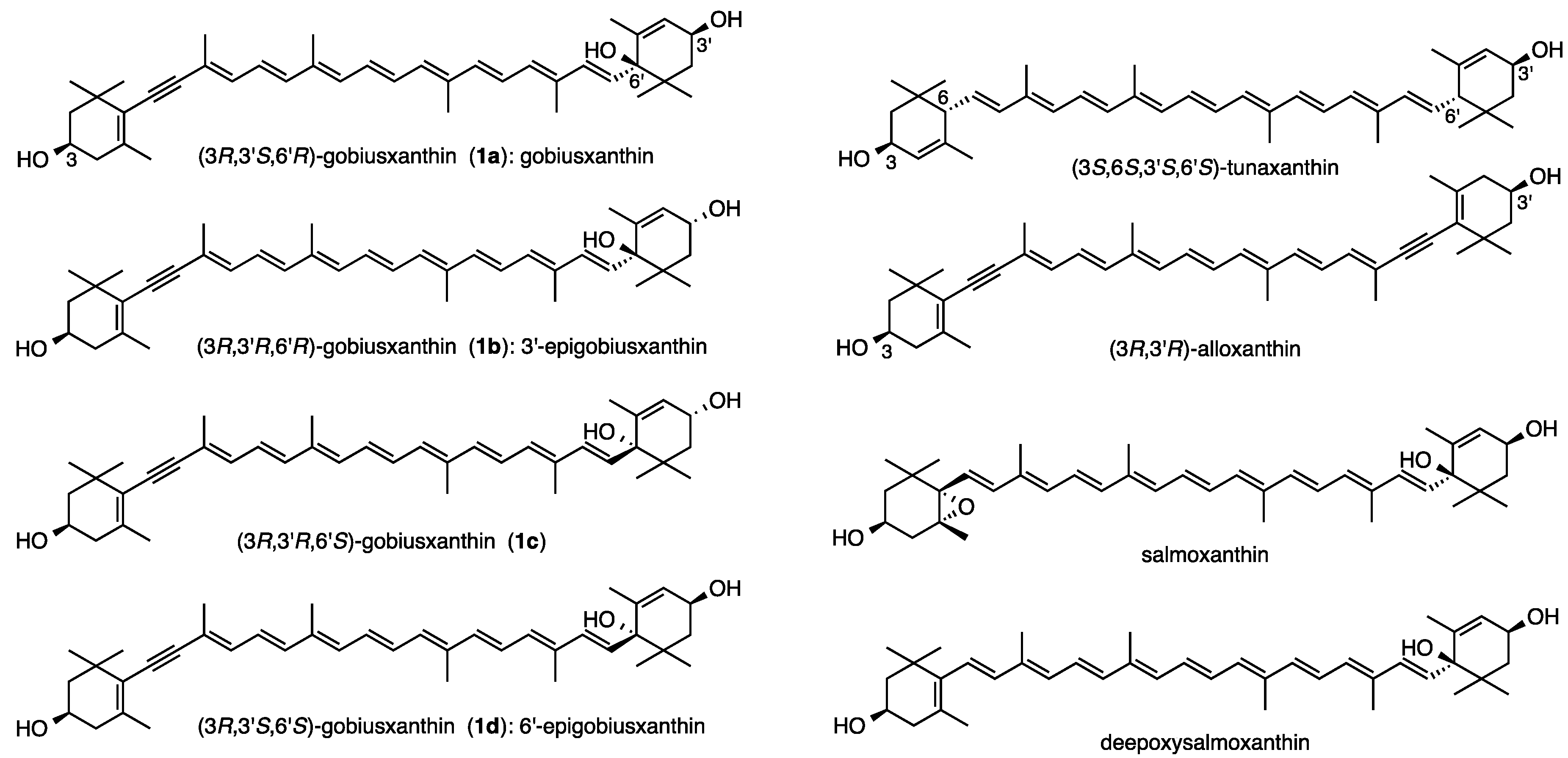
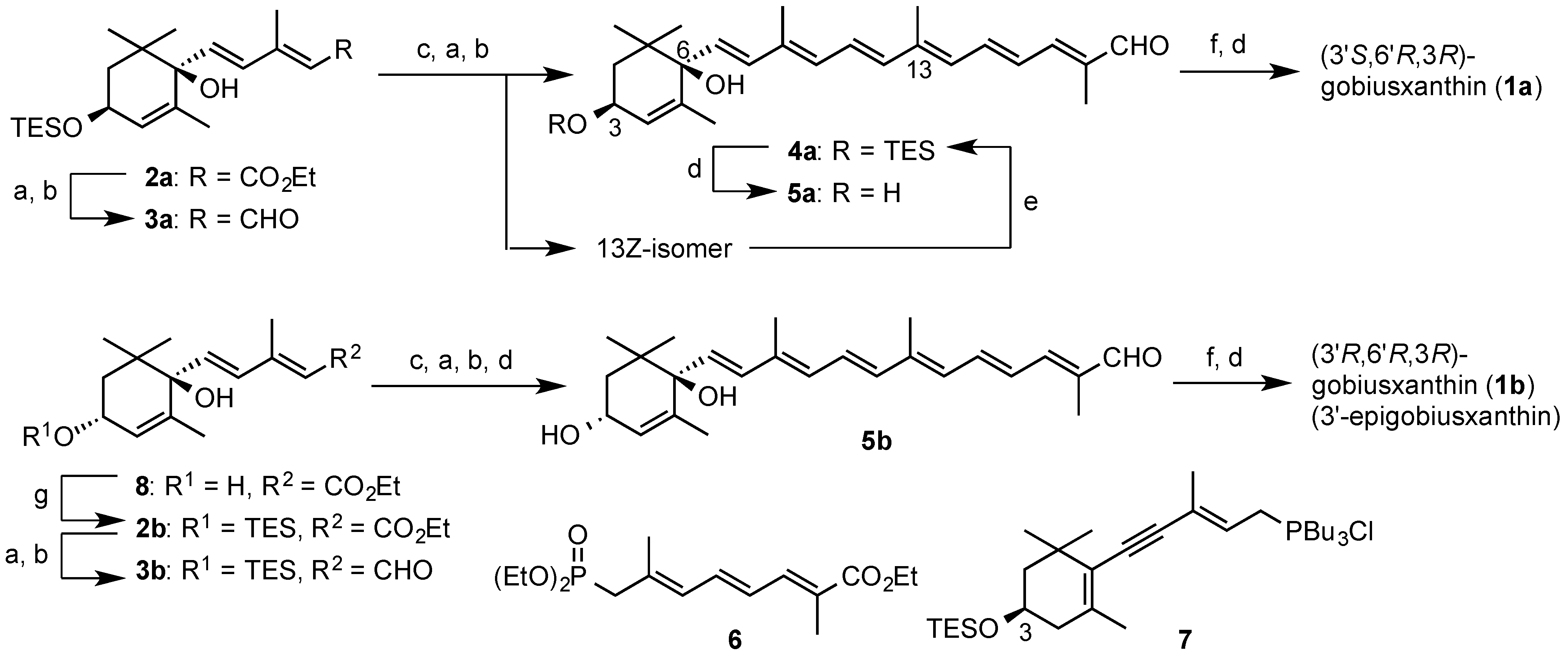
3.2. Synthesis of Gobiusxanthin (1a) and 3'-Epigobiusxanthin (1b)
(2
E,4
E)-5-[(1
R,4
S)-1-Hydroxy-2,6,6-trimethyl-4-triethylsilyloxycyclohex-2-en-1-yl]-3-methyl-penta-2,4-dienal (
3a). A solution of ester
2a [
4] (1.27 g, 3.11 mmol) in dry ether (15 mL) was added dropwise to a stirred suspension of LAH (118 mg, 3.11 mmol) in dry ether (20 mL) at 0 °C. After being stirred at 0 °C for 10 min, the excess of LAH was decomposed by dropwise addition of moist ether and the mixture was filtered through a pad of Celite. The filtrate was dried and evaporated to give the crude alcohol, which without purification, was dissolved in ether (15 mL) and hexane (15 mL) and stirred with MnO
2 (7.5 g) at rt for 30 min. After MnO
