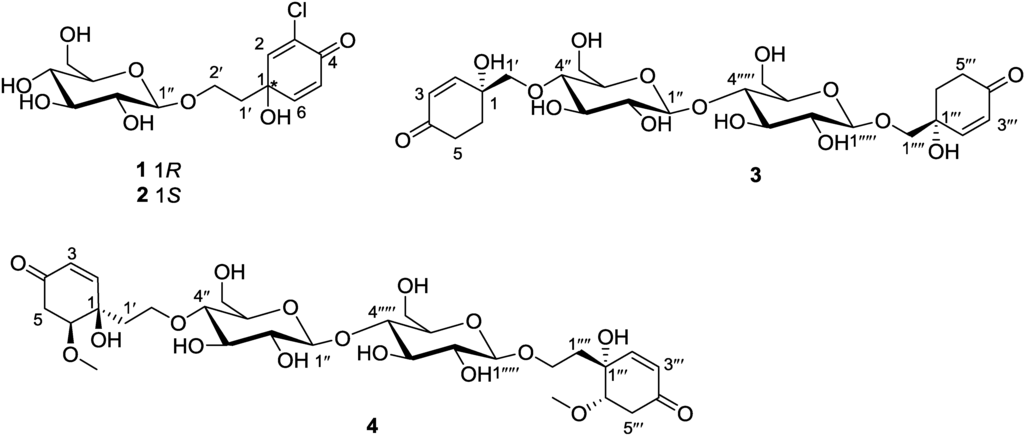
Figure 1.
Structures of marinoids F–I (1–4).
Figure 1.
Structures of marinoids F–I (1–4).
2. Results and Discussion
Marinoid F (1) was purified as a yellow oil with the molecular formula C14H19ClO8 as determined by HRESIMS (found [M + H]+ at m/z 351.0835, calcd [M + H]+, 351.0841) as well as 1H and 13C spectroscopic data (Table 1). 1H NMR spectra disclosed the presence of two methylene groups [(δH 3.94, ddd, J = 10.1, 7.0, 2.2 Hz, H-2′α) and 3.64, dd, J = 10.1, 2.2 Hz, H-2′β) and (δH 2.08, d, J = 7.0 Hz, H-1′)], an α,β-unsaturated carbonyl group [(UV λmax 220 nm; δH 7.16 (d, J = 2.8 Hz, H-2); 6.99 (dd, J = 10.0, 2.8 Hz, H-6) and 6.22 (d, J = 10.0 Hz, H-5)]. The characteristic chemical shift of the carbonyl resonance (δC 179.3, C-4), in addition to the presence of four olefinic groups [δC 152.8 (C-6), 148.9 (C-2), 130.5 (C-3) and 125.8 (C-5)], and a quaternary sp3 carbon (δC 70.2, C-1) (Table 1), demonstrated that 1 has a para-quinol-type partial structure [10,11]. NMR spectra also indicated the presence of a β-glucosyl group, i.e., one anomeric carbon resonance at δC 102.6 (C-1ʺ) and one anomeric proton at δH 4.18 (1H, d, J = 9.2 Hz, H-1ʺ). It was used as a starting point in the homonuclear correlated spectra to determine all glycosidic protons. The JH-1ʺ-H-2ʺ value (9.2 Hz) of compound 1, further confirmed that the sugar was a β-glucosyl group [12].

Table 1.
1H and 13C NMR data of marinoids F (1) and G (2) a.
| 1 | 2 | |||
|---|---|---|---|---|
| δC, Mult | δH (J in Hz) | δC, Mult | δH (J in Hz) | |
| 1 | 70.2, C | 70.2, C | ||
| 2 | 148.9, CH | 7.16 (d, 2.8) | 148.6, CH | 7.20 (d, 2.8) |
| 3 | 130.5, C | 130.0, C | ||
| 4 | 179.3, C | 179.1, C | ||
| 5 | 125.8, CH | 6.22 (d, 10.0) | 125.4, CH | 6.19 (d, 10.0) |
| 6 | 152.8, CH | 6.99 (dd, 10.0, 2.8) | 153.1, CH | 7.03 (dd, 10.0, 2.8) |
| 1′ | 39.6, CH2 | 2.08 (dd, 11.2, 7.0) | 39.7, CH2 | 2.07 (dd, 11.2, 7.0) |
| 2′α | 63.9, CH2 | 3.94 (ddd, 10.1, 7.0, 2.2) | 64.0, CH2 | 4.00 (dt, 10.4, 7.0, 2.4) |
| β | 3.64 (dd, 10.1, 2.2) | 3.65 (dd, 10.4, 2.4) | ||
| 1ʺ | 102.6, CH | 4.18 (d, 9.2) | 102.8, CH | 4.21 (d, 7.8) |
| 2ʺ | 73.6, CH | 3.13 (9.6, 9.2) | 73.6, CH | 3.14 (dd, 9.6, 7.8) |
| 3ʺ | 76.6, CH | 3.20 (m) | 76.6, CH | 3.12 (m) |
| 4ʺ | 70.1, CH | 3.24 (m) | 69.9, CH | 3.24 (m) |
| 5ʺ | 76.5, CH | 3.29 (m) | 76.6, CH | 3.31 (m) |
| 6ʺα | 61.3, CH2 | 3.81 (dd, 11.9, 2.0) | 61.4, CH2 | 3.84 (d, 11.8) |
| β | 3.62 (m) | 3.65 (m) | ||
a In CD3OD, 600 MHz for 1H and 150 MHz for 13C NMR.
