Echinochrome A Protects Mitochondrial Function in Cardiomyocytes against Cardiotoxic Drugs


 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

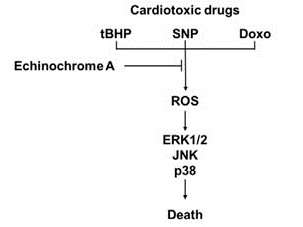
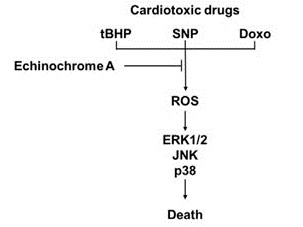
2. Results and Discussion
2.1. Ech A Inhibited Cardiotoxic Agent-Induced Cell Death

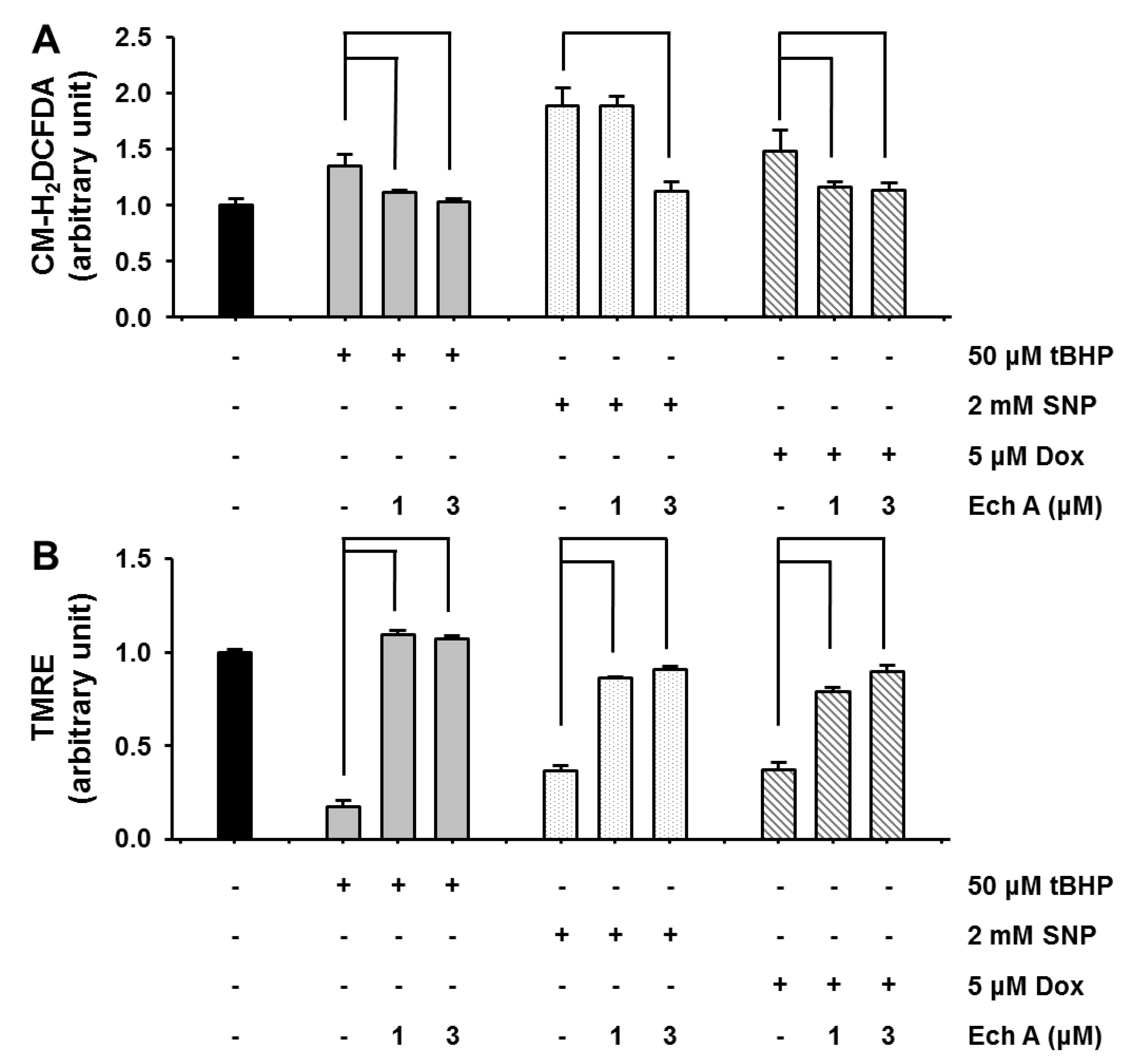
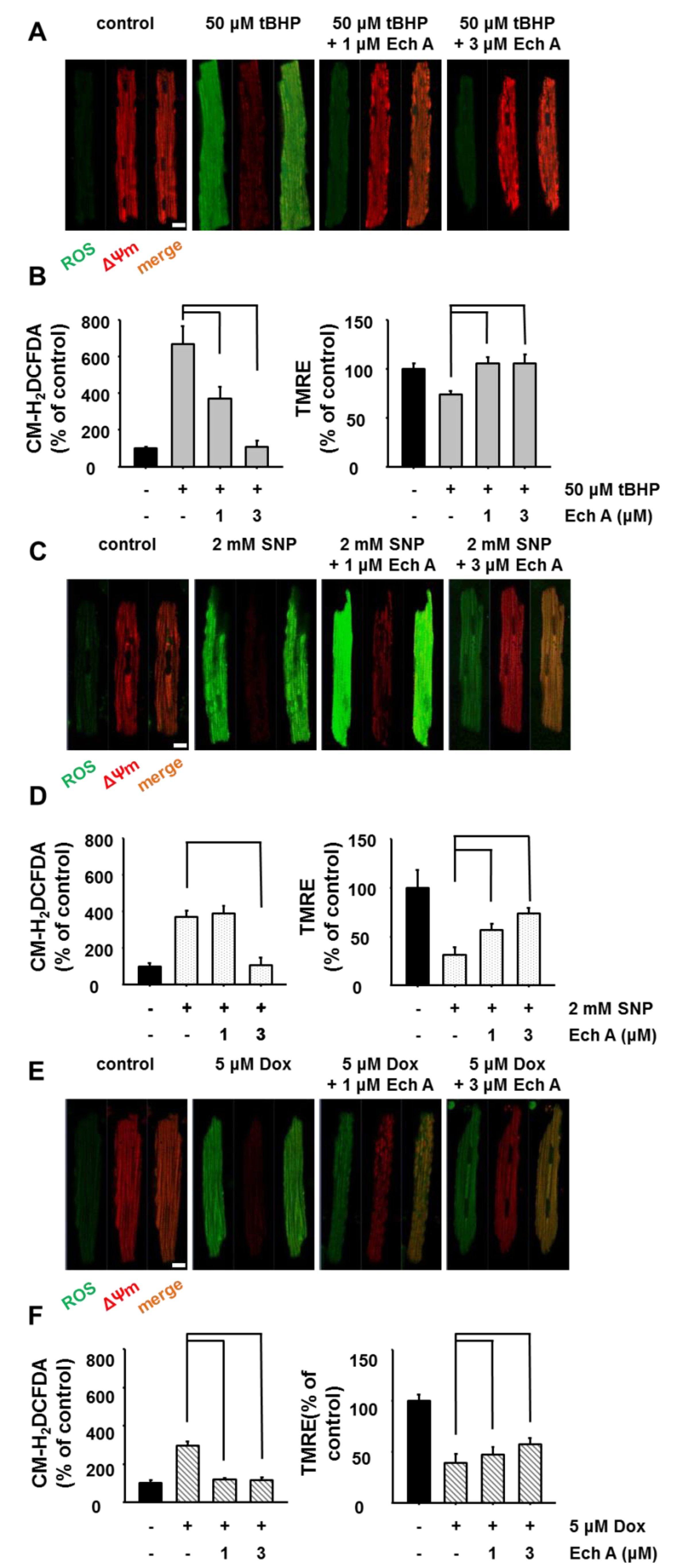
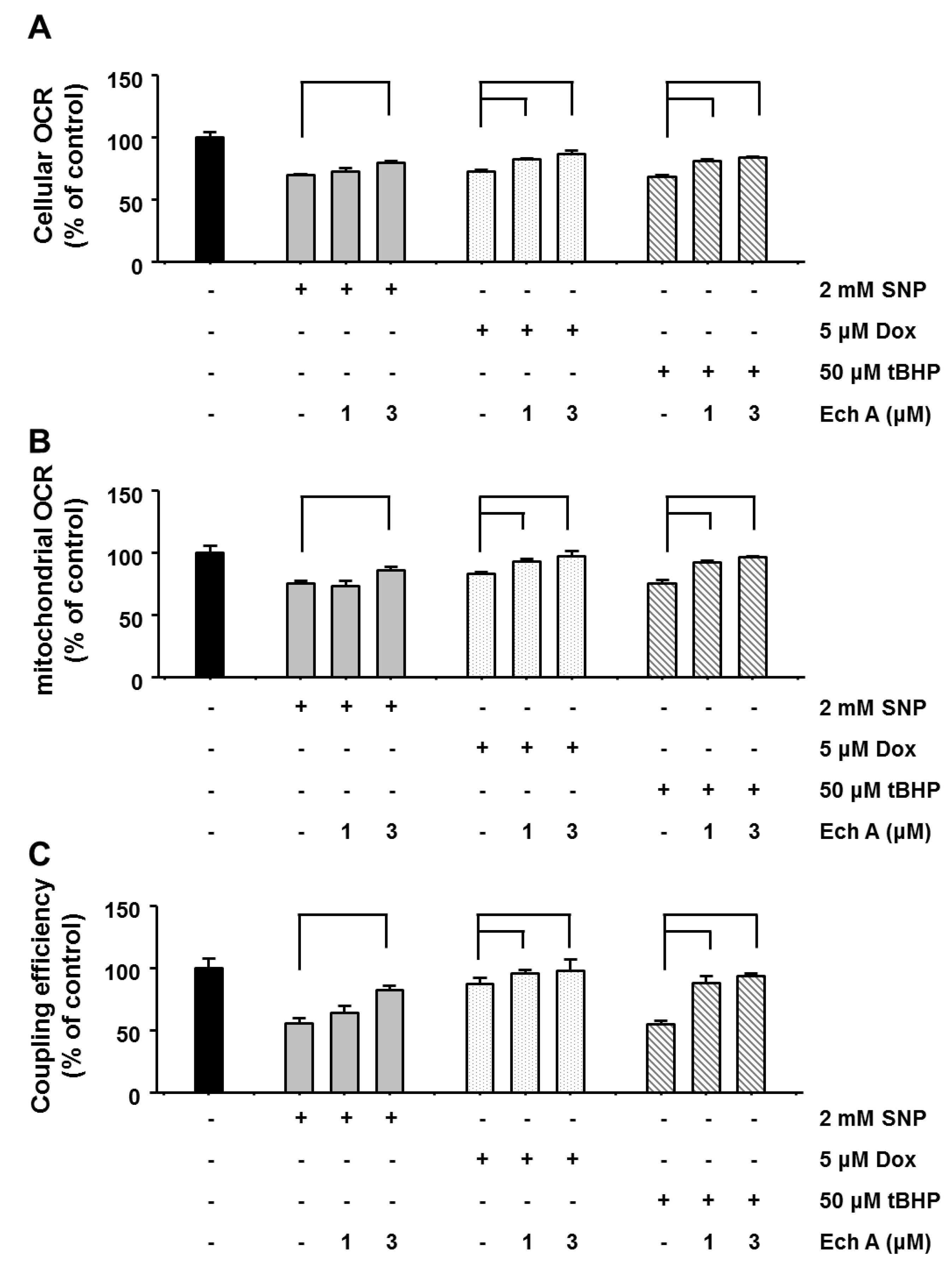
2.2. Ech A Attenuated Cardiotoxic Agent-Induced Mitochondrial Damage




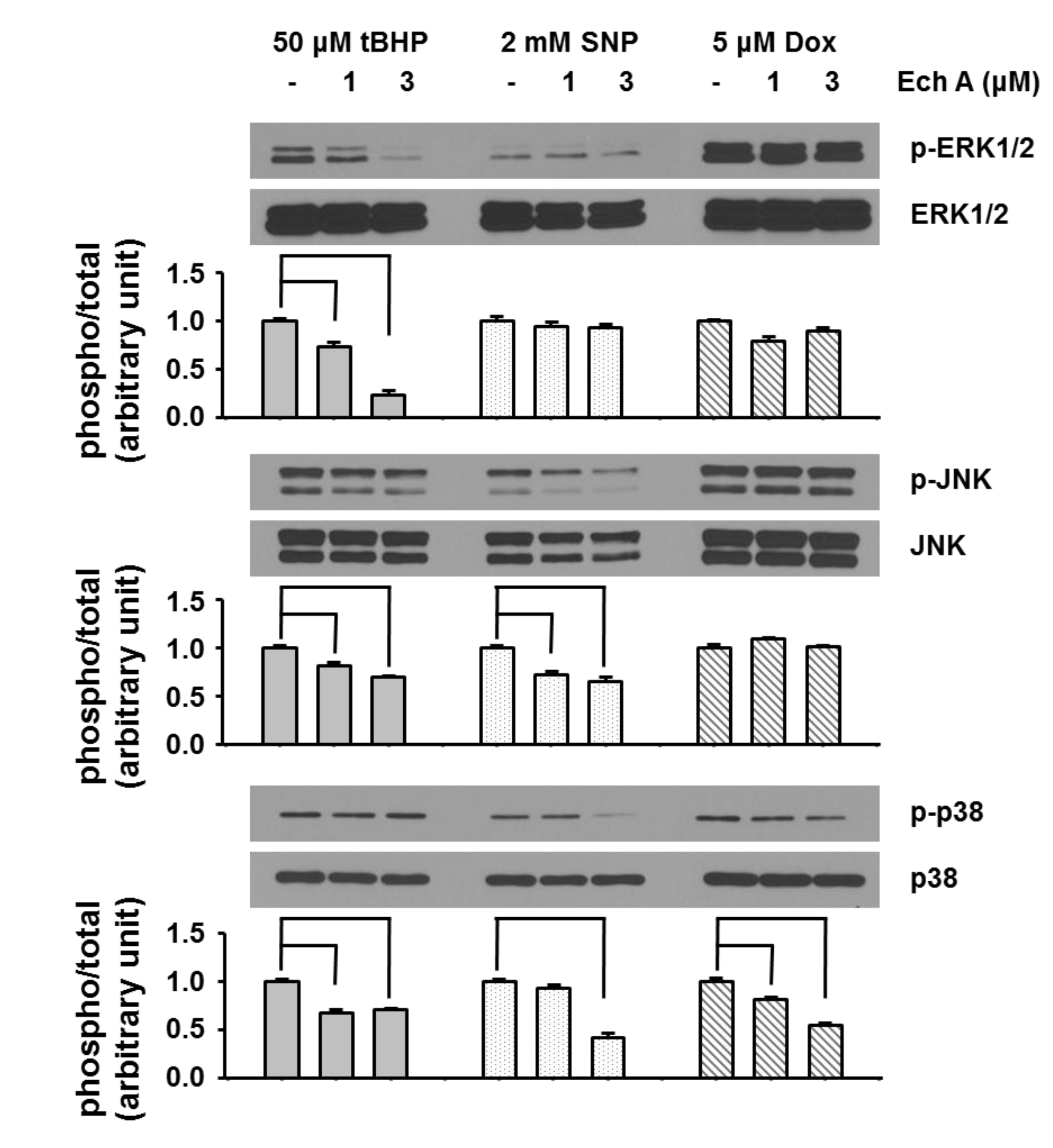
2.3. Ech A Regulated ERK1/2, JNK, and p38 Signaling Pathways

3. Experimental Section
3.1. Chemicals
3.2. Cell Culture
3.2.1. Measurement of Cell Viability
3.2.2. Measurement of Cytotoxicity
3.2.3. Measurement of ΔΨm and ROS Level
3.2.4. Measurement of Cellular and Mitochondrial ATP Level
3.2.5. Measurement of OCR
3.2.6. Western Blot Analysis
3.3. Isolated Single Rat Cardiomyocytes
3.3.1. Measurement of Mitochondrial Inner Membrane Potential
3.3.2. Measurement of ROS Level
3.4. Data Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Anderson, H.A.; Mathieson, J.W.; Thomson, R.H. Distribution of spinochrome pigments in echinoids. Comp. Biochem. Physiol. 1969, 28, 333–345. [Google Scholar] [CrossRef]
- Thomson, R.H. Distribution of naturally occurring quinones. Pharm. Weekbl. Sci. 1991, 13, 70–73. [Google Scholar] [CrossRef]
- Elyakov, G.B.; Maximov, O.B.; Mischenko, N.P.; Koltsova, E.A.; Fedoreev, S.A.; Glebko, L.I.; Krasovskaya, N.P.; Artjukov, A.A. Histochrome and its Therapeutic Use in Ophthalmology. US Patent 6,384,084, 7 May 2002. [Google Scholar]