2 was filtered off, the filtrate was concentrated. The resulting residue was purified by flash CC (AcOEt-hexane, 3:7) to afford the aldehyde
3a (697 mg, 61% from
2a): [α]
D22 −149.1 (
c 1.26, MeOH); IR ν 3600, 3447 (OH), 1661 (conj. CO), 1631, 1600 (C=C);
1H-NMR (300 MHz)
δ 0.63 (6H, q,
J 8, SiC
H2 × 3), 0.95, 0.97 (each 3H, s,
gem-Me), 0.98 (9H, t,
J 8, CH
2Me × 3), 1.64 (3H, t,
J 1.5, 5-Me), 1.68 (2H, m, 2-H
2), 2.27 (3H, d,
J 1, 9-Me), 4.26 (1H, m, 3-H), 5.56 (1H, br s, 4-H), 6.01 (1H, br d,
J 8, 10-H), 6.17 (1H, d,
J 16, 7-H), 6.56 (1H, d,
J 16, 8-H), 10.12 (1H, d,
J 8, CHO);
13C-NMR (75 MHz)
δ 4.79 (C × 3), 6.80 (C × 3), 13.32, 19.26, 24.38, 24.46, 38.52, 42.60, 65.29, 77.39, 129.18, 129.43, 133.29, 136.77, 138.82, 153.68, 191.49; HRMS (ESI)
m/
z calcd for C
21H
36O
3NaSi (M + Na)
+ 387.2326, found 387.2317.