The gross structure was further established by the aid of COSY and HMBC experiments (Figure 2). A careful comparison of 1 and cornoside revealed that 1 differs from cornoside by the presence of one chlorine atom attached at C-3 [13]. Compound 1 showed 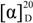 (MeOH) of −14.7°. The reported rotation value for cornoside is negative −10.5° [13], whose stereochemistry of the aglycone and the β-d-glucosyl residue have been established by enzymatic hydrolysis and other methods [13,14,15]. The reported rotation value after poly-acetylation of cornoside is also negative ( −10.2°) [16]. Moreover, the NMR data of the β-glucosyl residue in compound 1 are in accord with those observed in cornoside [13]. The substitution is simply that of one Cl atom for one H atom, and that many bonds away from the key chiral centre, therefore following those Literature data, we propose that the configuration of C-1 in compound 1 is the same as that found at C-1 in cornoside, namely R. Thus, the structure of 1 is predicted to be as shown in Figure 1.
(MeOH) of −14.7°. The reported rotation value for cornoside is negative −10.5° [13], whose stereochemistry of the aglycone and the β-d-glucosyl residue have been established by enzymatic hydrolysis and other methods [13,14,15]. The reported rotation value after poly-acetylation of cornoside is also negative ( −10.2°) [16]. Moreover, the NMR data of the β-glucosyl residue in compound 1 are in accord with those observed in cornoside [13]. The substitution is simply that of one Cl atom for one H atom, and that many bonds away from the key chiral centre, therefore following those Literature data, we propose that the configuration of C-1 in compound 1 is the same as that found at C-1 in cornoside, namely R. Thus, the structure of 1 is predicted to be as shown in Figure 1.
(MeOH) of −14.7°. The reported rotation value for cornoside is negative −10.5° [13], whose stereochemistry of the aglycone and the β-d-glucosyl residue have been established by enzymatic hydrolysis and other methods [13,14,15]. The reported rotation value after poly-acetylation of cornoside is also negative ( −10.2°) [16]. Moreover, the NMR data of the β-glucosyl residue in compound 1 are in accord with those observed in cornoside [13]. The substitution is simply that of one Cl atom for one H atom, and that many bonds away from the key chiral centre, therefore following those Literature data, we propose that the configuration of C-1 in compound 1 is the same as that found at C-1 in cornoside, namely R. Thus, the structure of 1 is predicted to be as shown in Figure 1.Marinoid G (2) was obtained as yellow oil. Its molecular formula was determined as C14H19ClO8 by HRESIMS (found [M + H]+ at m/z 351.0837, calcd [M + H]+, 351.0841) as well as 1H and 13C data (Table 1). The NMR spectra of 2 are very similar to those of 1. In addition, analysis of the COSY, HMBC and NOSEY correlations of 2 revealed identical spin systems and connections with those found in 1. Compound 2 showed (MeOH) of +10.2°, and the reported