
Synthesis and Antiproliferative Activity of Thiazolyl-bis-pyrrolo[2,3-b]pyridines and Indolyl-thiazolyl-pyrrolo[2,3-c]pyridines, Nortopsentin Analogues




Abstract
: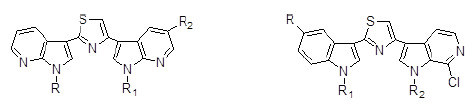
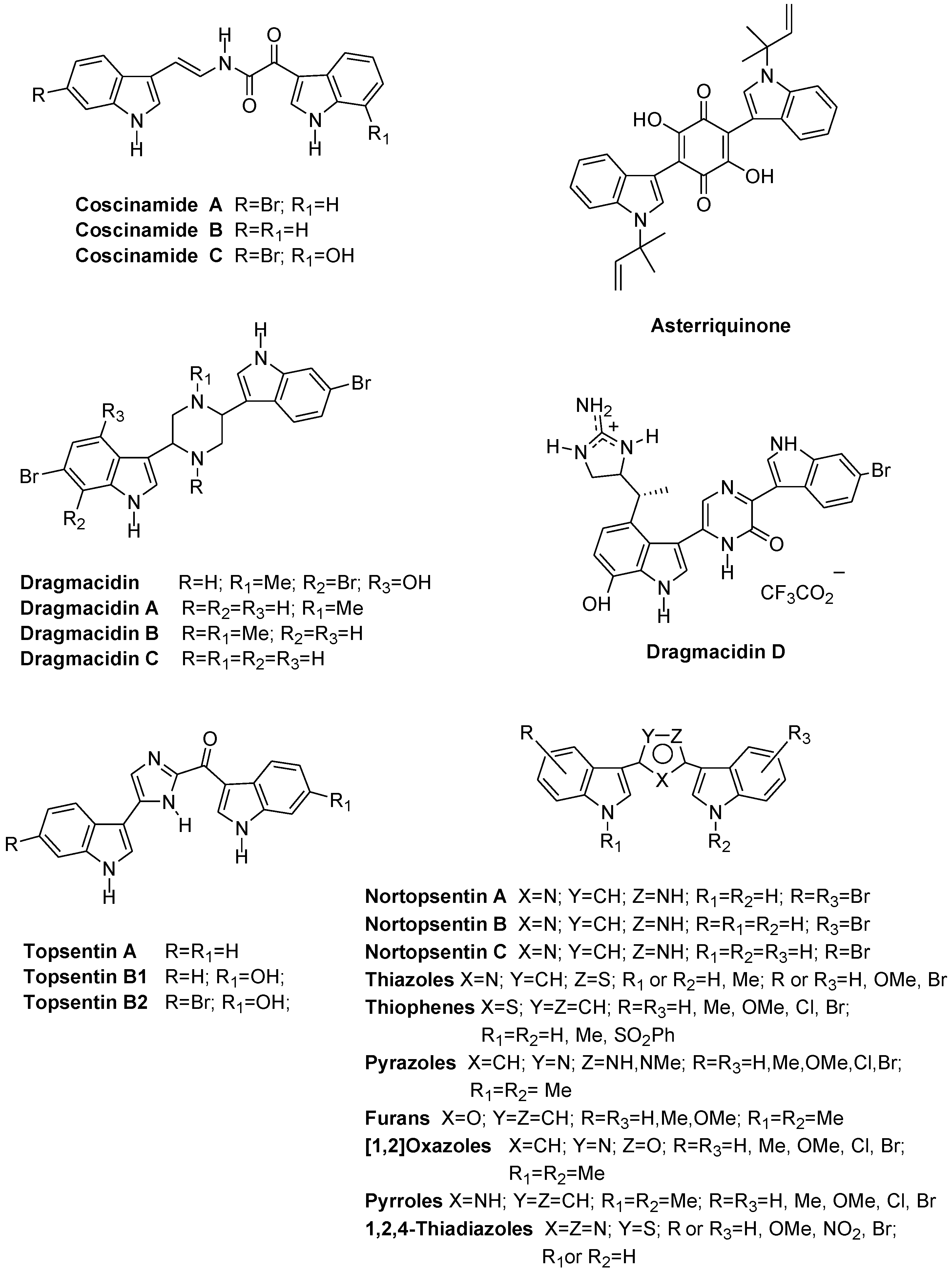
1. Introduction


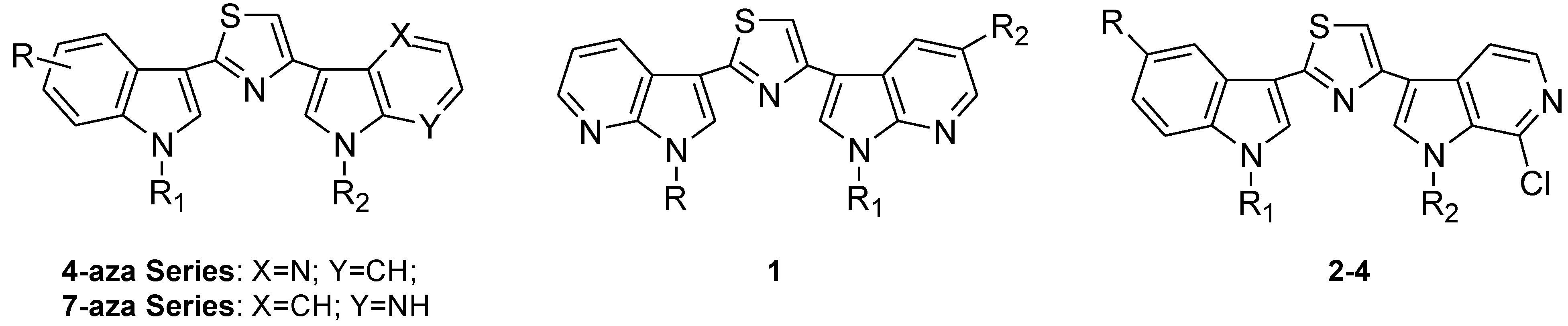
2. Results and Discussion
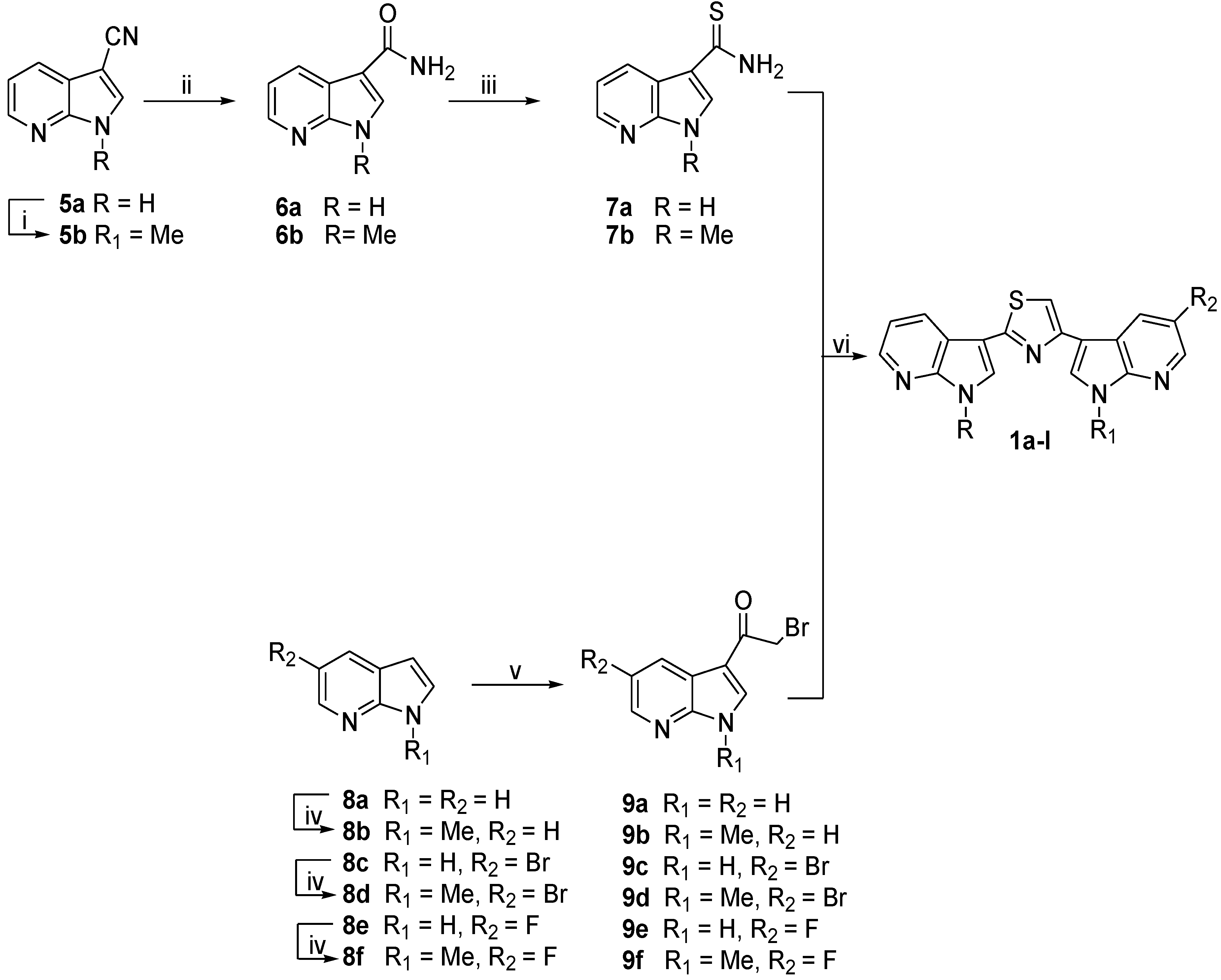
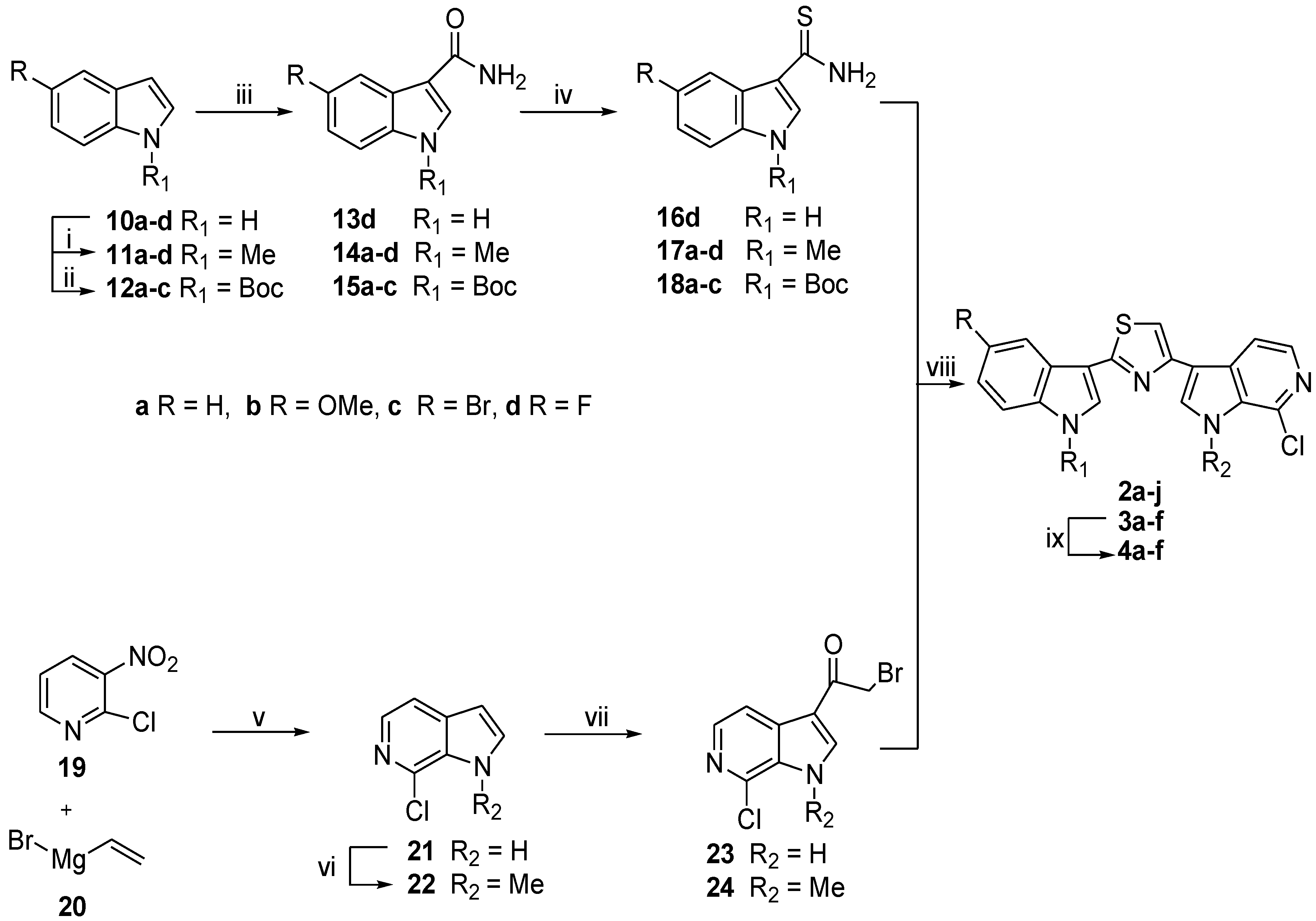
2.1. Chemistry



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | R | R1 | R2 | Yield% | Compd | R | R1 | R2 | Yield% |
|---|---|---|---|---|---|---|---|---|---|
| 1a | H | H | H | 63 | 2f | OMe | Me | Me | 79 |
| 1b | H | H | Br | 60 | 2g | Br | Me | Me | 98 |
| 1c | H | H | F | 75 | 2h | F | Me | Me | 95 |
| 1d | Me | H | H | 90 | 2i | F | H | Me | 82 |
| 1e | Me | H | Br | 94 | 2j | F | H | H | 66 |
| 1f | Me | H | F | 90 | 3a | H | Boc | H | 84 |
| 1g | Me | Me | H | 60 | 3b | OMe | Boc | H | 77 |
| 1h | Me | Me | Br | 90 | 3c | Br | Boc | H | 77 |
| 1i | Me | Me | F | 60 | 3d | H | Boc | Me | 91 |
| 1j | H | Me | H | 85 | 3e | OMe | Boc | Me | 92 |
| 1k | H | Me | Br | 80 | 3f | Br | Boc | Me | 91 |
| 1l | H | Me | F | 72 | 4a | H | H | H | 93 |
| 2a | H | Me | H | 98 | 4b | OMe | H | H | 62 |
| 2b | OMe | Me | H | 65 | 4c | Br | H | H | 98 |
| 2c | Br | Me | H | 93 | 4d | H | H | Me | 75 |
| 2d | F | Me | H | 97 | 4e | OMe | H | Me | 73 |
| 2e | H | Me | Me | 96 | 4f | Br | H | Me | 99 |
2.2. Biology
| Cell Lines | GI50 (µM) | Cell Lines | GI50 (µM) | Cell Lines | GI50 (µM) | |||
|---|---|---|---|---|---|---|---|---|
| 1k | 4c | 1k | 4c | 1k | 4c | |||
| Leukemia | CNS Cancer | Renal Cancer | ||||||
| CCRF-CEM | 6.81 | 3.16 | SF-268 | 6.01 | 4.06 | 786-0 | 9.65 | 1.37 |
| HL-60(TB) | >100 | 2.64 | SF-295 | 3.01 | 2.48 | A498 | 10.9 | 1.56 |
| K-562 | 8.76 | 2.73 | SF-539 | 27.7 | 1.87 | ACHN | 2.35 | 2.02 |
| MOLT-4 | >100 | 3.02 | SNB-19 | 6.74 | 3.25 | CAKI-1 | 1.56 | 1.96 |
| RPMI-8226 | >100 | 4.03 | SNB-75 | 2.18 | 2.37 | RXF393 | 2.05 | 1.48 |
| SR | ND b | 1.27 | U251 | 2.70 | 2.05 | SN12C | ND | 3.35 |
| TK-10 | 3.77 | 4.16 | ||||||
| Non-Small Cell Lung Cancer | Melanoma | UO-31 | ND | 0.93 | ||||
| A549/ATCC | 2.59 | 3.83 | LOX IMVI | 4.26 | 1.63 | |||
| EKVK | 1.27 | 3.11 | MALME-3M | 3.01 | ND | Prostate Cancer | ||
| HOP-62 | 2.39 | 2.11 | M14 | 4.06 | 2.22 | PC-3 | 4.35 | 3.86 |
| HOP-92 | 5.03 | 2.43 | MDA-MB-435 | 3.17 | 3.43 | DU-145 | 3.51 | 1.76 |
| NCI-H226 | 1.97 | 2.40 | SK-MEL-2 | 19.8 | 4.09 | |||
| NCI-H23 | 2.80 | 2.42 | SK-MEL-28 | ND | 1.85 | Breast Cancer | ||
| NCI-H322M | >100 | 3.54 | SK-MEL-5 | 2.05 | 2.61 | MCF7 | 6.77 | 2.20 |
| NCI-H460 | 2.98 | 2.16 | UACC-257 | ND | 2.68 | MDA-MB-231/ATCC | 3.02 | 1.68 |
| NCI-H522 | 4.86 | 2.28 | UACC-62 | 3.47 | 2.19 | HS 578T | 2.43 | 3.70 |
| BT-549 | 20.5 | 4.70 | ||||||
| T-47D | 1.80 | 3.12 | ||||||
| Colon Cancer | Ovarian Cancer | MDA-MB-468 | 0.81 | 1.18 | ||||
| COLO-205 | ND | 1.80 | IGROV1 | 2.21 | 2.51 | |||
| HCC-2998 | >100 | 2.22 | OVCAR-3 | 2.91 | 3.45 | |||
| HCT-116 | 2.91 | 2.35 | OVCAR-4 | 2.03 | 3.25 | |||
