
Cyanobactins from Cyanobacteria: Current Genetic and Chemical State of Knowledge



Abstract
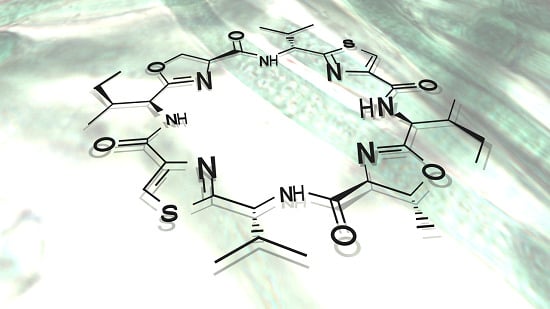
:
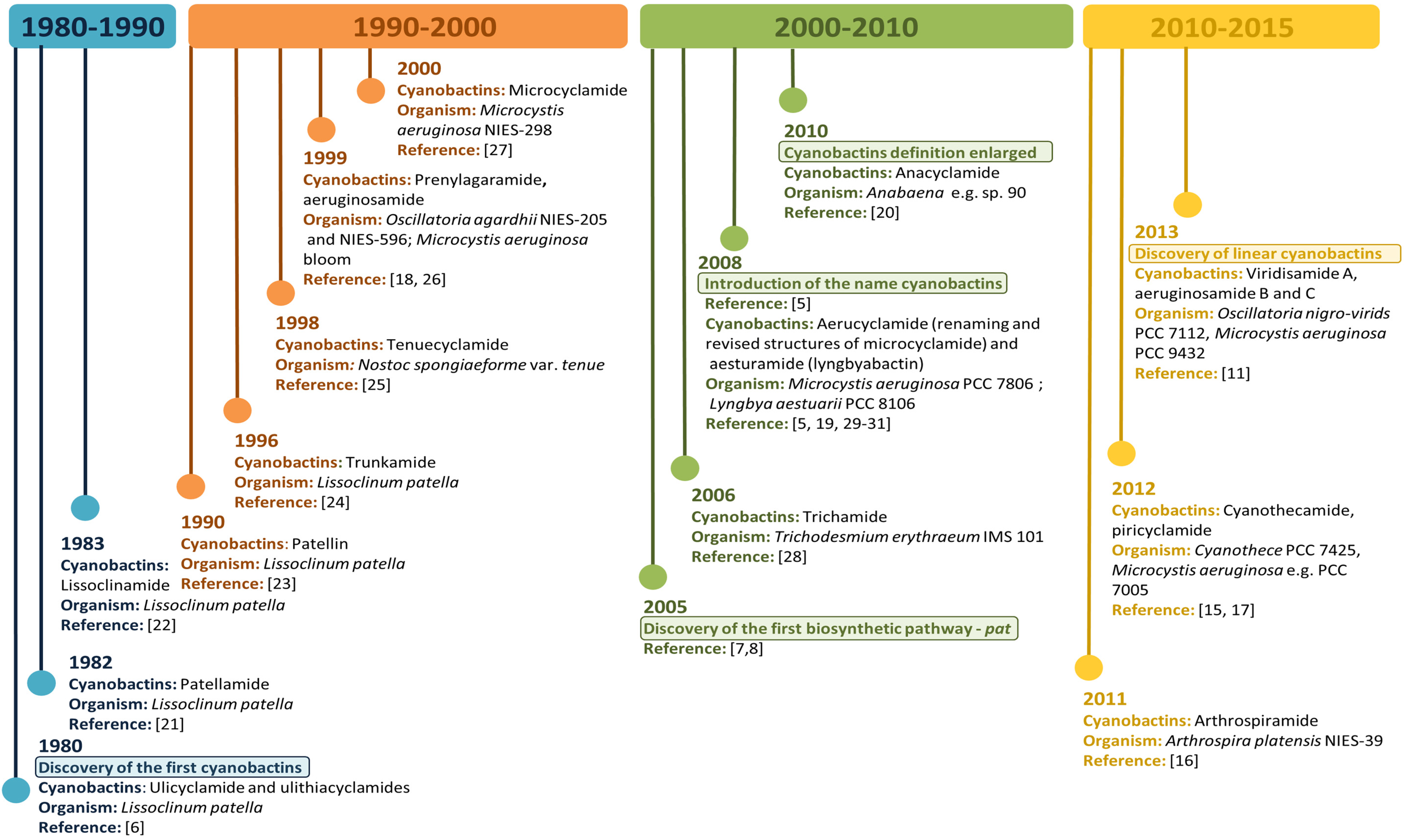
1. Introduction
2. Producing Cyanobacterial Strains

3. Chemical Structures
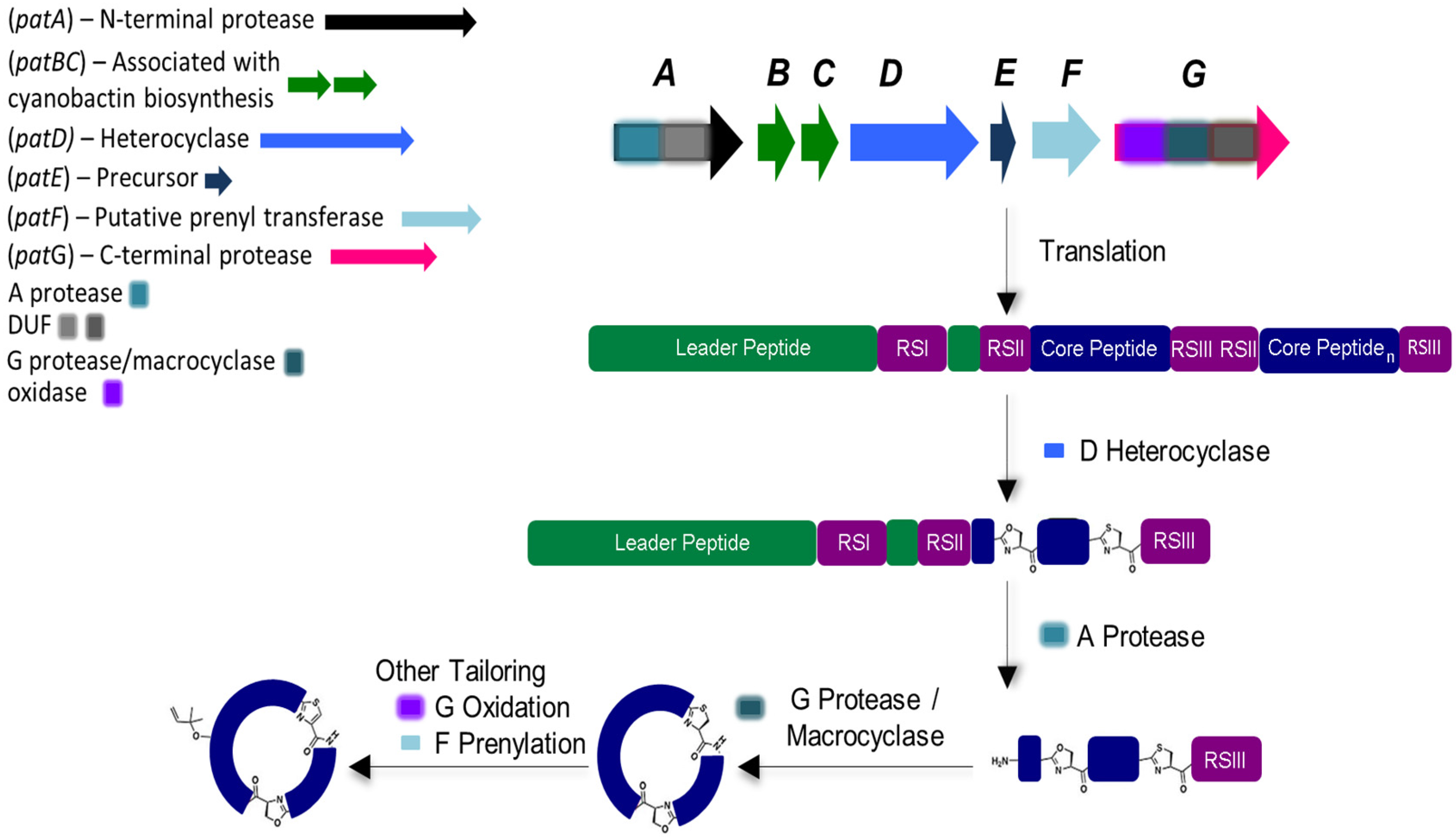
4. Biosynthetic Genetic Clusters

4.1. Heterocyclization
4.2. Cleavage and Macrocyclization
4.3. Prenylation, Oxidation and DUF
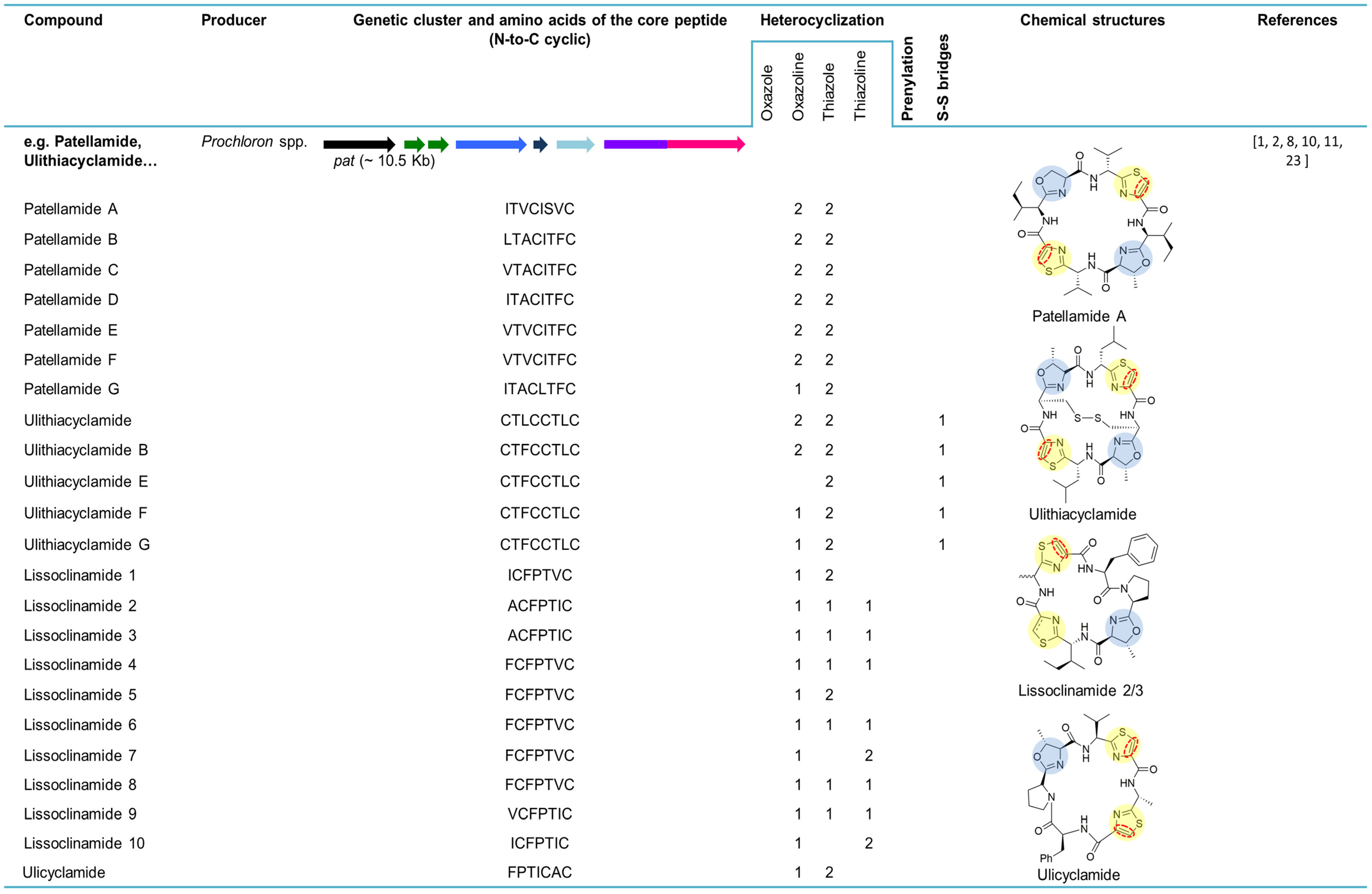
5. Cyanobactins Encoding Heterocyclization Enzymes
5.1. Ulicyclamide, Ulithiacyclamides, Patellamides and Lissoclinamides



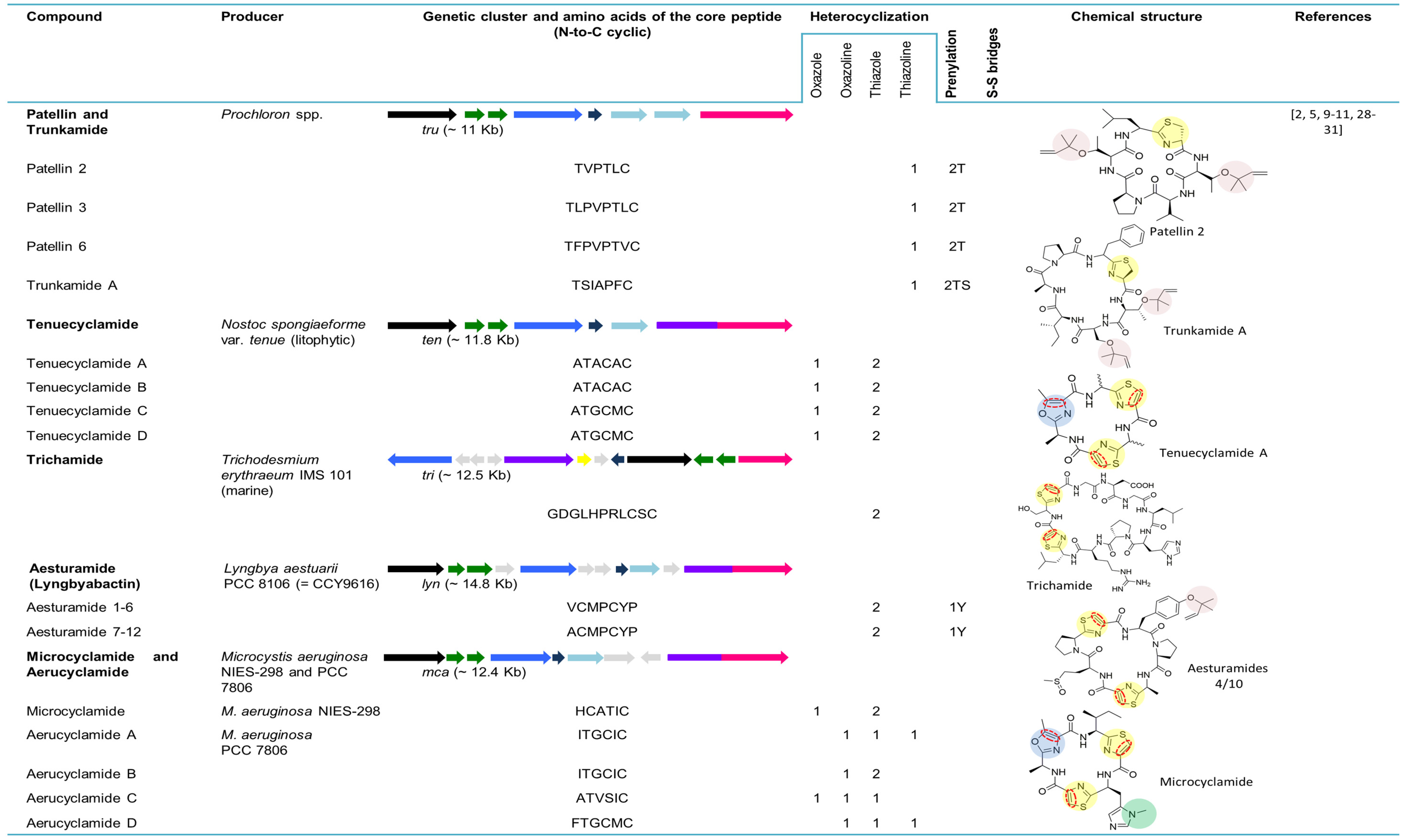
5.2. Patellins and Trunkamide
5.3. Tenuecyclamides
5.4. Trichamide
5.5. Aesturamides (Lyngbyabactins)
5.6. Microcyclamides and Aerucyclamides
5.7. Arthrospiramide
5.8. Cyanothecamides
5.9. Aeruginosamides and Viridisamide—Linear Cyanobactins

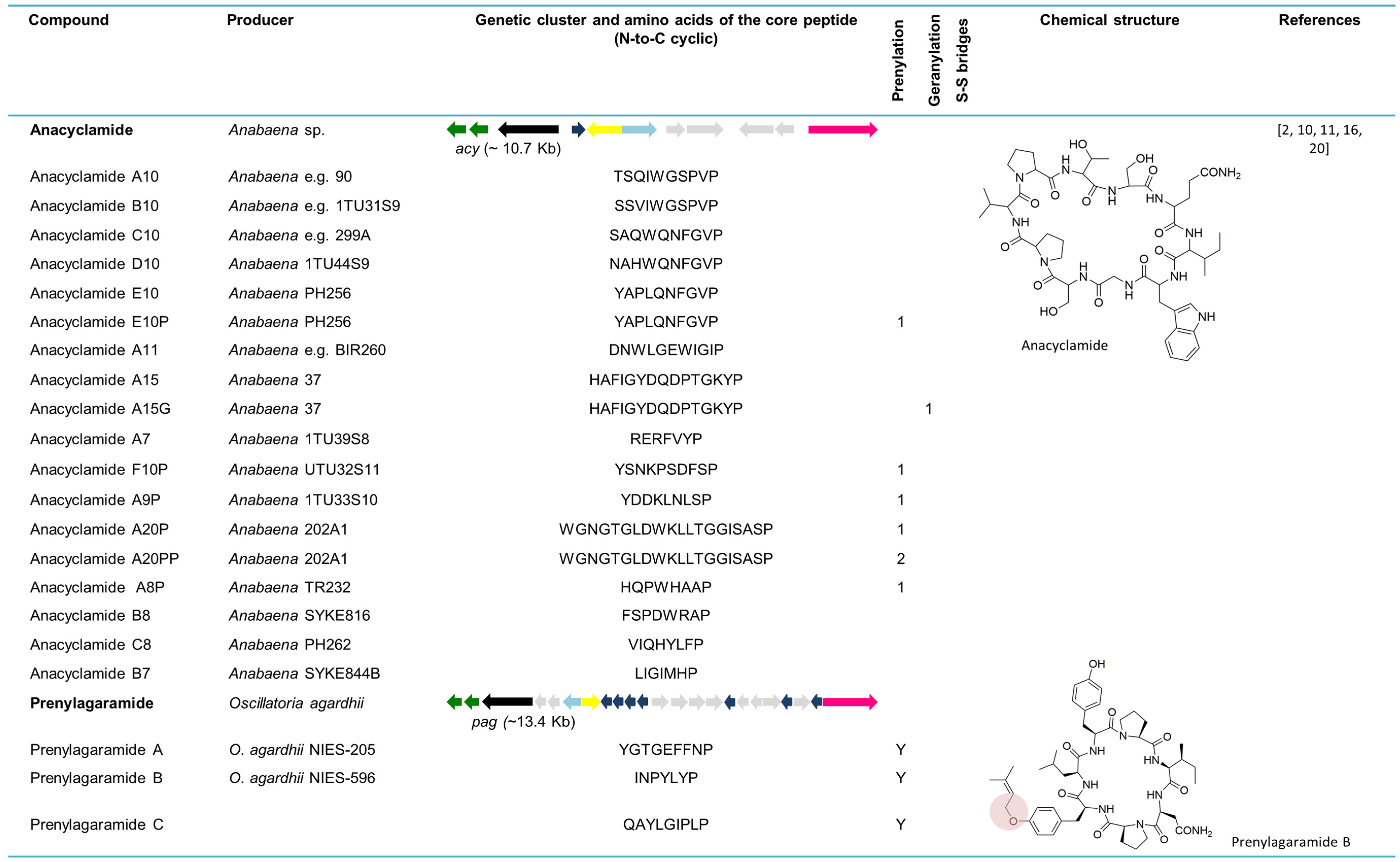
6. Cyanobactins Non-Encoding Heterocyclization or Oxidation Enzymes
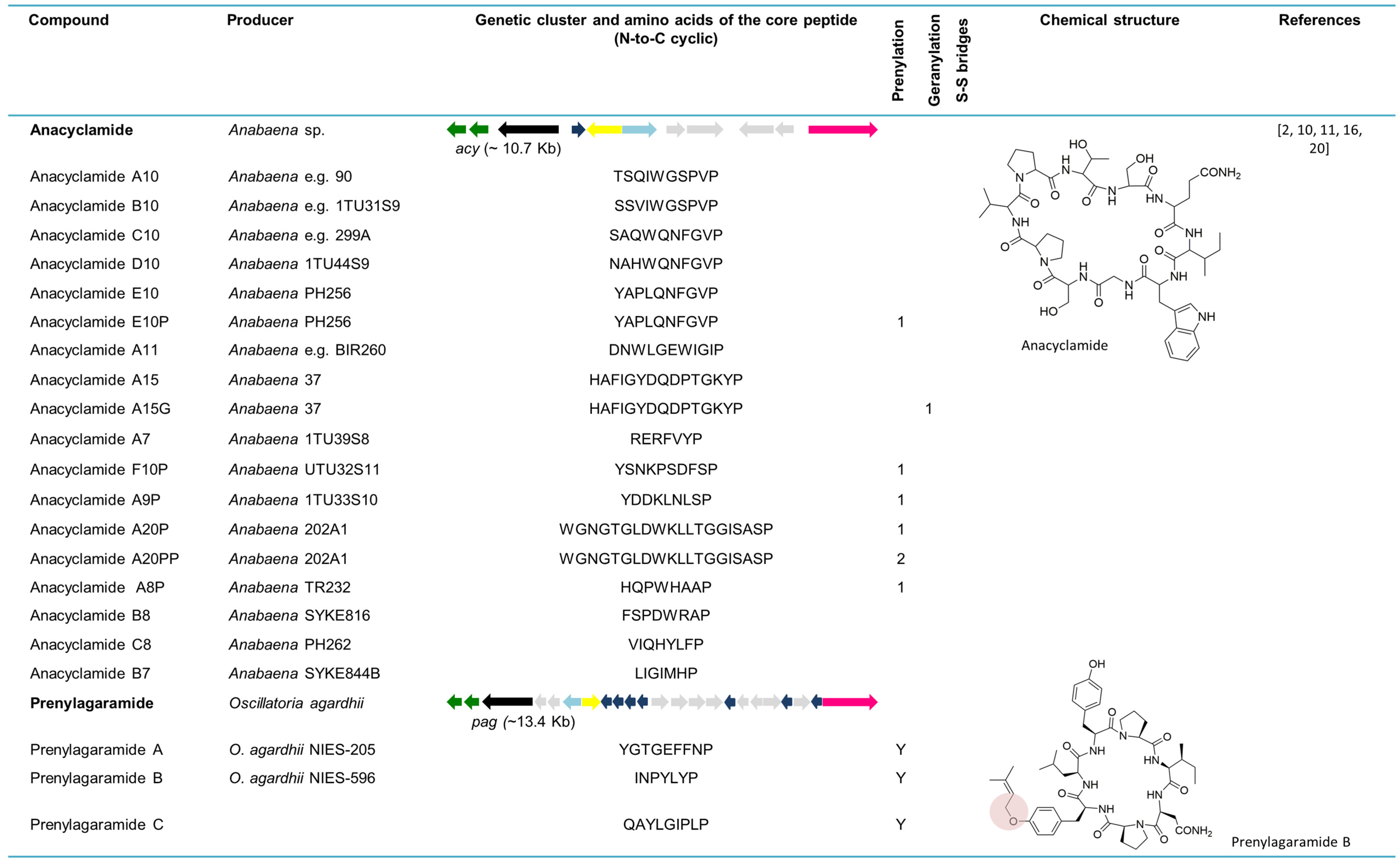
6.1. Anacyclamides
6.2. Prenylagaramides

6.3. Piricyclamides
7. Bioactivities