(2
E,4
E,6
E,8
E,10
E,12
E)-13-[(1
R,4
S)-1-Hydroxy-2,6,6-trimethyl-4-triethylsilyloxycyclohex-2-en-1-yl]-2,7,11-trimethyltrideca-2,4,6,8,10,12-hexaenal (
4a). To a solution of C
10-phosphonate
6 [
7] (1.90 g, 5.76 mmol) and
N,
N'-dimethylpropyleneurea (DMPU) (0.69 mL, 5.76 mmol) in dry THF (20 mL) was added
n-BuLi (1.63 M in hexane; 3.64 mL, 5.94 mmol) at −20 °C. After being stirred at −20 °C for 15 min, a solution of the aldehyde
3a (700 mg, 1.92 mmol) in dry THF (10 mL) was added to the reaction mixture and stirring was continued at −20 °C for 20 min. After being quenched with saturated aq. NH
4Cl, the mixture was extracted with AcOEt. The extracts were washed with brine, dried and evaporated to give a residue, which was purified by flash CC (ether-hexane, 3:7) to provide an isomeric mixture of the hexaenoate (786 mg, 76%) as an yellow oil: IR ν 3599, 3518 (OH), 1692 (conj. CO), 1614, 1602, 1555 (C=C); HRMS (ESI)
m/
z calcd for C
33H
52O
4NaSi (M + Na)
+ 563.3527, found 353.3516.
A solution of this isomeric mixture in dry ether (20 mL) was added dropwise to a stirred suspension of LAH (83 mg, 2.2 mmol) in dry ether (20 mL) at 0 °C. After being stirred at 0 °C for 15 min, the excess of LAH was decomposed by dropwise addition of moist ether and the mixture was filtered through a pad of Celite. The filtrate was dried and evaporated to give the crude diol, which without purification, was dissolved in THF (1 mL), ether (15 mL) and hexane (15 mL), and then stirred with MnO2 (5.3 g) at room temperature (rt) for 30 min. After MnO2 was filtered off, the filtrate was concentrated. The resulting residue was purified by flash CC (AcOEt-hexane, 3:7) and then preparative HPLC [LiChrosorb Si 60 (7 μm) 2 × 25 cm; ether-hexane, 27:73] to afford all-E-apocarotenal 4a (434 mg, 46% from 3a) and its 13Z-isomer (149 mg, 16% from 3a), as an orange foam, respectively.
all-E-Isomer 4a: UV-VIS λ 421; IR ν 3598, 3521 (OH), 1661 (conj. CO), 1611, 1601, 1550 (C=C); 1H-NMR (300 MHz) δ 0.63 (6H, q, J 8, SiCH2 × 3), 0.94, 0.98 (each 3H, s, gem-Me), 0.98 (9H, t, J 8, CH2Me × 3), 1.65 (3H, t, J 1.5, 5-Me), 1.68 (2H, m, 2-H2), 1.88, 1.94, 2.04 (each 3H, s, 9-Me, 13-Me, 13'-Me), 4.25 (1H, m, 3-H), 5.53 (1H, m, 4-H), 5.67 (1H, d, J 15.5, 7-H), 6.23 (1H, br d, J 11.5, 10-H), 6.31 (1H, br d, J 12, 14-H), 6.38 (1H, d, J 15, 12-H), 6.45 (1H, d, J 15.5, 8-H), 6.68 (1H, dd, J 12, 14.5, 15'-H), 6.77 (1H, dd, J 11.5, 15, 11-H), 6.96 (1H, br d, J 12, 14'-H), 7.03 (1H, dd, J 12, 14.5, 15-H), 9.45 (1H, s, CHO); 13C-NMR (75 MHz) δ 4.82 (C × 3), 6.83 (C × 3), 9.54, 13.00, 13.14, 19.35, 24.40, 24.45, 38.39, 42.73, 65.44, 77.42, 127.40 (C × 2), 128.49, 130.70, 131.03, 131.20, 134.45, 136.50, 136.85 (C × 2), 137.62, 137.82, 141.53, 148.85, 194.41; HRMS (ESI) m/z calcd for C31H48O3NaSi (M + Na)+ 519.3265, found 519.3257.
13Z-Isomer: UV-VIS λ 295, 410; IR ν 3598, 3521 (OH), 1660 (conj. CO), 1612, 1598, 1565 (C=C); 1H-NMR (300 MHz) δ 0.63 (6H, q, J 8, SiCH2 × 3), 0.94, 0.99 (each 3H, s, gem-Me), 0.99 (9H, t, J 8, CH2Me × 3), 1.66–1.69 (5H, overlapped, 5-Me, 2-Hα, 2-Hβ), 1.88, 1.95, 2.05 (each 3H, s, 9-Me, 13-Me, 13'-Me), 4.26 (1H, m, 3-H), 5.53 (1H, s, 4-H), 5.69 (1H, d, J 16, 7-H), 6.16 (1H, d, J 12, 14-H), 6.27 (1H, d, J 11, 10-H), 6.47 (1H, dd, J 16, 8-H), 6.62 (1H, dd, J 11.5, 14, 15'-H), 6.75 (1H, dd, J 11, 15, 11-H), 6.90 (1H, d, J 15, 12-H), 6.97 (1H, d, J 11.5, 14'-H), 7.03 (1H, dd, J 12, 14, 15-H), 9.45 (1H, s, CHO); 13C-NMR (75 MHz) δ 4.82, (C × 3) 6.84 (C × 3), 9.52, 13.20, 19.37, 20.92, 24.41, 24.47, 38.42, 42.73, 65.45, 77.42, 126.60, 128.38, 128.55, 128.57, 129.36, 130.96, 131.09, 134.39, 136.36, 136.91, 137.10, 137.80, 140.22, 148.96, 194.49; HRMS (ESI) m/z calcd for C31H48O3NaSi (M + Na)+ 519.3265, found 519.3255.