| HCT-15 | 13.7 | 1.40 | OVCAR-5 | >100 | 3.14 | |||
| HT29 | 6.75 | 2.66 | OVCAR-8 | 3.72 | 3.65 | |||
| KM12 | 5.70 | 2.15 | NCI/ADR-RES | 3.88 | 2.71 | |||
| SW-620 | 4.46 | 1.92 | SK-OV-3 | 3.25 | 2.78 | |||

2.2.1. Cell Cycle Alterations
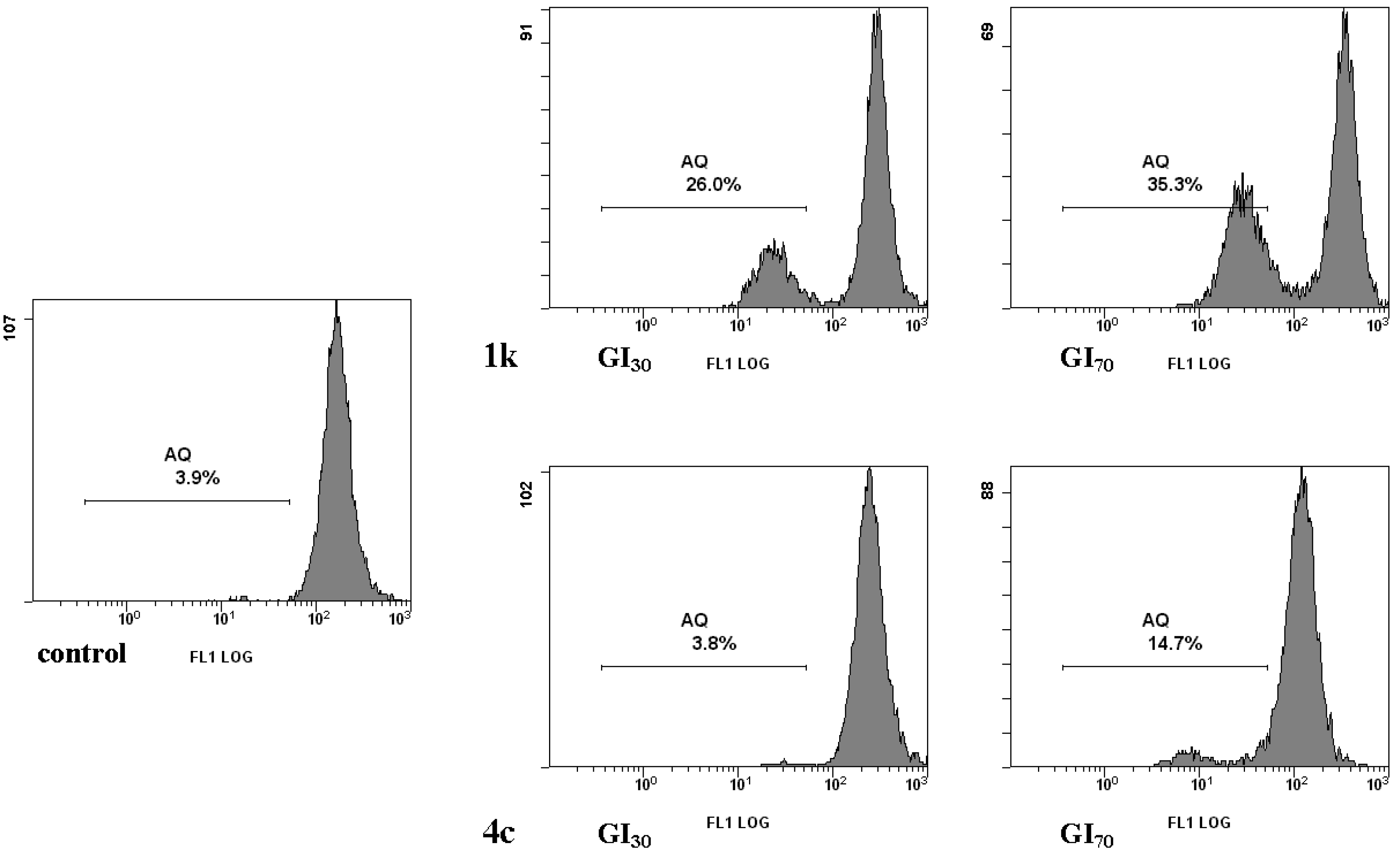
2.2.2. Cell Death



3. Experimental Section
3.1. Chemistry
3.1.1. General
3.1.2. Synthesis of 1-Methyl-1H-pyrrolo[2,3-b]pyridine-3-carbonitrile (5b)
3.1.3. General Procedure for the Synthesis of 1H-Pyrrolo[2,3-b]pyridine-3-carboxamides (6a,b)
1H-Pyrrolo[2,3-b]pyridine-3-carboxamide (6a)
1-Methyl-1H-pyrrolo[2,3-b]pyridine-3-carboxamide (6b)
3.1.4. General Procedure for the Synthesis of 1H-Pyrrolo[2,3-b]pyridine-3-carbothioamides (7a,b)
1H-Pyrrolo[2,3-b]pyridine-3-carbothioamide (7a)
1-Methyl-1H-Pyrrolo[2,3-b]pyridine-3-carbothioamide (7b)
3.1.5. General Procedure for the Synthesis of 1-Methyl-1H-pyrrolo[2,3-b]pyridines (8b,d,f)
1-Methyl-1H-pyrrolo[2,3-b]pyridine (8b)
5-Bromo-1-methyl-1H-pyrrolo[2,3-b]pyridine (8d)
5-Fluoro-1-methyl-1H-pyrrolo[2,3-b]pyridine (8f)
3.1.6. General Procedure for the Synthesis of 2-Bromo-1-(1H-pyrrolo[2,3-b]pyridin-3-yl)ethanones (9a–f)
2-Bromo-1-(1H-pyrrolo[2,3-b]pyridin-3-yl)pthanone (9a)
2-Bromo-1-(1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)ethanone (9b)
2-Bromo-1-(5-bromo-1H-pyrrolo[2,3-b]pyridin-3-yl)ethanone (9c)
2-Bromo-1-(5-bromo-1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)ethanone (9d)
2-Bromo-1-(5-fluoro-1H-pyrrolo[2,3-b]pyridin-3-yl)ethanone (9e)
2-Bromo-1-(5-fluoro-1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)ethanone (9f)
3.1.7. General Procedure for the Synthesis of Substituted-(1,3-thiazole-2,4-diyl)-bis(1H-pyrrolo[2,3-b]Pyridines (1a–l)
3,3′-(1,3-Thiazole-2,4-diyl)bis(1H-pyrrolo[2,3-b]pyridine) (1a)
5-Bromo-3-[2-(1H-pyrrolo[2,3-b]pyridin-3-yl)-1,3-thiazol-4-yl)-1H-pyrrolo[2,3-b]pyridine (1b)
5-Fluoro-3-[2-(1H-pyrrolo[2,3-b]pyridin-3-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[2,3-b]pyridine (1c)
1-Methyl-3-[4-(1H-pyrrolo[2,3-b]pyridin-3-yl)-1,3-thiazol-2-yl]-1H-pyrrolo[2,3-b]pyridine (1d)
3-[4-(5-Bromo-1H-pyrrolo[2,3-b]pyridin-3-yl)-1,3-thiazol-2-yl]-1-methyl-1H-pyrrolo[2,3-b]pyridine (1e)
3-[4-(5-Fluoro-1H-pyrrolo[2,3-b]pyridin-3-yl)-1,3-thiazol-2-yl]-1-methyl-1H-pyrrolo[2,3-b]pyridine (1f)
3,3′-(1,3-Thiazole-2,4-diyl)bis(1-methyl-1H-pyrrolo[2,3-b]pyridine) (1g)
5-Bromo-1-methyl-3-[2-(1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-1,3-thiazol-4-yl]-1H pyrrolo[2,3-b]pyridine (1h)
5-Fluoro-1-methyl-3-[2-(1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[2,3-b]pyridine (1i)
1-Methyl-3-[4-(1H-pyrrolo[2,3-b]pyridin-3-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[2,3-b]pyridine (1j)
5-Bromo-1-methyl-3-[2-(1H-pyrrolo[2,3-b]pyridin-3-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[2,3-b]pyridine (1k)
5-Fluoro-1-methyl-3-[2-(1H-pyrrolo[2,3-b]pyridin-3-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[2,3-b]pyridine (1l)
3.1.8. Synthesis of 7-Chloro-1-methyl-1H-pyrrolo[2,3-c]pyridine (22)
3.1.9. General Procedure for the Synthesis of 1-(7-Chloro-1H-pyrrolo[2,3-c]pyridin-3-yl) ethanones (23, 24)