value for cornoside ( −10.5°) and poly-acetylated cornoside ( −10.2°) are both negative [13], and the observed value for 1 is −14.7°. Although we do not have an explanation for the differences in the absolute values of the optical rotations of cornoside, poly-acetylated of cornoside, and compound 1, and the NMR data of the β-glucosyl residue in compound 2 are greatly similar to those of the β-glucosyl residue in cornoside and compound 1, and also compound 2 showed a positive Cotton effect at 220 nm (Δε +5.97), whereas the observed value for 1 was −4.28, the opposite optical rotation and Cotton effect indicate that 1 and 2 are diastereoisomers. Indeed, we propose that they are enantiomers of the aglycone each with β-glucosyl residues.
(MeOH) of +10.2°, and the reported value for cornoside ( −10.5°) and poly-acetylated cornoside ( −10.2°) are both negative [13], and the observed value for 1 is −14.7°. Although we do not have an explanation for the differences in the absolute values of the optical rotations of cornoside, poly-acetylated of cornoside, and compound 1, and the NMR data of the β-glucosyl residue in compound 2 are greatly similar to those of the β-glucosyl residue in cornoside and compound 1, and also compound 2 showed a positive Cotton effect at 220 nm (Δε +5.97), whereas the observed value for 1 was −4.28, the opposite optical rotation and Cotton effect indicate that 1 and 2 are diastereoisomers. Indeed, we propose that they are enantiomers of the aglycone each with β-glucosyl residues.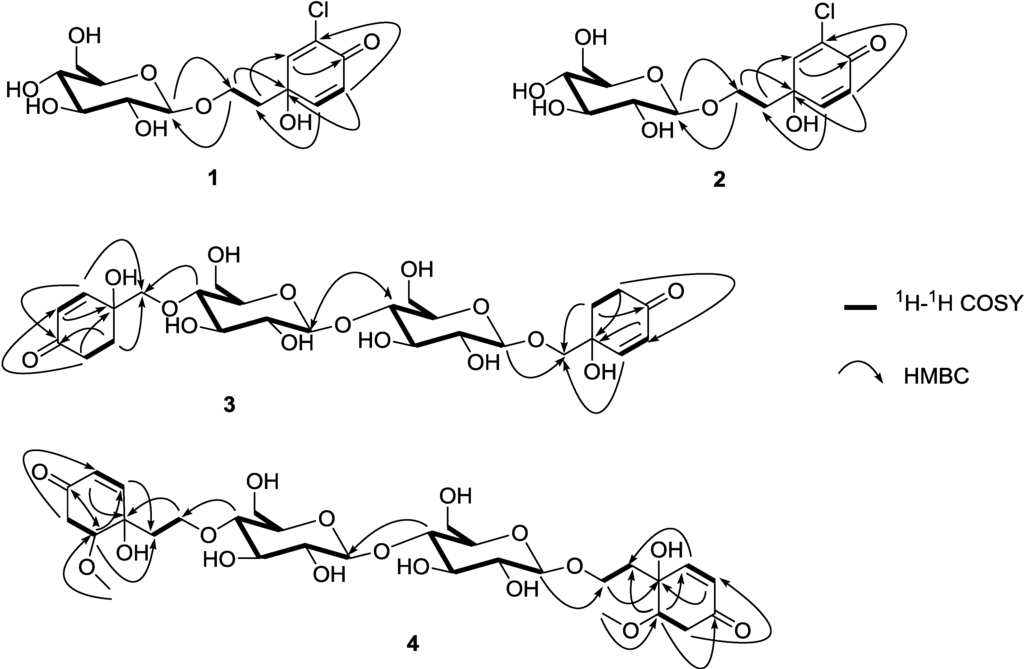
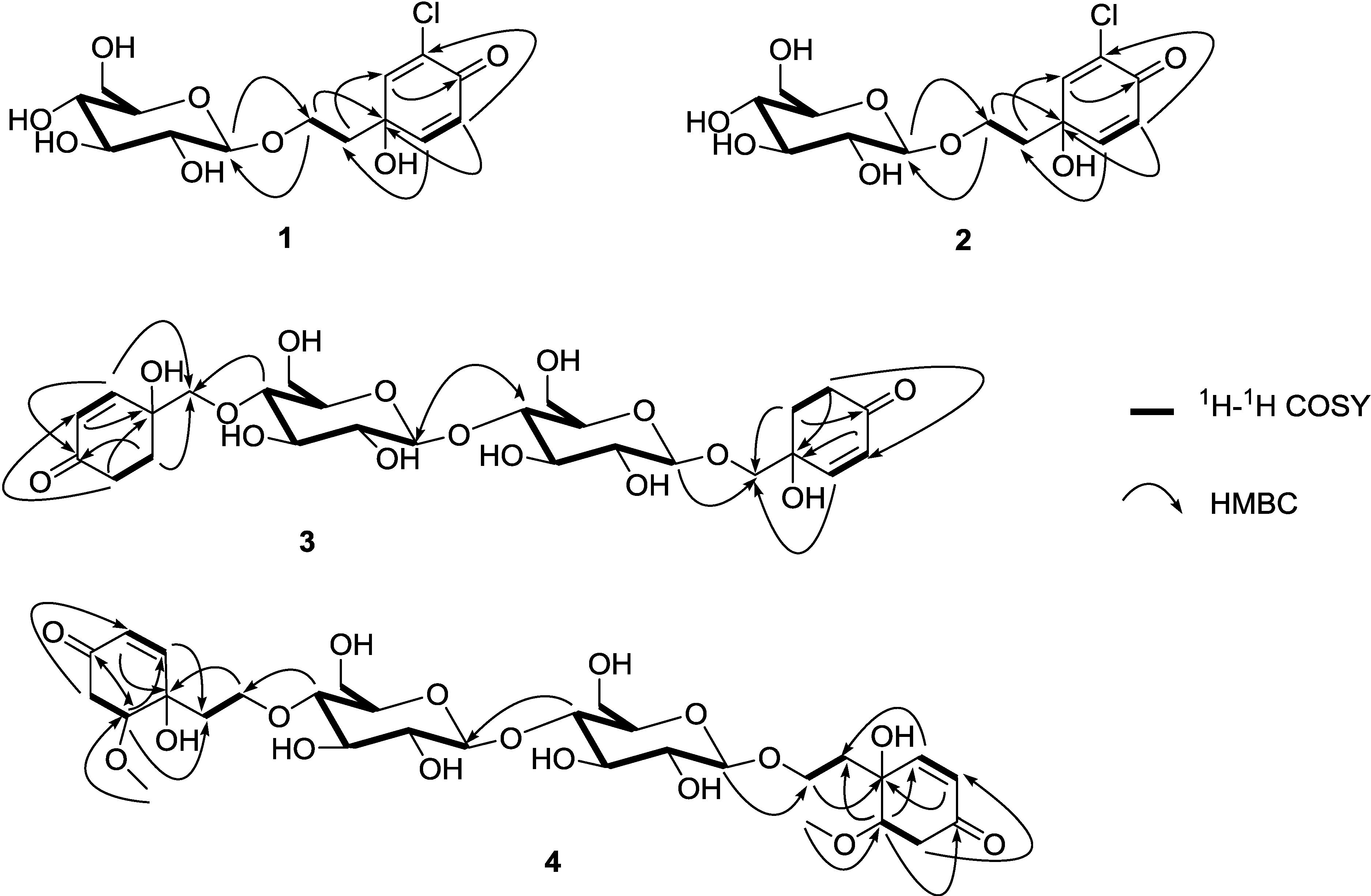
Figure 2.
Selected 1H-1H COSY and HMBC correlations of marinoids F–I (1–4).
Figure 2.
Selected 1H-1H COSY and HMBC correlations of marinoids F–I (1–4).
Marinoid H (3) was obtained as a colourless oil and its molecular formula was established as C26H38O15 by HRESIMS (found [M + H]+ at m/z 591.2281, calcd [M + H]+, 591.2283) as well as 1H and 13C data (Table 2). In the NMR spectra of compound 3, the proton signals at [δH 6.93 (1H, d, J = 15.1 Hz, H-2), 5.93 (1H, dd, J = 15.1, 2.6 Hz, H-3), 2.68 (1H, m, H-5α), 2.49 (1H, m, H-5β), 2.09 (1H, m, H-6α), and 2.03 (1H, m, H-6β)], and [δH 6.90 (1H, d, J = 15.1 Hz, H-2‴), 5.92 (1H, dd, J = 15.1, 2.6 Hz, H-3‴), 2.67 (2H, m, H-5‴), 2.08 (1H, m, H-6‴α), and 2.01 (1H, m, H-6‴β)] and the carbon signals at [δC 199.1 (C-4), 152.8 (C-2), 128.1 (C-3), 70.9 (C-1), 42.1 (C-5), and 37.3 (C-6)] and [δC 199.0 (C-4‴), 152.5 (C-2‴), 127.8 (C-3‴), 70.7 (C-1‴), 42.0 (C-5‴), and 37.2 (C-6‴)] indicated the existence of two cyclohexanone moieties [17], which were further confirmed by HMBC connections. NMR spectra also indicated the presence of two β-glucosyl groups by two anomeric carbon resonances at δC 102.9 (C-1ʺ) and 102.8 (C-1ʺ‴), and two anomeric protons at δH 4.27 (1H, d, J = 7.8 Hz, H-1ʺ) and 4.25 (1H, d, J = 7.8 Hz, H-1ʺ‴), which were further confirmed by the JH-1ʺ-H-2ʺ and JH-1ʺ‴-H-2ʺ‴ values [18]. The tentative molecular weight of the compound deduced from NMR analysis suggested compound 3 to be an unsymmetrical dimer.