Isomerization of 13Z-isomer of compound 4a. A solution (2 mL) prepared from PdCl2(MeCN)2 (13 mg), Et3N (7 mL) and water (1.2 mL) in MeCN (8.8 mL) was added to a solution of 13Z-isomer of compound 4a (103 mg) in MeCN (18 mL) and the mixture was stirred at rt for 3 h. The solvent was evaporated off to give a residue, which was purified by the same method described above to provide the all-E-isomer 4a (54 mg, 52%).
(2E,4E,6E,8E,10E,12E)-13-[(1R,4S)-1,4-Dihydroxy-2,6,6-trimethylcyclohex-2-en-1-yl]-2,7,11-trimethyltrideca-2,4,6,8,10,12-hexaenal (5a). To a stirred solution of TES ether 4a (434 mg, 0.86 mmol) in dry THF (9 mL) was added AcOH (1 M in THF; 0.30 mL, 0.30 mmol) and then TBAF (1 M in THF; 1.31 mL, 1.31 mmol) at rt. After being stirred at rt for 5 min, the mixture was evaporated to afford a residue, which was purified by flash CC (acetone-hexane, 35:65) to afford compound 5a (280 mg, 84%) as orange solids: UV-VIS λ 421; IR ν 3603, 3446 (OH), 1660 (conj. CO), 1611, 1601, 1550 (C=C); 1H-NMR (500 MHz) δ 0.94, 1.02 (each 3H, s, gem-Me), 1.53, 1.62 (each 1H, br s, OH × 2), 1.67 (1H, dd, J 7.5, 13.5, 2-H), 1.68 (3H, t, J 2, 5-Me), 1.81 (1H, dd, J 6.5, 13.5, 2-H), 1.89 (3H, s, 13'-Me), 1.94 (3H, s, 9-Me), 2.04 (3H, s, 13-Me), 4.25 (1H, m, 3-H), 5.64 (1H, br s, 4-H), 5.69 (1H, d, J 15.5, 7-H), 6.24 (1H, br d, J 11.5, 10-H), 6.31 (1H, br d, J 12, 14-H), 6.39 (1H, d, J 15, 12-H), 6.42 (1H, d, J 15.5, 8-H), 6.70 (1H, dd, J 11.5, 14.5, 15'-H), 6.76 (1H, dd, J 11.5, 15, 11-H), 6.96 (1H, br d, J 11.5, 14'-H), 7.02 (1H, dd, J 12, 14.5, 15-H), 9.45 (1H, s, CHO); 13C-NMR (125 MHz) δ 9.59 (13'-Me), 13.03 (13-Me), 13.22 (9-Me), 19.21 (5-Me), 24.37 (1-Me), 24.40 (1-Me), 38.19 (C1), 42.44 (C2), 65.24 (C3), 77.83 (C6), 127.29 (C11), 127.35 (C4), 127.55 (C15'), 130.74 (C7), 131.19 (C14), 131.50 (C10), 134.44 (C8), 136.31 (C9), 136.99 (C13'), 137.10 (C12), 137.59 (C15), 138.85 (C5), 141.48 (C13), 148.83 (C14'), 194.47 (CHO); HRMS (ESI) m/z calcd for C25H34O3NaSi (M + Na)+ 405.2400, found 405.2393.
(3
R,3'
S,6'
R)-Gobiusxanthin (
1a). NaOMe (1 M in MeOH; 0.45 mL, 0.45 mmol) was added to a solution of the phosphonium salt
7 [
6] (192 mg, 0.34 mmol) and the apocarotenal
5a (86 mg, 0.23 mmol) in CH
2Cl
2 (10 mL) at rt. After being stirred at rt for 10 min, the mixture was poured into saturated aq. NH
4Cl and extracted with AcOEt. The extracts were washed with brine, dried and evaporated to give a residue, which was purified by flash CC (acetone-hexane, 3:7) to provide the crude condensed products. This was dissolved in dry THF (9 mL) and then AcOH (1 M in THF; 0.30 mL, 0.30 mmol) and TBAF (1 M in THF; 0.42 mL, 0.42 mmol) were added to it at rt. After being stirred at rt for 30 min, the mixture was concentrated. The resulting residue was purified by flash CC (AcOEt-CH
2Cl
2-MeOH, 25:72:3) and then preparative HPLC [LiChrosorb Si 60 (7 μm) 2 × 25 cm; AcOEt-CH
2Cl
2-MeOH, 10:40:1] to give (3
R,3'
S,6'
R)-gobiusxanthin (
1a) (90 mg, 69% from
5a) as orange solids.