2-Bromo-1-(7-Chloro-1H-pyrrolo[2,3-c]pyridin-3-yl)ethanone (23)
2-Bromo-1-(7-chloro-1-methyl-1H-pyrrolo[2,3-c]pyridin-3-yl)ethanone (24)
3.1.10. General Procedure for the Synthesis of Indolyl-thiazolyl-pyrrolo[2,3-c]pyridines (2a–j), (3a–f)
7-Chloro-3-[2-(1-methyl-1H-indol-3-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[2,3-c]pyridine (2a)
7-Chloro-3-[2-(5-methoxy-1-methyl-1H-indol-3-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[2,3-c]pyridine (2b)
3-[2-(5-Bromo-1-methyl-1H-indol-3-yl)-1,3-thiazol-4-yl]-7-chloro-1H-pyrrolo[2,3-c]pyridine (2c)
7-Chloro-3-[2-(5-fluoro-1-methyl-1H-indol-3-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[2,3-c]pyridine (2d)
7-Chloro-1-methyl-3-[2-(1-methyl-1H-indol-3-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[2,3-c]pyridine (2e)
7-Chloro-3-[2-(5-methoxy-1-methyl-1H-indol-3-yl)-1,3-thiazol-4-yl]-1-methyl-1H-pyrrolo[2,3-c]pyridine (2f)
3-[2-(5-Bromo-1-methyl-1H-indol-3-yl)-1,3-thiazol-4-yl]-7-chloro-1-methyl-1H-pyrrolo[2,3-c]pyridine (2g)
7-Chloro-3-[2-(5-fluoro-1-methyl-1H-indol-3-yl)-1,3-thiazol-4-yl]-1-methyl-1H-pyrrolo[2,3-c]pyridine (2h)
7-Chloro-3-[2-(5-fluoro-1H-indol-3-yl)-1,3-thiazol-4-yl]-1-methyl-1H-pyrrolo[2,3-c]pyridine (2i)
7-Chloro-3-[2-(5-fluoro-1H-indol-3-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[2,3-c]pyridine (2j)
Tert-Butyl 3-[4-(7-Chloro-1H-pyrrolo[2,3-c]pyridin-3-yl)-1,3-thiazol-2-yl]-1H-indole-1-carboxylate (3a)
Tert-Butyl 3-[4-(7-chloro-1H-pyrrolo[2,3-c]pyridin-3-yl)-1,3-thiazol-2-yl]-5-methoxy-1H-indole-1-carboxylate (3b)
Tert-Butyl 5-bromo-3-[4-(7-chloro-1H-pyrrolo[2,3-c]pyridin-3-yl)-1,3-thiazol-2-yl]-1H-indole-1-carboxylate (3c)
Tert-Butyl 3-[4-(7-chloro-1-methyl-1H-pyrrolo[2,3-c]pyridin-3-yl)-1,3-thiazol-2-yl]-1H-indole-1-carboxylate (3d)
Tert-Butyl 3-[4-(7-chloro-1-methyl-1H-pyrrolo[2,3-c]pyridin-3-yl)-1,3-thiazol-2-yl]-5-methoxy-1H-indozle-1-carboxylate (3e)
Tert-Butyl 5-bromo-3-[4-(7-chloro-1-methyl-1H-pyrrolo[2,3-c]pyridin-3-yl)-1,3-thiazol-2-yl]-1H-indole-1-carboxylate (3f)
3.1.11. General Procedure for the Synthesis of Indolyl-thiazolyl-pyrrolo[2,3-c]pyridines (4a–f)
7-Chloro-3-[2-(1H-indol-3-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[2,3-c]pyridine (4a)
7-Chloro-3-[2-(5-methoxy-1H-indol-3-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[2,3-c]pyridine (4b)
3-[2-(5-Bromo-1H-indol-3-yl)-1,3-thiazol-4-yl]-7-chloro-1H-pyrrolo[2,3-c]pyridine (4c)
7-Chloro-3-[2-(1H-indol-3-yl)-1,3-thiazol-4-yl]-1-methyl-1H-pyrrolo[2,3-c]pyridine (4d)
7-Chloro-3-[2-(5-methoxy-1H-indol-3-yl)-1,3-thiazol-4-yl]-1-methyl-1H-pyrrolo[2,3-c]pyridine (4e)
3-[2-(5-Bromo-1H-indol-3-yl)-1,3-thiazol-4-yl]-7-chloro-1-methyl-1H-pyrrolo[2,3-c]pyridine (4f)
3.2. Biology
3.2.1. Viability Assay in Vitro
3.2.2. Cell Cycle Analysis
3.2.3. Measurement of Phosphatidylserine (PS) Exposure
3.2.4. Measurement of Mitochondrial Transmembrane Potential
3.2.5. Morphology
3.2.6. Quantification of Acidic Vesicular Organelles (AVO) by Acridine Orange (AO) Staining
3.2.7. Statistics