Table 2.
1H and 13C NMR data of marinoids H (3) and I (4) a.
| 3 | 4 | |||
|---|---|---|---|---|
| δC, Mult | δH (J in Hz) | δC, Mult | δH (J in Hz) | |
| 1 | 70.9, C | 72.4, C | ||
| 2 | 152.8, CH | 6.93 (d, 15.1) | 154.7, CH | 6.95 (d, 10.0) |
| 3 | 128.1, CH | 5.93 (dd, 15.1, 2.6) | 127.1, CH | 5.87 (d, 10.0) |
| 4 | 199.1, C | 198.6, C | ||
| 5α | 42.1, CH2 | 2.68 (m) | 38.9, CH2 | 2.82 (dd, 11.1, 3.6) |
| β | 2.49 (m) | 2.49 (dd, 11.1, 7.0) | ||
| 6α | 37.3, CH2 | 2.09 (m) | 82.8, CH | 3.67 (dd, 7.0, 3.6) |
| β | 2.03 (m) | |||
| 1ʹα | 61.3, CH2 | 3.83 (d, 10.1) | 34.4, CH2 | 2.18 (dd, 11.1, 5.3) |
| β | 3.85 (d, 10.1) | 2.00 (dd, 11.1, 6.1) | ||
| 2ʹα | 61.3, CH2 | 3.86 (d, 10.1) | ||
| β | 3.79 (d, 10.1) | |||
| 1ʺ | 102.9, CH | 4.27 (d, 7.8) | 103.0, CH | 4.28 (d, 7.5) |
| 2ʺ | 73.6, CH | 3.13 (dd, 9.4, 7.8) | 73.6, CH | 3.14 (dd, 9.4, 7.8) |
| 3ʺ | 70.9, CH | 4.08 (m) | 70.2, CH | 3.26 (m) |
| 4ʺ | 76.6, CH | 3.32 (m) | 76.7, CH | 3.33 (m) |
| 5ʺ | 76.6, CH | 3.25 (m) | 76.7, CH | 3.26 (m) |
| 6ʺα | 64.9, CH2 | 4.13 (dt, 9.5, 5.0) | 65.1, CH2 | 4.16 (dt, 9.5, 5.0) |
| β | 4.09 (dt, 9.5, 5.0) | 4.14 (dt, 9.5, 5.0) | ||
| 1‴ | 70.7, C | 72.4, C | ||
| 2‴ | 152.5, CH | 6.90 (d, 15.1) | 154.0, CH | 6.93 (d, 10.0) |
| 3‴ | 127.8, CH | 5.92 (dd, 15.1, 2.6) | 127.0, CH | 5.85 (d, 10.0) |
| 4‴ | 199.0, C | 198.6, C | ||
| 5‴α | 42.0, CH2 | 2.67 (m) | 38.9, CH2 | 2.82 (dd, 11.1, 3.6) |
| β | 2.49 (dd, 11.1, 7.0) | |||
| 6‴α | 37.2, CH2 | 2.08 (m)2.01 (m) | 82.7, CH | 3.64 (dd, 7.0, 3.6) |
| β | 2.01 (m) | |||
| 1ʺʺα | 61.3, CH2 | 3.63 (d, 10.1) | 34.3, CH2 | 2.17 (dd, 11.1, 5.3) |
| β | 3.65(d, 10.1) | 1.98 (dd, 11.1, 6.1) | ||
| 2ʺʺα | 61.3, CH2 | 3.84 (d, 10.1) | ||
| β | 3.64(d, 10.1) | |||
| 1ʺ‴ | 102.8, CH | 4.25 (d, 7.8) | 102.8, CH | 4.27 (d, 7.5) |
| 2ʺ‴ | 73.6, CH | 3.13 (dd, 9.4, 7.8) | 73.6, CH | 3.14 (dd, 9.4, 7.8) |
| 3ʺ‴ | 70.2, CH | 3.25 (m) | 70.2, CH | 3.24 (m) |
| 4ʺ‴ | 76.6, CH | 3.31 (m) | 76.6, CH | 3.31 (m) |
| 5ʺ‴ | 76.6, CH | 3.31 (m) | 76.6, CH | 3.24 (m) |
| 6ʺ‴α | 64.9, CH2 | 3.78 (dt, 9.8, 5.5) | 64.8, CH2 | 3.82 (dt, 9.8, 5.5) |
| β | 3.78 (dt, 9.8, 5.5) | 3.80 (dt, 9.8, 5.5) | ||
| OCH3 | 56.9, CH3 | 3.43 (s) | ||
| OCH3 | 56.9, CH3 | 3.43 (s) | ||
a In CD3OD, 600 MHz for 1H and 150 MHz for 13C NMR.