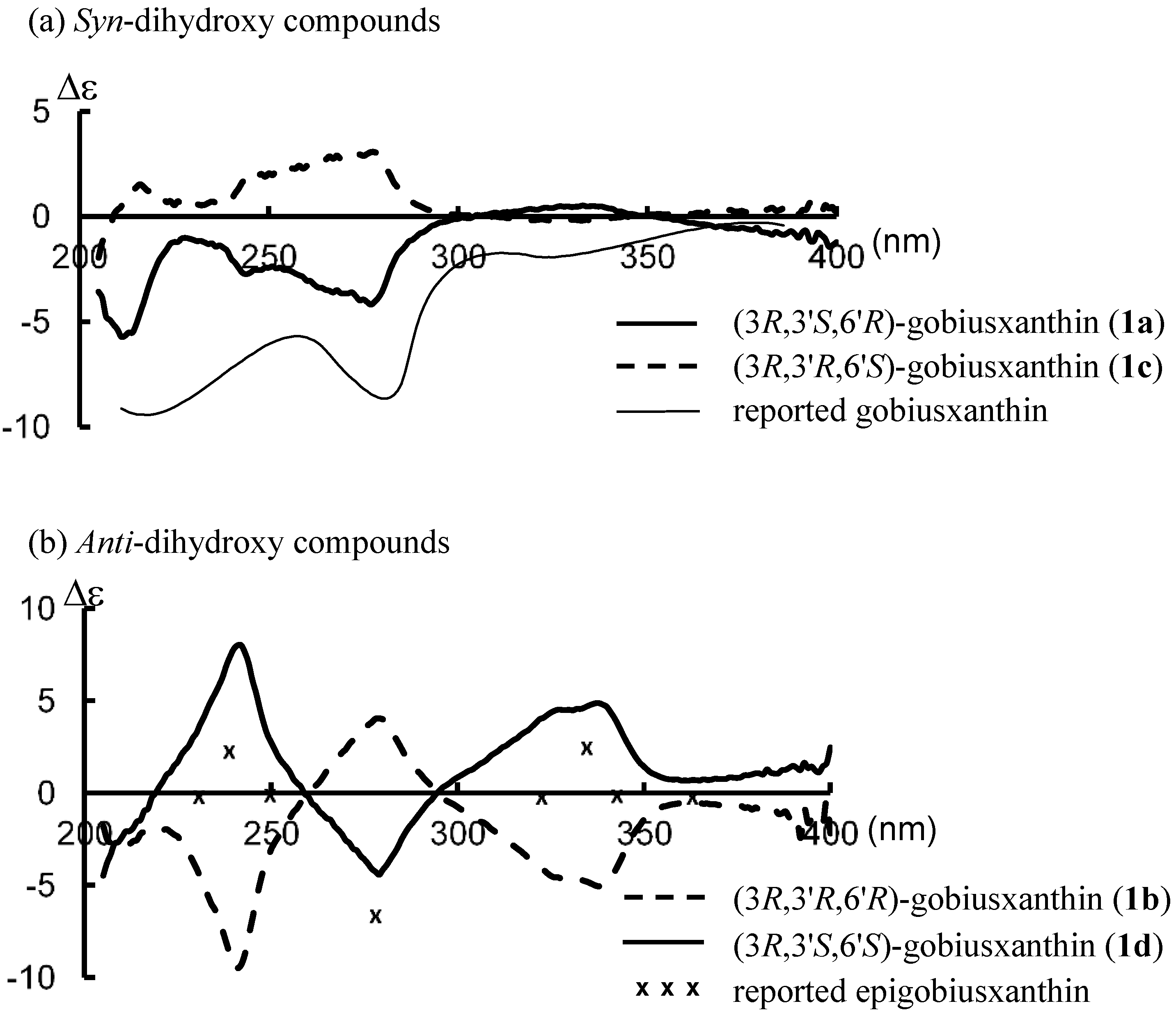
1H-NMR spectral data of this synthetic
1a were identical with those reported [
1]: UV-VIS λ 277, 424, 447, 477; CD (9.83 × 10
−5 mol/L, EPA) λ(∆ε) 211 (−5.7), 228 (−1.0), 244 (−2.7), 277 (−4.2), 304 (0), 333 (+0.5), 351 (0); IR ν 3604, 3448 (OH), 2172 (C≡C), 1568 (C=C);
1H-NMR (500 MHz);
δ 0.94 (3H, s, 1'-Meα), 1.02 (3H, s, 1'-Meβ), 1.15 (3H, s, 1-Meα), 1.20 (3H, s, 1-Meβ), 1.45 (1H, t,
J 12, 2-Hβ), 1.50, 1.61. 1.63 (each 1H, br s, OH × 3), 1.67 (1H, dd,
J 7, 13.5, 2'-H), 1.67 (3H, t,
J 1.5, 5'-Me), 1.81 (1H, dd,
J 6, 13.5, 2'-H), 1.83 (1H, ddd,
J 2, 3.5, 12, 2-Hα), 1.92 (6H, s, 5-Me, 9'-Me), 1.95, 1.97 (each 3H, s, 13-Me, 13'-Me), 2.01 (3H, s, 9-Me), 2.07 (1H, ddd,
J 1.5, 9.5, 18, 4-Hβ), 2.43 (1H, ddd,
J 1.5, 5.5, 18, 4-Hα), 3.99 (1H, m, 3-H), 4.24 (1H, m, 3'-H), 5.63 (1H, d,
J 15.5, 7'-H), 5.63 (1H, m, 4'-H), 6.22 (1H, br d
J 11.5, 10'-H), 6.27 (2H, br d,
J 9, 14-H, 14'H), 6.35 (1H, d,
J 14, 12-H), 6.37 (1H, d,
J 15, 12'-H), 6.39 (1H, d,
J 15.5, 8'-H), 6.45 (1H, dd-like,
J 1, 11.5, 10-H), 6.51 (1H, dd,
J 11.5, 14, 11-H), 6.62 (1H, dd,
J 11.5, 15, 11'-H), 6.64 (2H, m, 15-H, 15'-H);
13C-NMR (125 MHz)
δ 12.73, 12.81 (13-M, 13'-Me), 13.13 (9'-Me), 18.04 (9-Me), 19.22 (5'-Me), 22.47 (5-Me), 24.37 (1'-Me), 24.40 (1'-Me), 28.76 (1-Me), 30.50 (1-Me), 36.61 (C1), 38.16 (C1'), 41.46 (C4), 42.47 (C2'), 46.68 (C2), 64.86 (C3), 65.28 (C3'), 77.87 (C6'), 89.00 (C7), 98.62 (C8), 118.96 (C9), 124.16 (C11), 124.22 (C6), 124.84 (C11'), 127.22 (C4'), 129.67 (C7'), 130.08, 130.42 (C15, C15'), 131.98 (C10'), 132.74, 133.44 (C14, C14'), 134.53 (C9'), 134.60 (C8'), 135.17 (C10), 136.22, 136.63 (C13, C13'), 137.26 (C5), 137.98, 138.05 (C12, C12'), 139.01 (C5'); HRMS (ESI)