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source Organism | Compound | Bioactivity | References |
|---|---|---|---|
| Lissoclinum patella (Prochloron spp.) | Lissoclinamides 1–3 | Borderline cytotoxicity against L1210 murine leukemia cells (IC50 > 10 µg/mL) | [22] |
| Lissoclinamides 4–6 | Slight cytotoxicity against PS lymphocytic leukemia cells (ID50 = 10, 12 and 6.9 µg/mL for lissoclinamides 4, 5 and 6, respectively) | [22] | |
| Patellamide A | Mild cytotoxicity against L1210 murine leukemia cells (IC50 = 3.9 µg/mL) | [21,57] | |
| Poor cytotoxicity against KB cell line (IC50 = 3000 ng/mL) | |||
| Patellamide B | Mild cytotoxicity against L1210 murine leukemia cells (IC50 = 2.0 µg/mL) | [21,57,59,60] | |
| Poor cytotoxicity against KB cell line (IC50 > 4000 ng/mL) | |||
| General cytotoxicity in NCI’s 60 human tumor cell line panel (Average LC50 = 48 µM) | |||
| Multidrug reversing activity | |||
| Patellamide C | Mild cytotoxicity against L1210 murine leukemia cells (IC50 = 3.2 µg/mL) | [21,57,60] | |
| Poor cytotoxicity against KB cell line (IC50 = 6000 ng/mL) | |||