- Elyakov, G.B.; Maximov, O.B.; Mischenko, N.P.; Koltsova, E.A.; Fedoreev, S.A.; Glebko, L.I.; Krasovskaya, N.P.; Artjukov, A.A. Composition Comprising di-and Trisodium Salts of Echinochrome for Treating Ocular Conditions. European Patent 1121929, 3 November 2004. [Google Scholar]
- Egorov, E.A.; Alekhina, V.A.; Volobueva, T.M.; Fedoreev, S.A.; Mishchenko, N.P.; Kol’tsova, E.A. Histochrome, a new antioxidant, in the treatment of ocular disease. Vestn. Oftalmol. 1999, 115, 34–35. [Google Scholar]
- Mishchenko, N.P.; Fedoreev, S.A.; Bagirova, V.L. Histochrome: A new original domestic drug. Pharm. Chem. J. 2003, 37, 48–52. [Google Scholar] [CrossRef]
- Elyakov, G.B.; Maximov, O.B.; Mischenko, N.P.; Koltsova, E.A.; Fedoreev, S.A.; Glebko, L.I.; Krasovskaya, N.P.; Artjukov, A.A. Histochrome and its Therapeutic Use in Acute Myocardial Infarction and Ischemic Heart Disease. US Patent 6,410,601, 11 October 2001. [Google Scholar]
- Elyakov, G.B.; Maximov, O.B.; Mischenko, N.P.; Koltsova, E.A.; Fedoreev, S.A.; Glebko, L.I.; Krasovskaya, N.P.; Artjukov, A.A. Drug preparation “histochrome” for treating acute myocardial infarction and ischaemic heart diseases. European Patent 1121930, 14 November 2007. [Google Scholar]
- Shvilkin, A.V.; Serebriakov, L.I.; Tskitishvili, O.V.; Sadretdinov, S.M.; Kol’tsova, E.A.; Maksimov, O.B.; Mishchenko, N.P.; Novikov, V.L.; Levitskii, D.O.; Ruda, M. Effect of echinochrom on experimental myocardial reperfusion injury. Kardiologiia 1991, 31, 79–81. [Google Scholar]