m/
z calcd for C
40H
54O
3Na (M + Na)
+ 605.3965, found 605.3962.
Ethyl (2
E,4
E)-5-[(1
R,4
R)-1-Hydroxy-2,6,6-trimethyl-4-triethylsilyloxycyclohex-2-en-1-yl]-3-methylpenta-2,4-dienoate (
2b). To a stirred solution of
anti-diol
8 [
4] (465 mg, 1.58 mmol), Et
3N (0.66 mL, 5.4 mmol) and
N,N-dimethyl-4-aminopyridine (19 mg, 0.16 mmol) in dry CH
2Cl
2 (7 mL) was added TESCl (0.40 mL, 2.4 mmol) at 0 °C. The mixture was stirred at 0 °C for 30 min, poured into saturated aq. NH
4Cl and extracted AcOEt. The extracts were washed with brine, dried and evaporated to give a residue, which was purified by flash CC (AcOEt- hexane, 1:4) to afford TES ether
2b (645 mg, quant.) as a colorless oil: [α]
D26 157.89 (
c 0.91, MeOH); IR ν 3605, 3473 (OH), 1704 (conj. CO), 1632, 1612 (C=C);
1H-NMR (300 MHz)
δ 0.62 (6H, q,
J 8, SiC
H2 × 3), 0.87, 1.02 (each 3H, s,
gem-Me), 0.98 (9H, t,
J 8, SiCH
2Me × 3), 1.28 (3H, t,
J 7.5, OCH
2Me), 1.60 (3H, m, 5-Me), 1.66 (2H, d-like,
J 8, 2-H
2), 2.27 (3H, br s, 9-Me), 4.17 (2H, q,
J 7.5, OCH
2), 4.28 (1H, m, 3-H), 5.47 (1H, br s, 4-H), 5.81 (1H, br s, 10-H), 6.14 (1H, d,
J 16, 7-H), 6.34 (1H, d,
J 16, 8-H);
13C-NMR (75 MHz)
δ 4.82 (C × 3), 6.87 (C × 3), 14.11, 14.31, 17.53, 22.66, 25.16, 39.65, 44.53, 59.71, 65.91, 79.09, 119.38, 128.70, 132.51, 136.94, 138.15, 151.57, 167.14; HRMS (ESI)
m/
z calcd for C
23H
40O
4Si (MH)
+ 409.2769, found 409.2764.