The gross structure was further established by the aid of COSY and HMBC experiments (Figure 2). Six spin systems could be revealed by analysis of COSY correlations corresponding to the H-2/H-3, H2-5/H2-6, H-1ʺ/H-2ʺ/H-3ʺ/H-4ʺ/H-5ʺ/H2-6ʺ, H-2‴/H-3‴, H2-5‴/H2-6‴, and H-1ʺ‴/H-2ʺ‴/H-3ʺ‴/H-4ʺ‴/H-5ʺ‴/H2-6ʺ‴. The connectivity of the two cyclohexanone moieties to C-1′ and C-1ʺʺ were established by the HMBC correlations of H-2 to C-1ʹ and H-6 to C-1ʹ, and of H-2‴ to C-1ʺʺ and H-6‴ to C-1ʺʺ, respectively. The presence of the two β-glucosyl groups in 3 could be proposed on the basis of HMBC correlations from H-4ʺ to C-1′ and H-1ʺ‴ to C-1ʺʺ, respectively. The connection of the two β-glucosyl groups was confirmed by the presence of HMBC correlation from H-4ʺ‴ to C-1ʺ. The configuration of C-1 and C-1‴ were assigned as R because a negative was observed, which was in accord with that observed in cornoside ( −10.5°), and analysis of the 13C NMR data of C-1 and C-1‴ in 3 indicated that they were greatly similar to that of C-1 (δC 68.9) in 4-[2-(β-d-glucopyranosyloxy)ethyl]-4-hydroxy-2-cyclohexen-1-one obtained from Millingtonia hortensis[19]. Consequently, the structure of 3 was determined as showed in Figure 1.
was observed, which was in accord with that observed in cornoside ( −10.5°), and analysis of the 13C NMR data of C-1 and C-1‴ in 3 indicated that they were greatly similar to that of C-1 (δC 68.9) in 4-[2-(β-d-glucopyranosyloxy)ethyl]-4-hydroxy-2-cyclohexen-1-one obtained from Millingtonia hortensis[19]. Consequently, the structure of 3 was determined as showed in Figure 1.Marinoid I (4) was obtained as colourless oil. The presence of a molecular ion at m/z 701.2630 [M + Na]+ (calcd [M + Na]+, 701.2633) in the HRESIMS spectrum suggested a molecular formula of C30H46O17. Analysis of the 1H NMR data (Table 2) showed the presence of two cyclohexanone moieties in 4[14] by the presence of the proton signals at δH 6.95 (1H, d, J = 10.0 Hz, H-2), 5.87 (1H, d, J = 10.0 Hz, H-3), 3.67 (1H, dd, J = 7.0, 3.6 Hz, H-6), 2.82 (1H, m, H-5α), and 2.49 (1H, m, H-5β)], and [δH 6.93 (1H, d, J = 10.0 Hz, H-2‴), 5.85 (1H, d, J = 10.0 Hz, H-3‴), 3.64 (1H, dd, J = 7.0, 3.6 Hz, H-6‴), 2.82 (1H, m, H-5‴α), and 2.49 (1H, m, H-5‴β), and the carbon signals at δC 198.6 (C-4), 154.7 (C-2), 127.1 (C-3), 72.4 (C-1), 82.8 (C-6), and 38.9 (C-5) and δC 198.6 (C-4‴), 154.0 (C-2‴), 127.0 (C-3‴), 72.4 (C-1‴), 82.7 (C-6‴), and 38.9 (C-5‴). The proton signals at δH 3.67 (1H, dd, J = 7.0, 3.6 Hz, H-6) and 3.64 (1H, dd, J = 7.0, 3.6 Hz, H-6‴), and corresponding carbon signals at δC 82.8 (C-6), 82.7 (C-6‴), 38.9 (C-5) and 38.9 (C-5‴) revealed that 4 was the CH3OH adduct of cornoside [13]. Moreover, the HMBC correlation from δH 3.43 (3H, s) to C-6 and δH 3.43 (3H, s) to C-6‴ we therefore assign the methoxyl to C-6 and C-6‴, respectively. NMR spectra also indicated the presence of two β-glucosyl groups, i.e., two anomeric carbon resonances at δC 103.0 (C-1ʺ) and 102.8 (C-1ʺʺ), and two anomeric protons at δH 4.28 (1H, d, J = 7.5 Hz, H-1ʺ) and 4.27 (1H, d, J = 7.5 Hz, H-1ʺʺ). They were used as a starting point in the homonuclear correlated spectra to determine all the glycosidic protons. The JH-1ʺ-H-2ʺ value (7.5 Hz) and of JH-1ʺʺ-H-2ʺʺ value (7.5 Hz) compound 4, further confirmed that the sugars were β-glucosyl groups [12]. The tentative molecular weight of the compound deduced from NMR analysis suggested compound 4 to be an unsymmetrical dimer of cornoside analog.
1H-1H COSY and HMBC correlations (Figure 2) were used to establish the molecular skeleton of 4. Spin systems were revealed by analysis of COSY correlations corresponding to the H-2/H-3, H2-5/H-6, H2-1′/H2-2′, H-1ʺ/H-2ʺ/H-3ʺ/H-4ʺ/H-5ʺ/H2-6ʺ, H-2‴/H-3‴, H2-5‴/H-6‴, H2-1ʺʺ/H2-2ʺʺ, and H-1ʺ‴/H-2ʺ‴/H-3ʺ‴/H-4ʺ‴/H-5ʺ‴/H2-6ʺ‴. The connectivity of the two cyclohexanone moieties to C-1′ and C-1ʺʺ were established by the HMBC correlations of H-2 to C-1ʹ and H-6 to C-1ʹ, and of H-2‴ to C-1ʺʺ and H-6‴ to C-1ʺʺ, respectively. The presence of the two β-glucosyl groups in 4 could be proposed on the basis of HMBC correlations from H-4ʺ to C-2′ and H-1ʺ‴ to C-2ʺ, respectively. The connection of the two β-glucosyl groups was confirmed by the presence of HMBC correlation from H-4ʺ‴ to C-1ʺ.