- Holley, J.E.; Butler, J.W.; Mahoney, J.M. Carbon monoxide poisoning in racing car drivers. J. Sports Med. Phys. Fitness 1999, 39, 20–23. [Google Scholar]
- Buimov, G.A.; Maksimov, I.V.; Perchatkin, V.A.; Repin, A.N.; Afanas’ev, S.A.; Markov, V.A.; Karpov, R.S. Effect of the bioantioxidant histochrome on myocardial injury in reperfusion therapy on patients with myocardial infarction. Ter Arkh 2002, 74, 12–16. [Google Scholar]
- Clark, D., III; Tesseneer, S.; Tribble, C.G. Nitroglycerin and sodium nitroprusside: Potential contributors to postoperative bleeding? Heart Surg. Forum 2012, 15, E92–E96. [Google Scholar] [CrossRef]
- Amoroso, S.; Tortiglione, A.; Secondo, A.; Catalano, A.; Montagnani, S.; Di Renzo, G.; Annunziato, L. Sodium nitroprusside prevents chemical hypoxia-induced cell death through iron ions stimulating the activity of the Na+-Ca2+ exchanger in C6 glioma cells. J. Neurochem. 2000, 74, 1505–1513. [Google Scholar]
- Bernabe, J.C.; Tejedo, J.R.; Rincon, P.; Cahuana, G.M.; Ramirez, R.; Sobrino, F.; Bedoya, F.J. Sodium nitroprusside-induced mitochondrial apoptotic events in insulin-secreting RINm5F cells are associated with MAP kinases activation. Exp. Cell Res. 2001, 269, 222–229. [Google Scholar] [CrossRef]
- Rabkin, S.W.; Kong, J.Y. Nitroprusside induces cardiomyocyte death: Interaction with hydrogen peroxide. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H3089–H3100. [Google Scholar]