| Multidrug reversing activity | |||
| Patellamide D | Slight cytotoxicity against PS lymphocytic leukemia cells (ID50 = 11 µg/mL) | [22] | |
| Multidrug reversing activity | |||
| Patellamide E | Weak cytotoxicity against human colon tumor cells (IC50 = 125 µg/mL) | [58] | |
| Patellamide F | General cytotoxicity in NCI’s 60 human tumor cell line panel (Average LC50 = 13 µM) | [59] | |
| Patellin 6 | Moderate cytotoxic against P388, A549, HT29 and CVI cells (Average IC50 = 2 µg/mL) and inhibition of topoisomerase II activity (IC50 = 2.5µg/mL) | [24] | |
| Trunkamide A | Active against P-388 mouse lymphoma, A-549 human lung carcinoma, HT-29 human colon carcinoma (IC50 = 0.5 µg/mL) and MEL-28 human melanoma (IC50 = 1.0 µg/mL) cell lines. | [69] | |
| Ulicyclamide | Poor cytotoxicity against L1210 murine leukemia cells (IC50 = 7.2 µg/mL) | [21] | |
| Ulithiacyclamide | Cytotoxicity against L1210 murine leukemia (IC50 = 0.35 µg/mL) and KB (IC50 = 35 ng/mL) cell lines | [21,57,59] | |
| General cytotoxicity in NCI’s 60 human tumor cell line panel (Average LC50 = 3 µM) | |||