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Barraja, P.; Spanò, V.; Diana, P.; Carbone, A.; Cirrincione, G.; Vedaldi, D.; Salvador, A.; Viola, G.; Dall’Acqua, G. Pyrano[2,3-e]isoindol-2-ones, new angelicin heteroanalogue. Bioorg. Med. Chem. Lett. 2009, 19, 1711–1714. [Google Scholar] [CrossRef] [PubMed]
- Diana, P.; Martorana, A.; Barraja, P.; Montalbano, A.; Dattolo, G.; Cirrincione, G.; Dall’Acqua, F.; Salvador, A.; Vedaldi, D.; Basso, G.; et al. Isoindolo[2,1-a]quinoxaline derivatives, novel potent antitumor agents with dual inhibition of tubulin polymerization and topoisomerase I. J. Med. Chem. 2008, 51, 2387–2399. [Google Scholar] [CrossRef] [PubMed]
- Barraja, P.; Spanò, V.; Diana, P.; Carbone, A.; Cirrincione, G. Synthesis of the new ring system 6,8-dihydro-5H-pyrrolo[3,4-h]-quinazoline. Tetrahedron Lett. 2009, 50, 5389–5391. [Google Scholar] [CrossRef]
- Barraja, P.; Caracausi, L.; Diana, P.; Carbone, A.; Montalbano, A.; Cirrincione, G.; Brun, P.; Palù, G.; Castagliuolo, I.; Dall’Acqua, F.; et al. Synthesis of pyrrolo[3,2-h]quinolinones with good photochemotherapeutic activity and no DNA damage. Bioorg. Med. Chem. 2010, 18, 4830–4843. [Google Scholar] [CrossRef] [PubMed]
- Diana, P.; Stagno, A.; Barraja, P.; Montalbano, A.; Carbone, A.; Parrino, B.; Cirrincione, G. Synthesis of the new ring system pyrrolizino[2,3-b]indol-4(5H)-one. Tetrahedron 2011, 67, 3374–3379. [Google Scholar] [CrossRef]
- Barraja, P.; Spanò, V.; Giallombardo, D.; Diana, P.; Montalbano, A.; Carbone, A.; Parrino, B.; Cirrincione, G. Synthesis of [1,2]oxazolo[5,4-e]indazoles as antitumour agents. Tetrahedron 2013, 69, 6474–6477. [Google Scholar]
- Diana, P.; Barraja, P.; Lauria, A.; Montalbano, A.; Almerico, A.M.; Dattolo, G.; Cirrincione, G. Pyrrolo[2,1-c][1,2,4,]triazines from 2-diazopyrroles: Synthesis and antiproliferative activity. Eur. J. Med. Chem. 2002, 37, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Barraja, P.; Diana, P.; Spanò, V.; Montalbano, A.; Carbone, A.; Parrino, B.; Cirrincione, G. An efficient synthesis of pyrrolo[3′,2′:4,5]thiopyrano[3,2-b]pyridin-2-one: A new ring system of pharmaceutical interest. Tetrahedron 2012, 68, 5087–5094. [Google Scholar] [CrossRef]
- Barraja, P.; Caracausi, L.; Diana, P.; Montalbano, A.; Carbone, A.; Salvador, A.; Brun, P.; Castagliuolo, I.; Tisi, S.; Dall’Acqua, F.; et al. Pyrrolo[3,2-h]quinazolines as photochemotherapeutic agents. ChemMedChem 2011, 6, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Diana, P.; Martorana, A.; Barraja, P.; Lauria, A.; Montalbano, A.; Almerico, A.M.; Dattolo, G.; Cirrincione, G. Isoindolo[2,1-c]benzo[1,2,4]triazines: A new ring system with antiproliferative activity. Bioorg. Med. Chem. 2007, 15, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Barraja, P.; Diana, P.; Montalbano, A.; Carbone, A.; Viola, G.; Basso, G.; Salvador, A.; Vedaldi, D.; Dall’Acqua, F.; Cirrincione, G. Pyrrolo[3,4-h]quinolinones a new class of photochemotherapeutic agents. Bioorg. Med. Chem. 2011, 19, 2326–2341. [Google Scholar] [CrossRef] [PubMed]