- Binaschi, M.; Bigioni, M.; Cipollone, A.; Rossi, C.; Goso, C.; Maggi, C.A.; Capranico, G.; Animati, F. Anthracyclines: Selected new developments. Curr. Med. Chem. Anticancer Agents 2001, 1, 113–130. [Google Scholar] [CrossRef]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef]
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P.I. Doxorubicin: The good, the bad and the ugly effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef]
- Westhoff, T.H.; Scheid, S.; Tolle, M.; Kaynak, B.; Schmidt, S.; Zidek, W.; Sperling, S.; van der Giet, M. A physiogenomic approach to study the regulation of blood pressure. Physiol. Genomics 2005, 23, 46–53. [Google Scholar] [CrossRef]
- Ferrans, V.J.; Clark, J.R.; Zhang, J.; Yu, Z.X.; Herman, E.H. Pathogenesis and prevention of doxorubicin cardiomyopathy. Tsitologiia 1997, 39, 928–937. [Google Scholar]
- Jones, R.L.; Swanton, C.; Ewer, M.S. Anthracycline cardiotoxicity. Expert Opin. Drug Saf. 2006, 5, 791–809. [Google Scholar] [CrossRef]
- Lee, B.S.; Kim, S.H.; Jin, T.; Choi, E.Y.; Oh, J.; Park, S.; Lee, S.H.; Chung, J.H.; Kang, S.M. Protective effect of survivin in Doxorubicin-induced cell death in h9c2 cardiac myocytes. Korean Circ. J. 2013, 43, 400–407. [Google Scholar] [CrossRef]
- Kang, J.Y.; Costyn, L.J.; Nagy, T.; Cowan, E.A.; Oldham, C.D.; May, S.W.; Arnold, R.D. The antioxidant phenylaminoethyl selenide reduces doxorubicin-induced cardiotoxicity in a xenograft model of human prostate cancer. Arch. Biochem. Biophys. 2011, 515, 112–119. [Google Scholar] [CrossRef]