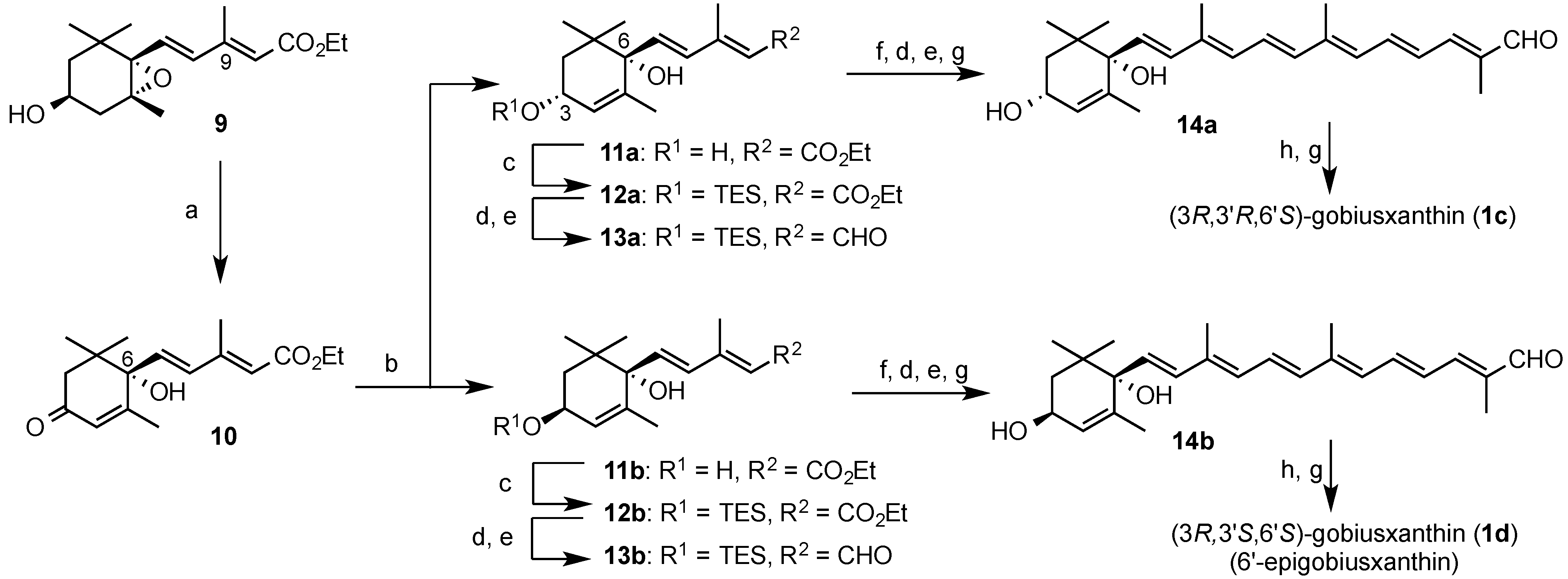
In the same procedure as preparation of (3R,3'S,6'R)-gobiusxanthin (1a), (3R,3'R,6'R)-gobiusxanthin (1b) was prepared from the above (3R,6R)-dienoate 2b.
(3R,6R)-Dienal 3b: [α]D27 −194.8 (c 0.98, MeOH); IR ν 3608, 3477 (OH), 1662 (conj. CO), 1627, 1597, 1580 (C=C); 1H-NMR (300 MHz) δ 0.63 (6H, q, J 8, SiCH2 × 3), 0.88, 1.03 (each 3H, s, gem-Me), 0.97 (9H, t, J 8, CH2Me × 3), 1.60 (3H, t, J 2, 5-Me), 1.68 (2H, d-like, J 8, 2-H2), 2.25 (3H, d, J 1, 9-Me), 4.30 (1H, m, 3-H), 5.48 (1H, m, 4-H), 5.98 (1H, br d, J 8, 10-H), 6.31, 6.46 (each 1H, d, J 15.5, 7-H, 8-H), 10.10 (1H, J 8, CHO); 13C-NMR (75 MHz) δ 4.83 (C × 3), 6.83 (C × 3), 13.38, 17.48, 22.64, 25.18, 39.77, 44.62, 65.85, 79.16, 128.95, 129.65, 132.13, 136.70, 140.21, 154.02, 191.51; HRMS (ESI) m/z calcd for C21H36O3NaSi (M + Na)+ 387.2326, found 387.2319.