- Barraja, P.; Caracausi, L.; Diana, P.; Spanò, V.; Montalbano, A.; Carbone, A.; Parrino, B.; Cirrincione, G. Synthesis and antiproliferative activity of the ring system [1,2]oxazolo[4,5-g]indole. ChemMedChem 2012, 7, 1901–1904. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Seo, Y.; Cho, K.W.; Rho, J.R.; Sim, C.J. New bis(indole)alkaloids of the topsentin class from the sponge Spongosorites genitrix. J. Nat. Prod. 1999, 62, 647–649. [Google Scholar] [CrossRef] [PubMed]
- Casapullo, A.; Bifulco, G.; Bruno, I.; Riccio, R. New bisindole alkaloids of the topsentin and hamacanthin classes from the Mediterranean marina sponge Rhaphisia lacazei. J. Nat. Prod. 2000, 63, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Sun, Q.; Yao, X.; Hong, J.; Lee, C.; Sim, C.J.; Im, K.S.; Jung, J.H. Cytotoxic bisindole alkaloids from a marine sponge Spongosorites sp. J. Nat. Prod. 2005, 68, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Gul, W.; Hamann, M.T. Indole alkaloid marine natural products: An established source of cancer drug leads with considerable promise for the control of parasitic, neurological and other diseases. Life Sci. 2005, 78, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Bokesch, H.R.; Pannell, L.K.; McKee, T.C.; Boyd, M.R. Coscinamides A, B, and C, three new bis indole alkaloids from the marine sponge Coscinoderma sp. Tetrahedron Lett. 2000, 41, 6305–6308. [Google Scholar] [CrossRef]
- Shimizu, S.; Yamamoto, Y.; Inagaki, L.; Koshimura, S. Antitumor effect and structure-activity relationship of asterriquinone analogs. Gann 1982, 73, 642–648. [Google Scholar] [PubMed]
- Kohmoto, S.; Kashman, Y.; McConnel, O.J.; Rinehart, K.L., Jr.; Wrigh, A.; Koehn, F. Dragmacidin, a new cytotoxic bis(indole)alkaloid from a deep water marine sponge, dragmacidon sp. J. Org. Chem. 1988, 53, 3116–3118. [Google Scholar] [CrossRef]
- Morris, S.A.; Andersen, R.J. Brominated bis(indole)alkaloids from the marine sponge Hexadella sp. Tetrahedron 1990, 46, 715–720. [Google Scholar] [CrossRef]
- Fahy, E.; Potts, B.C. M.; Faulkner, D.J.; Smith, K. 6-Bromotryptamine derivatives from the Gulf of California tunicate Didemnum candidum. J. Nat. Prod. 1991, 54, 564–569. [Google Scholar] [CrossRef]
- Wright, A.E.; Pomponi, S.A.; Cross, S.S.; McCarthy, P. A new bis-(indole)alkaloids from a deep-water marine sponge of the genus Spongosorites. J. Org. Chem. 1992, 57, 4772–4775. [Google Scholar] [CrossRef]
- Capon, R.J.; Rooney, F.; Murray, L.M.; Collins, E.; Sim, A.T. R.; Rostas, J.A.P.; Butler, M.S.; Carrol, A.R. Dragmacidins: New protein phosphatase inhibitors from a Southern Australian deep-water marine sponge Spongosorites sp. J. Nat. Prod. 1998, 61, 660–662. [Google Scholar] [CrossRef] [PubMed]
- Bartik, K.; Braekman, J.C.; Daloze, D.; Stoller, C.; Huysecom, J.; Vandevyver, G.; Ottinger, R. Topsentin, new toxic bis-indole alkaloids from the marine sponge Topsentia genitrix. Can. J. Chem. 1987, 65, 2118–2121. [Google Scholar] [CrossRef]
- Tsujii, S.; Rinehart, K.L.; Gunasekera, S.P.; Kashman, Y.; Cross, S.S.; Lui, M.S.; Pomponi, S.A.; Diaz, M.C. Topsentin, bromotopsentin, and dihydrodeoxybromotopsentin: Antiviral and antitumor bis(indolyl)imidazoles from Caribbean deep-sea sponge of the family Halichondriidae. Structural and synthetic studies. J. Org. Chem. 1988, 53, 5446–5453. [Google Scholar] [CrossRef]