- Sun, Y. Myocardial repair/remodelling following infarction: Roles of local factors. Cardiovasc. Res. 2009, 81, 482–490. [Google Scholar] [CrossRef]
- Lee, S.R.; Lee, S.J.; Kim, S.H.; Ko, K.S.; Rhee, B.D.; Xu, Z.; Kim, N.; Han, J. NecroX-5 suppresses sodium nitroprusside-induced cardiac cell death through inhibition of JNK and caspase-3 activation. Cell Biol. Int. 2014, in press. [Google Scholar]
- Kim, H.J.; Koo, S.Y.; Ahn, B.H.; Park, O.; Park, D.H.; Seo, D.O.; Won, J.H.; Yim, H.J.; Kwak, H.S.; Park, H.S.; et al. NecroX as a novel class of mitochondrial reactive oxygen species and ONOO(−) scavenger. Arch. Pharm. Res. 2010, 33, 1813–1823. [Google Scholar] [CrossRef]
- Kitamura, Y.; Koide, M.; Akakabe, Y.; Matsuo, K.; Shimoda, Y.; Soma, Y.; Ogata, T.; Ueyama, T.; Matoba, S.; Yamada, H.; Ikeda, K. Manipulation of Cardiac Phosphatidylinositol 3-Kinase (PI3K)/Akt Signaling by Apoptosis Regulator through Modulating IAP Expression (ARIA) Regulates Cardiomyocyte Death during Doxorubicin-induced Cardiomyopathy. J. Biol. Chem. 2014, 289, 2788–2800. [Google Scholar] [CrossRef]
- Meloche, S.; Pouyssegur, J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 2007, 26, 3227–3239. [Google Scholar] [CrossRef]
- Roux, P.P.; Blenis, J. ERK and p38 MAPK-activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef]
- Chen, M.B.; Wu, X.Y.; Gu, J.H.; Guo, Q.T.; Shen, W.X.; Lu, P.H. Activation of AMP-activated protein kinase contributes to doxorubicin-induced cell death and apoptosis in cultured myocardial H9c2 cells. Cell Biochem. Biophys. 2011, 60, 311–322. [Google Scholar] [CrossRef]