| Ulithiacyclamide B | Cytotoxicity against KB cell line (IC50 = 17 ng/mL) | [57] | |
| Microcystis aeruginosa | Aerucyclamides | Toxic to freshwater crustacean Thamnocephalus platyurus (LC50 = 30.5 µM for aerucyclamide A and LC50 = 33.8 µM for aerucyclamide B) | [29,30,31] |
| Antimalarial (aerucyclamide B presented IC50 = 0.7 µM, aerucyclamide C presented IC50 = 2.3 µM and aerucyclamide D presented IC50 = 6.3 µM) | |||
| Aerucyclamide C—moderate activity against Trypanosoma brucei rhodesiense (IC50 = 9.2 µM) | |||
| No inhibitory activity against HeLa cells and standard antiproliferative, antibacterial and antifungal assays | |||
| Microcystis aeruginosa NIES-298 (freshwater) | Microcyclamide | Slight cytotoxicity against P388 murine leukemia cells (IC50 = 1.2 µg/mL) | [27] |
| Microcystis sp. | Microcyclamide | Microcyclamide MZ602—mild cytotoxicity against Molt4 leukemia cell line (20% cell grow inhibition) and mild inhibition of chymotrypsin (IC50 = 75 µM) | [70] |
| Microcyclamide MZ568—strong cytotoxicity against Molt4 leukemia cell line (36% cell grow inhibition) and no inhibition of serine proteases | |||