- Alvarez, M.; Salas, M.; Joule, J.A. Marine, nitrogen-containing heterocyclic natural products. Structures and syntheses of compounds containing indole units. Heterocycles 1991, 32, 1391–1429. [Google Scholar] [CrossRef]
- Sakemi, S.; Sun, H.H. Nortopsentins A, B and C. Cytotoxic and antifungal imidazolediylbis[indoles] from the sponge Spongosorites ruetzleri. J. Org. Chem. 1991, 56, 4304–4307. [Google Scholar] [CrossRef]
- Kawasaki, I.; Yamashita, M.; Ohta, S. Total synthesis of nortopsentins A–D marine alkaloids. Chem. Pharm. Bull. 1996, 44, 1831–1839. [Google Scholar] [CrossRef]
- Moody, C.J.; Roffey, J.R.A. Synthesis of N-protected Nortopsentins B and D. Arkivoc 2000, 1, 393–401. [Google Scholar] [CrossRef]
- Miyake, F.Y.; Yakushijin, K.; Horne, D.A. A concise synthesis of Topsentin A and Nortopsentin B and D. Org. Lett. 2000, 2, 2121–2123. [Google Scholar] [CrossRef] [PubMed]
- Fresneda, P.M.; Molina, P.; Sanz, M.A. Microwave-assisted regioselective synthesis of 2,4-disubstituted imidazoles: Nortopsentin D synthesized by minimal effort. Synlett 2001, 2, 218–221. [Google Scholar]
- Jiang, B.; Gu, X.-H. Syntheses and cytotoxicity evaluation of bis(indolyl)thiazole, bis(indolyl)pyrazinone and bis(indolyl)pyrazine: Analogues of cytotoxic marine bis(indole) alkaloid. Bioorg. Med. Chem. 2000, 8, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Yang, C.G.; Xiong, W.N.; Wang, J. Synthesis and cytotoxicity evaluation of novel indolylpyrimidines and indolylpyrazines as potential antitumor agents. Bioorg. Med. Chem. 2001, 9, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Xiong, W.N.; Yang, C.G. Synthesis and antitumor evaluation of novel monoindolyl-4-trifluoromethylpyridines and bisindolyl-4-trifluoromethylpyridines. Bioorg. Med. Chem. Lett. 2001, 11, 475–477. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.N.; Yang, C.G.; Jiang, B. Synthesis of novel analogues of marine indole alkaloids: Mono(indolyl)-4-trifluoromethylpyridines and bis(indolyl)-4-trifluoromethylpyridines as potential anticancer agents. Bioorg. Med. Chem. 2001, 9, 1773–1780. [Google Scholar] [CrossRef] [PubMed]
- Diana, P.; Carbone, A.; Barraja, P.; Montalbano, A.; Martorana, A.; Dattolo, G.; Gia, O.; Dalla Via, L.; Cirrincione, G. Synthesis and antitumor properties of 2,5-bis(3′-indolyl)thiophenes: Analogues of marine alkaloid nortopsentin. Bioorg. Med. Chem. Lett. 2007, 17, 2342–2346. [Google Scholar] [CrossRef] [PubMed]
- Diana, P.; Carbone, A.; Barraja, P.; Martorana, A.; Gia, O.; Dalla Via, L.; Cirrincione, G. 3,5-Bis(3′-indolyl)pyrazoles, analogues of marine alkaloid nortopsentin: Synthesis and antitumor properties. Bioorg. Med. Chem. Lett. 2007, 17, 6134–6137. [Google Scholar] [CrossRef] [PubMed]