- Chae, H.J.; So, H.S.; Chae, S.W.; Park, J.S.; Kim, M.S.; Oh, J.M.; Chung, Y.T.; Yang, S.H.; Jeong, E.T.; Kim, H.M.; et al. Sodium nitroprusside induces apoptosis of H9C2 cardiac muscle cells in a c-Jun N-terminal kinase-dependent manner. Int. Immunopharmacol. 2001, 1, 967–978. [Google Scholar] [CrossRef]
- Rasbach, K.A.; Schnellmann, R.G. Signaling of mitochondrial biogenesis following oxidant injury. J. Biol. Chem. 2007, 282, 2355–2362. [Google Scholar] [CrossRef]
- Wang, X.; Ye, X.L.; Liu, R.; Chen, H.L.; Bai, H.; Liang, X.; Zhang, X.D.; Wang, Z.; Li, W.L.; Hai, C.X. Antioxidant activities of oleanolic acid in vitro: Possible role of Nrf2 and MAP kinases. Chem. Biol. Interact. 2010, 184, 328–337. [Google Scholar] [CrossRef]
- Yang, Y.C.; Lii, C.K.; Lin, A.H.; Yeh, Y.W.; Yao, H.T.; Li, C.C.; Liu, K.L.; Chen, H.W. Induction of glutathione synthesis and heme oxygenase 1 by the flavonoids butein and phloretin is mediated through the ERK/Nrf2 pathway and protects against oxidative stress. Free Radic. Biol. Med. 2011, 51, 2073–2081. [Google Scholar] [CrossRef]
- Li, J.M.; Wheatcroft, S.; Fan, L.M.; Kearney, M.T.; Shah, A.M. Opposing roles of p47phox in basal versus angiotensin II-stimulated alterations in vascular O2− production, vascular tone, and mitogen-activated protein kinase activation. Circulation 2004, 109, 1307–1313. [Google Scholar] [CrossRef]