| Nostoc spongiaeforme var. tenue (litophytic) | Tenuecyclamide A, C and D | Inhibited division of sea urchin embryos Paracentrotus lividus (ED100 = 108 µM, for tenuecyclamide A, ED100 = 9.0 µM for C and ED100 = 19.1 µM for D). B not tested. | [25] |
| Trichodesmium erythraeum IMS 101 (marine) | Trichamide | No effects found (tested for cytotoxicity, antifungal, antibacterial and antiviral activities) | [28] |
8. Ecological Roles
9. Biotechnological Importance
10. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jaspars, M. The origins of cyanobactin chemistry and biology. Chem. Commun. 2014, 50, 10174–10176. [Google Scholar] [CrossRef] [PubMed]
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J.; et al. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [Google Scholar] [CrossRef] [PubMed]
- Oman, T.J.; van der Donk, W.A. Follow the leader: The use of leader peptides to guide natural product biosynthesis. Nat. Chem. Biol. 2010, 6, 9–18. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.A.; Donia, M.S.; Schmidt, E.W. Ribosomal peptide natural products : Bridging the ribosomal and nonribosomal worlds. Nat. Prod. Rep. 2009, 26, 537–559. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.S.; Ravel, J.; Schmidt, E.W. A global assembly line for cyanobactins. Nat. Chem. Biol. 2008, 4, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Ireland, C.; Scheuer, P.J. Ulicyclamide and Ulithiacyclamide, Two New Small Peptides from a Marine Tunicate. J. Am. Chem. Soc. 1980, 102, 5688–5691. [Google Scholar] [CrossRef]
- Long, P.F.; Dunlap, W.C.; Battershill, C.N.; Jaspars, M. Shotgun Cloning and Heterologous Expression of the Patellamide Gene Cluster as a Strategy to Achieving Sustained Metabolite Production. ChemBioChem 2005, 6, 1760–1765. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.W.; Nelson, J.T.; Rasko, D.A.; Sudek, S.; Eisen, J.A.; Haygood, M.G.; Ravel, J. Patellamide A and C biosynthesis by a microcin-like pathway in Prochloron didemni, the cyanobacterial symbiont of Lissoclinum patella. PNAS 2005, 102, 7315–7320. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.S.; Schmidt, E.W. Cyanobactins—Ubiquitous Cyanobacterial Ribosomal Peptide Metabolites. In Comprehensive Natural Products II. Chemistry and Biology; Mander, L., Lui, H.-W., Eds.; Elsevier: Oxford, UK, 2010; Volume 2, pp. 539–558. [Google Scholar]