The configuration of C-6, C-6‴, C-1 and C-1‴ were determined using the optical rotation and analysis of the coupling constants (J). The coupling patterns of the H-6 (dd, J = 7.0, 3.6 Hz) and H-5α (dd, J = 11, 3.6 Hz); H-6‴ (dd, J = 7.0, 3.6 Hz) and H-5‴α (dd, J = 11, 3.6 Hz) led to confirmation of the cis-orientation of H-6/H-5α and H-6‴/H-5‴α [20,21], which are in line with those of 4-[2-(β-d-glucopyranosyloxy)ethyl]-4-hydroxy-5-methoxy-2-cyclohexen-1-one that was obtained from M. hortensis [19], and 1,6-dihydroxy-4-oxo-2-cyclohexene-1-cetic acid ethyl ester isolated from Senecio scandens [17], and thus the configuration of C-6 and C-6‴ were determined as S and S. Analysis of the 13C NMR data of C-1 and C-1‴ in 4 indicated that they were greatly similar to that of C-1 (δC 71.8) in 4-[2-(β-d-glucopyranosyloxy)ethyl]-4-hydroxy-5-methoxy-2-cyclohexen-1-one [19], and our compound 4 showed in MeOH of −26.4°, which is in accord with that observed in 1,6-dihydroxy-4-oxo-2-cyclohexene-1-acetic acid ethyl ester ( −12.5°) [17]. From the aforementioned analyses, the configurations of 4 were assumed to be 1R, 6S, 1‴R and 6‴S. On the basis of this cumulative analysis, the structure of 4 was thus determined as shown in Figure 1.
in MeOH of −26.4°, which is in accord with that observed in 1,6-dihydroxy-4-oxo-2-cyclohexene-1-acetic acid ethyl ester ( −12.5°) [17]. From the aforementioned analyses, the configurations of 4 were assumed to be 1R, 6S, 1‴R and 6‴S. On the basis of this cumulative analysis, the structure of 4 was thus determined as shown in Figure 1.The cellular antioxidant assay (CAA) is a new approach to quantify antioxidants under physiological conditions when compared to chemical antioxidant activity assays [22,23,24]. The CAA assay has been widely used for fruits and vegetables recently, but not yet in marine natural products research. The EC50 values of compounds 1–3 were weak, 598, 4971, and 1103 μM, respectively. However, the EC50 value of compound 4 was 26 μM, of the same order of the positive control quercetin (EC50 = 11 μM).
3. Experimental Section
3.1. General Experimental Procedures
UV spectra were recorded in MeOH on a Perkin-Elmer Lambda 35 UV-Vis spectrophotometer (Wellesley, MA, USA). The IR spectra were measured in KBr on a WQF-410 FT-IR spectrophotometer (Beifen-Ruili, Beijing, China). NMR spectra were recorded on a Bruker AV 600 NMR spectrometer (Bruker, Bremen, Germany) with TMS as an internal standard. HR-ESI-MS data were obtained from Bruker Maxis mass spectrometer (Bruker, Bremen, Germany). Waters-2695 HPLC system (Waters, Milford, MA, USA), using a Sunfire™ C18 column (150 × 10 mm i.d., 10 μm, Waters, Milford, MA, USA) coupled to a Waters 2998 photodiode array detector (Waters, Milford, MA, USA). Optical rotation data were measured by Perkin-Elmer Model 341 polarimeter (Wellesley, MA, USA). CD spectra were recorded on a spectropolarimeter (MODEL J-810-150S, Tokyo, Japan). The silica gel GF254 used for TLC were supplied by the Qingdao Marine Chemical Factory, Qingdao, China. Spots were detected on TLC under UV light or by heating after spraying with 5% H2SO4 in EtOH. All solvent ratios are measured v/v.
3.2. Plant Material
The fruits of A. marina were collected from Beihai city, Guangxi province, China, in September, 2011. The specimen was identified by Professor Hangqing Fan who is from Guangxi Mangrove Research Center, Guangxi Academy of Sciences. A voucher specimen (2011-GXAS-008) was deposited in Guangxi Key Laboratory of Marine Environmental Science, Guangxi Academy of Sciences, China.
3.3. Extraction and Isolation
The fruits of A. marina (35.4 kg, wet weight) were exhaustively extracted with EtOH-CH2Cl2 (2:1, v/v). The solvent was evaporated in vacuo to afford a syrupy residue that was suspended in distilled water and fractionated successively with petroleum ether, ethyl acetate, and n-butanol. The n-butanol soluble portion (269 g) was subjected to column chromatography (CC) on silica gel, using CHCl3–MeOH (from 10:0 to 0:10) as eluent, giving eleven fractions (A–K). Fraction D was subjected to column chromatography to afford four subfractions (D1–D4). Fraction D3 was separated by HPLC, using MeOH–H2O (MeOH:H2O = 15:85, 25:75, 60:40) to yield 4 (3.9 mg, Rt = 10.2 min), 1 (3.5 mg, Rt = 12.5 min) and 2 (2.3 mg, Rt = 13.9 min), respectively. Fraction D4 was separated by HPLC, using MeOH–H2O (MeOH:H2O = 5:95) to yield 3 (6.0 mg, Rt = 9.5 min).
Marinoid F (1): Yellow oil; UV (MeOH) λmax (log εmax) 220 (2.45) and 242 (3.31) nm. −14.7° (c 0.18, MeOH); CD (MeOH) Δε220nm −4.28, IR (KBr) νmax 3425, 1720 and 1682 cm−1. 1H (CD3OD, 600 MHz) and 13C (CD3OD, 150 MHz) NMR data, see Table 1; HRESIMS: m/z 351.0835 (calcd. for C14H19ClO8 + H, 351.0841).