- Chu, E.S.; Yow, C.M. Modulation of telomerase and signal transduction proteins by hexyl-ALA-photodynamic therapy (PDT) in human doxorubicin resistant cancer cell models. Photodiagnosis Photodyn. Ther. 2012, 9, 243–255. [Google Scholar] [CrossRef]
- Velez, J.M.; Miriyala, S.; Nithipongvanitch, R.; Noel, T.; Plabplueng, C.D.; Oberley, T.; Jungsuwadee, P.; van Remmen, H.; Vore, M.; St Clair, D.K. p53 Regulates oxidative stress-mediated retrograde signaling: A novel mechanism for chemotherapy-induced cardiac injury. PLoS One 2011, 6, e18005. [Google Scholar] [CrossRef]
- Mischenko, N.P.; Fedoreyev, S.A.; Pokhilo, N.D.; Anufriev, V.P.; Denisenko, V.A.; Glazunov, V.P. Echinamines A and B, first aminated hydroxynaphthazarins from the sea urchin Scaphechinus mirabilis. J. Nat. Prod. 2005, 68, 1390–1393. [Google Scholar] [CrossRef]
- Jeong, S.H.; Song, I.S.; Kim, H.K.; Lee, S.R.; Song, S.; Suh, H.; Yoon, Y.G.; Yoo, Y.H.; Kim, N.; Rhee, B.D.; et al. An analogue of resveratrol HS-1793 exhibits anticancer activity against MCF-7 cells via inhibition of mitochondrial biogenesis gene expression. Mol. Cells 2012, 34, 357–365. [Google Scholar] [CrossRef]
- Jeong, S.H.; Hanh, T.M.; Kim, H.K.; Lee, S.R.; Song, I.S.; Noh, S.J.; Song, S.; Suh, H.; Kim, N.; Rhee, B.D.; et al. HS-1793, a recently developed resveratrol analogue protects rat heart against hypoxia/reoxygenation injury via attenuating mitochondrial damage. Bioorg. Med. Chem. Lett. 2013, 23, 4225–4229. [Google Scholar] [CrossRef]