(3R,6R)-Hexaenel 5b: UV-VIS λ 420; IR ν 3604, 3446 (OH), 1660 (conj. CO), 1611, 1600, 1550 (C=C); 1H-NMR (500 MHz) δ 0.92, 1.04 (each 3H, s, gem-Me), 1.46 (1H, br s, 3-OH), 1.53 (1H, br d, J 2, 6-OH), 1.58 (1H, dd, J 10, 13.5, 2-H), 1.66 (3H, t, J 2, 5-Me), 1.79 (1H, ddd, J 1.5, 6.5, 13.5, 2-H), 1.88 (3H, d, J 0.5, 13'-Me), 1.94 (3H, d, J 0.5, 9-Me), 2.03 (3H, s, 13-Me), 4.30 (1H, m, 3-H), 5.57 (1H, quint-like, J 1.5, 4-H), 5.79 (1H, d, J 16, 7-H), 6,23 (1H, br d, J 11, 10-H), 6.31 (1H, br d, J 12, 14-H), 6.32 (1H, d, J 16, 8-H), 6.38 (1H, d, J 15, 12-H), 6.69 (1H, dd, J 11.5, 14.5, 15'-H), 6.75 (1H, dd, J 11 and 15, 11-H), 6.96 (1H, br d, J 11.5, 14'-H), 7.02 (1H, dd, J 12, 15, 15-H), 9.45 (1H, s, CHO); 13C-NMR (125 MHz) δ 9.59 (13'-Me), 13.03 (13-Me), 13.27 (9-Me), 17.89 (5-Me), 22.45, 25.05 (gem-Me), 39.91 (C1), 44.15 (C2), 65.80 (C3), 79.41 (C6), 127.25, 127.27 (C4, C4'), 127.58 (C15'), 131.23 (C14), 131.75 (C10), 132.15 (C7), 134.18 (C8), 136.39 (C9), 137.03 (C13'), 137.17 (C12), 137.58 (C15), 138.76 (C5), 141.48 (C13), 148.76 (C14'), 194.43 (CHO); HRMS (ESI) m/z calcd for C25H34O3Na (M + Na)+ 405.2400, found 405.2396.
(3R,3'R,6'R)-Gobiusxanthin (1b): UV-VIS λ 278, 424, 448, 478; CD (1.08 × 10−4 mol/L, EPA) λ(∆ε) 209 (−2.9), 219 (−1.9), 241 (−9.5), 259 (0), 279 (+4.0), 294 (0), 337 (−5.0), 369 (−0.6); IR ν 3606, 3446 (OH), 2172 (C≡C), 1568 (C=C); 1H-NMR (500 MHz) δ 0.92 (3H, s, 1'-Meα), 1.03 (H, s, 1'-Meβ), 1.14 (3H, s, 1-Meα), 1.20 (3H, s, 1-Meβ), 1.45 (1H, t, J 12, 2-Hβ), 1.57 (1H, dd, J 10 and 13, 2'-Hα), 1.66 (3H, t, J 1.5, 5'-Me), 1.79 (1H, ddd, J 1, 6, 13, 2'-Hβ), 1.84 (1H, ddd, J 1.5, 3.5, 12, 2-Hα), 1.92 (6H, s, 5-Me, 9'-Me), 1.95 (3H, s, 13'-Me), 1.97 (3H, s, 13-Me), 2.00 (3H, s, 9-Me), 2.07 (1H, br dd, J 9, 18, 4-Hβ), 2.43 (1H, ddd, J 1.5, 5.5, 18, 4-Hα), 3.99 (1H, m, 3-H), 4.29 (1H, m, 3'-H), 5.57 (1H, m, 4'-H), 5.73 (1H, d, J 15.5, 7'-H), 6.21 (1H, br d, J 11.5, 10'-H), 6.26, 6.27 (each 1H, br d, J 9, 14-H, 14'-H), 6.30 (1H, d, J 15.5, 8'-H), 6.35 (1H, d, J 14.5, 12-H), 6.36 (1H, d, J 15, 12'-H), 6.45 (1H, dd-like, J 1, 11.5, 10-H), 6.51 (1H, dd, J 11.5, 14.5, 11-H), 6.61 (1H, dd, J 11.5, 15, 11'-H), 6.64 (2H, m, 15-H, 15'-H); 13C-NMR (125 MHz) δ 12.75, 12.83 (13-Me, 13'-Me), 13.20 (9'-Me), 17.95 (5'-Me), 18.06 (9-Me), 22.48, 22.50 (5-Me, 1'-Meβ), 25.07 (1'-Meα), 28.79 (1-Meα), 30.53 (1-Meβ), 36.64 (C1), 39.93 (C1'), 41.51 (C4), 44.19 (C2'), 46.74 (C2), 64.91 (C3), 65.88 (C3'), 79.45 (C6'), 89.04 (C7), 98.66 (C8), 119.00 (C9), 124.19 (C11), 124.29 (C6), 124.86 (C11'), 127.14 (C4'), 130.13, 130.45, (C15, C15') 131.12 (C7'), 132.27 (C10'), 132.81 (C14'), 133.47 (C14), 134.43 (C8'), 134.62 (C9'), 135.20 (C10), 136.25 (C13), 136.67 (C13'), 137.26 (C5), 138.07, 138.09 (C12, C12'), 138.92 (C5'); HRMS (ESI) m/z calcd for C40H55O3 (M + H)+ 583.4146, found 583.4140.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