−14.7° (c 0.18, MeOH); CD (MeOH) Δε220nm −4.28, IR (KBr) νmax 3425, 1720 and 1682 cm−1. 1H (CD3OD, 600 MHz) and 13C (CD3OD, 150 MHz) NMR data, see Table 1; HRESIMS: m/z 351.0835 (calcd. for C14H19ClO8 + H, 351.0841).Marinoid G (2): Yellow oil; UV (MeOH) λmax (log εmax) 220 (2.39) and 240 (2.75) nm. +10.2° (c 0.21, MeOH); CD (MeOH) Δε220nm +5.97; IR (KBr) νmax 3424, 1711 and 1684 cm−1. 1H (CD3OD, 600 MHz) and 13C (CD3OD, 150 MHz) NMR data, see Table 1; HRESIMS: m/z 351.0837 (calcd. for C14H19ClO8 + H, 351.0841).
+10.2° (c 0.21, MeOH); CD (MeOH) Δε220nm +5.97; IR (KBr) νmax 3424, 1711 and 1684 cm−1. 1H (CD3OD, 600 MHz) and 13C (CD3OD, 150 MHz) NMR data, see Table 1; HRESIMS: m/z 351.0837 (calcd. for C14H19ClO8 + H, 351.0841).Marinoid H (3): Colourless oil; UV (MeOH) λmax (log εmax) 218 (2.35) and 237 (2.94) nm. −15.8° (c 0.25, MeOH); CD (MeOH) Δε220nm −6.32; IR (KBr) νmax 3452, 1701 and 1675 cm−1. 1H (CD3OD, 600 MHz) and 13C (CD3OD, 150 MHz) NMR data, see Table 1; HRESIMS: m/z 591.2281 (calcd. for C26H38lO15 + H, 591.2283).
−15.8° (c 0.25, MeOH); CD (MeOH) Δε220nm −6.32; IR (KBr) νmax 3452, 1701 and 1675 cm−1. 1H (CD3OD, 600 MHz) and 13C (CD3OD, 150 MHz) NMR data, see Table 1; HRESIMS: m/z 591.2281 (calcd. for C26H38lO15 + H, 591.2283).Marinoid I (4): Colourless oil; UV (MeOH) λmax (log εmax) 219 (2.15) and 238 (3.05) nm. −26.4° (c 0.31, MeOH); CD (MeOH) Δε220nm −5.42; IR (KBr) νmax 3447, 1705 and 1679 cm−1. 1H (CD3OD, 600 MHz) and 13C (CD3OD, 150 MHz) NMR data, see Table 1; HRESIMS: m/z 701.2630 (calcd. for C30H46O17 + Na, 701.2633).
−26.4° (c 0.31, MeOH); CD (MeOH) Δε220nm −5.42; IR (KBr) νmax 3447, 1705 and 1679 cm−1. 1H (CD3OD, 600 MHz) and 13C (CD3OD, 150 MHz) NMR data, see Table 1; HRESIMS: m/z 701.2630 (calcd. for C30H46O17 + Na, 701.2633).3.4. Cellular Antioxidant Assay
Following the reported method [21,22,23], the cellular antioxidant activity was determined.
4. Conclusions
In conclusion, four new jacaranone analogs, marinoids F–I (1–4 respectively), were isolated from a Beibu Gulf mangrove A. marina and identified. Marinoids F and G are shown to be diastereoisomers of chlorocornoside, a new halogen containing marine secondary metabolite. The CAA assay is considered to be a more physiologically relevant assay in the measurement of antioxidant activity of food when compared to the common chemistry antioxidant activity assays [25,26]. Until today, there have been no reports of the use of this CAA assay in the marine research area. Using the assay, the antioxidant activity of the isolates was therefore determined. This is the first report of chlorocornoside and of the dimeric disaccharide 4 which showed good antioxidant activity (EC50 = 26 μM), comparable with the positive control quercetin.
Acknowledgments
This study was supported by grants from National Natural Science Foundation of China (No. 31100260, 81260480, 81260673 and 81060344), Knowledge Innovation Program of Chinese Academy of Sciences (KSCX2-EW-J-28), Natural Science Foundation of Guangxi (2011GXNSFE018002, 2011GXNSFB018035, 2011GXNSFA018108, 2012GXNSFAA053160 and 2012GXNSFEA053001). R-MH acknowledges a CAS Academic Visitor Fellowship and thanks Dr Ian S Blagbrough, University of Bath, for helpful discussions.