- Kang, S.; Kim, N.; Joo, H.; Youm, J.B.; Park, W.S.; Warda, M.; Kim, H.; Cuong, D.V.; Kim, T.; Kim, E.; et al. Changes of Cytosolic Ca2+ under Metabolic Inhibition in Isolated Rat Ventricular Myocytes. Korean J. Physiol. Pharmacol. 2005, 9, 291–298. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jeong, S.H.; Kim, H.K.; Song, I.-S.; Lee, S.J.; Ko, K.S.; Rhee, B.D.; Kim, N.; Mishchenko, N.P.; Fedoryev, S.A.; Stonik, V.A.; et al. Echinochrome A Protects Mitochondrial Function in Cardiomyocytes against Cardiotoxic Drugs. Mar. Drugs 2014, 12, 2922-2936. https://doi.org/10.3390/md12052922
Jeong SH, Kim HK, Song I-S, Lee SJ, Ko KS, Rhee BD, Kim N, Mishchenko NP, Fedoryev SA, Stonik VA, et al. Echinochrome A Protects Mitochondrial Function in Cardiomyocytes against Cardiotoxic Drugs. Marine Drugs. 2014; 12(5):2922-2936. https://doi.org/10.3390/md12052922
Chicago/Turabian StyleJeong, Seung Hun, Hyoung Kyu Kim, In-Sung Song, Seon Joong Lee, Kyung Soo Ko, Byoung Doo Rhee, Nari Kim, Natalia P. Mishchenko, Sergey A. Fedoryev, Valentin A. Stonik, and et al. 2014. "Echinochrome A Protects Mitochondrial Function in Cardiomyocytes against Cardiotoxic Drugs" Marine Drugs 12, no. 5: 2922-2936. https://doi.org/10.3390/md12052922
APA StyleJeong, S. H., Kim, H. K., Song, I.-S., Lee, S. J., Ko, K. S., Rhee, B. D., Kim, N., Mishchenko, N. P., Fedoryev, S. A., Stonik, V. A., & Han, J. (2014). Echinochrome A Protects Mitochondrial Function in Cardiomyocytes against Cardiotoxic Drugs. Marine Drugs, 12(5), 2922-2936. https://doi.org/10.3390/md12052922