- Diana, P.; Carbone, A.; Barraja, P.; Kelter, G.; Fiebig, H.H.; Cirrincione, G. Synthesis and antitumor activity of 2,5-bis(3′-indolyl)-furans and 3,5-bis(3′-indolyl)-isoxazoles, nortopsentin analogues. Bioorg. Med. Chem. 2010, 18, 4524–4529. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Parrino, B.; Barraja, P.; Spanò, V.; Cirrincione, G.; Diana, P.; Maier, A.; Kelter, G.; Fiebig, H.H. Synthesis and antiproliferative activity of 2,5-bis(3′-indolyl)pyrroles, analogues of the marine alkaloid nortopsentin. Marine Drugs 2013, 11, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Kumar, N.M.; Chang, K.H.; Gupta, R.; Shah, K. Synthesis and in vitro anticancer activity of 3,5-bis(indolyl)-1,2,4-thiadiazoles. Bioorg. Med. Chem. Lett. 2011, 21, 5897–5900. [Google Scholar] [CrossRef] [PubMed]
- Jacquemard, U.; Dias, N.; Lansiaux, C.; Bailly, C.; Logè, C.; Robert, J.M.; Lozach, O.; Meijer, L.; Merour, J.Y.; Routier, S. Synthesis of 3,5-bis(2-indolyl)pyridine and 3-[(2-indolyl)-5-phenyl]pyridine derivatives as CDK inhibitors and cytotoxic agents. Bioorg. Med. Chem. 2008, 16, 4932–4953. [Google Scholar] [CrossRef] [PubMed]
- Diana, P.; Carbone, A.; Barraja, P.; Montalbano, A.; Parrino, B.; Lopergolo, A.; Pennati, M.; Zaffaroni, N.; Cirrincione, G. Synthesis and antitumor activity of 3-(2-phenyl-1,3-thiazol-4-yl)-1H-indoles and 3-(2-phenyl-1,3-thiazol-4-yl)-1H-7-azaindoles. ChemMedChem 2011, 6, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Pennati, M.; Barraja, P.; Montalbano, A.; Parrino, B.; Spanò, V.; Lopergolo, A.; Sbarra, S.; Doldi, V.; Zaffaroni, N.; et al. Synthesis and antiproliferative activity of substituted 3[2-(1H-indol-3-yl)-1,3-thiazol-4-yl]-1H-pyrrolo[3,2-b]pyridines, marine alkaloid nortopsentin analogues. Curr. Med. Chem. 2014, 21, 1654–1666. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Pennati, M.; Parrino, B.; Lopergolo, A.; Barraja, P.; Montalbano, A.; Spanò, V.; Sbarra, S.; Doldi, V.; de Cesare, M.; et al. Novel 1H-pyrrolo[2,3-b]pyridine derivatives nortopsentin analogues: Synthesis and antitumor activity in peritoneal mesothelioma experimental models. J. Med. Chem. 2013, 56, 7060–7072. [Google Scholar] [CrossRef] [PubMed]
- Parrino, B.; Carbone, A.; Muscarella, M.; Spanò, V.; Montalbano, A.; Barraja, P.; Salvador, A.; Vedaldi, D.; Cirrincione, G.; Diana, P. 11H-Pyrido[3′,2′:4,5]pyrrolo[3,2-c]cinnoline and pyrido[3′,2′:4,5]pyrrolo[1,2-c][1,2,3]benzotriazine: Two new ring systems with antitumor activity. J. Med. Chem. 2014, 57, 9495–9511. [Google Scholar] [CrossRef] [PubMed]
- Spanò, V.; Montalbano, A.; Carbone, A.; Parrino, B.; Diana, P.; Cirrincione, G.; Castagliuolo, I.; Brun, P.; Issinger, O.G.; Tisi, S.; et al. Synthesis of a new class of pyrrolo[3,4-h]quinazolines with antimitotic activity. Eur. J. Med. Chem. 2014, 74, 340–357. [Google Scholar] [CrossRef] [PubMed]
- Diana, P.; Stagno, A.; Barraja, P.; Carbone, A.; Parrino, B.; Dall’Acqua, F.; Vedaldi, D.; Salvador, A.; Brun, P.; Castagliuolo, I.; et al. Synthesis of triazenoazaindoles: A new class of triazenes with antitumor activity. ChemMedChem 2011, 6, 1291–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirrincione, G.; Almerico, A.M.; Barraja, P.; Diana, P.; Lauria, A.; Passannanti, A.; Musiu, C.; Pani, A.; Murtas, P.; Minnei, C.; et al. Derivatives of the new ring system indolo[1,2-c]benzo[1,2,3]triazine with potent antitumor and antimicrobial activity. J. Med. Chem. 1999, 42, 2561–2568. [Google Scholar] [CrossRef] [PubMed]