Author Contributions
In this paper, Xiang-Xi Yi was in charge of writing the manuscript; Yong Chen and Ming-Ben Xu was responsible for the isolation of the compounds; Yin-Ning Chen was responsible for structures identification; Wen-Pei Xie was in charge of biological activity; Cheng-Hai Gao is the corresponding author who was responsible for the analysis of the data of biological activity; and Ri-Ming Huang is the corresponding author who was responsible for arranging, checking and revising the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sun, Y.; Ouyang, J.; Deng, Z.W.; Li, Q.S.; Lin, W.H. Structure elucidation of five new iridoid glucosides from the leaves of Avicennia marina. Magn. Reson. Chem. 2008, 46, 638–642. [Google Scholar] [CrossRef]
- Fauvel, M.T.; Taoubi, K.; Gleye, J.; Fourasté, I. Phenylpropanoid glycosides from Avicennia marina. Planta Med. 1993, 59, 387. [Google Scholar]
- Konig, G.; Rimpler, H. Iridoid glucosides in Avicennia marina. Phytochemistry 1985, 24, 1245–1248. [Google Scholar] [CrossRef]
- Feng, Y.; Li, X.M.; Duan, X.J.; Wang, B.G. Iridoid glucosides and flavones from the aerial parts of Avicennia marina. Chem. Biodivers. 2006, 3, 799–806. [Google Scholar] [CrossRef]
- Fauvel, M.T.; Bousquetmelou, A.; Moulis, C.; Gleye, J.; Jensen, S.R. Iridoid glucosides from Avicennia germinans. Phytochemistry 1995, 38, 893–894. [Google Scholar]
- Ito, C.; Katsuno, S.; Kondo, Y.; Tan, H.T.W.; Furukawa, H. Chemical constituents of Avicennia alba. Isolation and structural elucidation of new naphthoquinones and their analogues. Chem. Pharm. Bull. 2000, 48, 339–343. [Google Scholar] [CrossRef]
- Han, L.; Huang, X.S.; Dahse, H.M.; Moellmann, U.; Fu, H.Z.; Grabley, S.; Sattler, I.; Lin, W.H. Unusual naphthoquinone derivatives from the twigs of Avicennia marina. J. Nat. Prod. 2007, 70, 923–927. [Google Scholar] [CrossRef]
- Sharaf, M.; El-Ansari, M.A.; Saleh, N.A.M. New flavonoids from Avicennia marina. Fitoterapia 2000, 71, 274–277. [Google Scholar] [CrossRef]
- Han, L.; Huang, X.S.; Dahse, H.M.; Moellmann, U.; Grabley, S.; Lin, W.H.; Sattler, I. New abietane diterpenoids from the mangrove Avicennia marina. Planta Med. 2008, 74, 432–437. [Google Scholar] [CrossRef]
- Parker, K.A.; Kang, S.K. Regiospecific nucleophilic aromatic-substitution—Conjugate addition of active methylene-compounds to quinone monoacetals and aromatization of the adducts. J. Org. Chem. 1980, 45, 1218–1224. [Google Scholar] [CrossRef]
- Hollenst, R.; Philipsb, W.V. Carbon magnetic-resonance spectra of α, β, γ, σ- and α, β, α′, β′-unsaturated-ketones. Helv. Chim. Acta 1972, 55, 2030–2044. [Google Scholar] [CrossRef]
- Breitmaier, E.; Voelter, W. Carbon-13 NMR Spectroscopy. VCH Publishers: New York, NY, USA, 1987; pp. 380–394. [Google Scholar]
- Endo, K.; Hikino, H. Structures of rengyol, rengyoxide, and rengyolone, new cyclohexylethane derivatives from Forsythia suspensa fruits. Can. J. Chem. 1984, 62, 2011–2014. [Google Scholar] [CrossRef]
- Bianco, A.; Scalzo, R.L.; Scarpati, M.L. Isolation of cornoside from Olea europaea and its transformation into halleridone. Phytochemistry 1993, 32, 445–457. [Google Scholar] [CrossRef]
- Jensen, S.R.; Kjaer, A.; Nielsen, B.J. A quinol glucoside isolated from Cornus species. Acta Chem. Scand. 1973, 27, 367–369. [Google Scholar] [CrossRef]
- Sasaki, H.; Taguchi, H.; Endo, T.; Yosioka, I. The glycosides of Martynia louisiana Mill. A new phenylpropanoid glycoside, martynoside. Chem. Pharm. Bull. 1978, 26, 2111–2121. [Google Scholar] [CrossRef]
- Tian, X.Y.; Wang, Y.H.; Yang, Q.Y.; Yu, S.S.; Fang, W.S. Jacaranone analogs from Senecio scandens. J. Asian Nat. Prod. Res. 2009, 11, 63–68. [Google Scholar] [CrossRef]
- Seo, S.; Tomita, Y.; Tori, K.; Yoshimura, Y. Determination of the absolute configuration of a secondary hydroxy group in a chiral secondary alcohol using glycosidation shifts in carbon-13 nuclear magnetic resonance sepctroscopy. J. Am. Chem. Soc. 1978, 100, 3331–3339. [Google Scholar] [CrossRef]
- Hase, T.; Kawamoto, Y.; Ohtani, K.; Kasai, R.; Yamasaki, K.; Picheansoonthon, C. Cyclohexylethanoids and related glucosides from Millingtonia hortensis. Phytochemistry 1995, 39, 235–241. [Google Scholar] [CrossRef]
- Huang, R.M.; Ma, W.; Dong, J.D.; Zhou, X.F.; Xu, T.H.; Lee, K.J.; Yang, X.W.; Xu, S.H.; Liu, Y.H. A new 1,4-diazepine from South China Sea marine sponge Callyspongia species. Molecules 2010, 15, 871–877. [Google Scholar] [CrossRef]
- Wang, Y.L.; Li, Z.L.; Zhang, H.L.; Sha, Y.; Pei, Y.H.; Hua, H.M. New germacrane sequiterpenes from Salvia chinensis. Chem. Pharm. Bull. 2008, 61, 843–846. [Google Scholar]
- Faller, A.L.K.; Fialho, E.; Liu, R.H. Cellular antioxidant activity of Feijoada whole meal coupled with an in vitro digestion. J. Agric. Food Chem. 2012, 60, 4826–4832. [Google Scholar] [CrossRef]
- Song, W.; Derito, C.M.; Liu, M.K.; He, X.J.; Dong, M.; Liu, R.H. Cellular antioxidant activity of common vegetables. J. Agric. Food Chem. 2010, 58, 6621–6629. [Google Scholar] [CrossRef]
- Wolfe, K.L.; Kang, X.M.; He, X.J.; Dong, M.; Zhang, Q.Y.; Liu, R.H. Cellular antioxidant activity of common fruits. J. Agric. Food Chem. 2008, 56, 8418–8426. [Google Scholar]
- Wolfe, K.L.; Liu, R.H. Cellular antioxidant activity (CAA) assay for assessing antioxidants, foods, and dietary supplements. J. Agric. Food Chem. 2007, 55, 8896–8907. [Google Scholar] [CrossRef]
- Felice, D.L.; Sun, J.; Liu, R.H. A modified methylene blue assay for accurate cell counting. J. Funct. Foods 2009, 1, 109–118. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).