- Barraja, P.; Diana, P.; Carbone, A.; Cirrincione, G. Nucleophilic reactions in the indole series: Displacement of bromine under phase transfer catalysis. Tetrahedron 2008, 64, 11625–11631. [Google Scholar] [CrossRef]
- Ganser, C.; Lauermann, E.; Maderer, A.; Stauder, T.; Kramb, J.P.; Plutizki, S.; Kindler, T.; Moehler, M.; Dannhardt, G. Novel 3-azaindolyl-4-arylmaleimides exhibiting potent antiangiogenic efficacy, protein kinase inhibition, and antiproliferative activity. J. Med. Chem. 2012, 55, 9531–9540. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nature Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Paglin, S.; Hollister, T.; Delohery, T.; Hackett, N.; McMahill, M.; Sphicas, E.; Domingo, D.; Yahalom, J. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001, 61, 439–444. [Google Scholar] [PubMed]
- Sun, D.X.; Lennernas, H.; Welage, L.S.; Barnett, J.L.; Landowski, C.P.; Foster, D.; Fleischer, D.; Lee, K.D.; Amidon, G.L. Comparison of human duodenum and Caco-2 gene expression profiles for 12,000 gene sequence tags and correlation with permeability of 26 drugs. Pharm. Res. 2002, 19, 1400–1416. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.; Maiuri, M.C.; Tajeddine, N.; Vitale, I.; Criollo, A.; Vicencio, J.M.; Hickman, J.A.; Geneste, O.; Kroemer, G. Cell cycle-dependent induction of autophagy, mitophagy and reticulophagy. Cell Cycle 2007, 6, 2263–2267. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carbone, A.; Parrino, B.; Vita, G.D.; Attanzio, A.; Spanò, V.; Montalbano, A.; Barraja, P.; Tesoriere, L.; Livrea, M.A.; Diana, P.; et al. Synthesis and Antiproliferative Activity of Thiazolyl-bis-pyrrolo[2,3-b]pyridines and Indolyl-thiazolyl-pyrrolo[2,3-c]pyridines, Nortopsentin Analogues. Mar. Drugs 2015, 13, 460-492. https://doi.org/10.3390/md13010460
Carbone A, Parrino B, Vita GD, Attanzio A, Spanò V, Montalbano A, Barraja P, Tesoriere L, Livrea MA, Diana P, et al. Synthesis and Antiproliferative Activity of Thiazolyl-bis-pyrrolo[2,3-b]pyridines and Indolyl-thiazolyl-pyrrolo[2,3-c]pyridines, Nortopsentin Analogues. Marine Drugs. 2015; 13(1):460-492. https://doi.org/10.3390/md13010460
Chicago/Turabian StyleCarbone, Anna, Barbara Parrino, Gloria Di Vita, Alessandro Attanzio, Virginia Spanò, Alessandra Montalbano, Paola Barraja, Luisa Tesoriere, Maria Antonia Livrea, Patrizia Diana, and et al. 2015. "Synthesis and Antiproliferative Activity of Thiazolyl-bis-pyrrolo[2,3-b]pyridines and Indolyl-thiazolyl-pyrrolo[2,3-c]pyridines, Nortopsentin Analogues" Marine Drugs 13, no. 1: 460-492. https://doi.org/10.3390/md13010460
APA StyleCarbone, A., Parrino, B., Vita, G. D., Attanzio, A., Spanò, V., Montalbano, A., Barraja, P., Tesoriere, L., Livrea, M. A., Diana, P., & Cirrincione, G. (2015). Synthesis and Antiproliferative Activity of Thiazolyl-bis-pyrrolo[2,3-b]pyridines and Indolyl-thiazolyl-pyrrolo[2,3-c]pyridines, Nortopsentin Analogues. Marine Drugs, 13(1), 460-492. https://doi.org/10.3390/md13010460