- Sivonen, K.; Leikoski, N.; Fewer, D.P.; Jokela, J. Cyanobactins—Ribosomal cyclic peptides produced by cyanobacteria. Appl. Microbiol. Biotechnol. 2010, 86, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Leikoski, N.; Liu, L.; Jokela, J.; Wahlsten, M.; Gugger, M.; Calteau, A.; Permi, P.; Kerfeld, C.A.; Sivonen, K.; Fewer, D.P. Genome Mining Expands the Chemical Diversity of the Cyanobactin Family to Include Highly Modified Linear Peptides. Chem. Biol. 2013, 20, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Shih, P.M.; Wu, D.; Latifi, A.; Axen, S.D.; Fewer, D.P.; Talla, E.; Calteau, A.; Cai, F.; Tandeau de Marsac, N.; Rippka, R.; et al. Improving the coverage of the cyanobacterial phylum using diversity-driven genome sequencing. PNAS 2013, 110, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Leikoski, N.; Fewer, D.P.; Sivonen, K. Widespread Occurrence and Lateral Transfer of the Cyanobactin Biosynthesis Gene Cluster in Cyanobacteria. Appl. Environ. Microbiol. 2009, 75, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.; Leão, P.N.; Ramos, V.; Vasconcelos, V. N-terminal Protease Gene Phylogeny Reveals the Potential for Novel Cyanobactin Diversity in Cyanobacteria. Mar. Drugs 2013, 11, 4902–4916. [Google Scholar] [CrossRef] [PubMed]
- Leikoski, N.; Fewer, D.P.; Jokela, J.; Alakoski, P.; Wahlsten, M.; Sivonen, K. Analysis of an Inactive Cyanobactin Biosynthetic Gene Cluster Leads to Discovery of New Natural Products from Strains of the Genus Microcystis. PLoS ONE 2012, 7, e43002. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.S.; Schmidt, E. Linking chemistry and genetics in the growing cyanobactin natural products family. Chem. Biol. 2012, 18, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Houssen, W.E.; Koehnke, J.; Zollman, D.; Vendome, J.; Raab, A.; Smith, M.C.M.; Naismith, J.H.; Jaspars, M. The Discovery of New Cyanobactins from Cyanothece PCC 7425 Defines a New Signature for Processing of Patellamides. ChemBioChem 2012, 13, 2683–2689. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Itou, Y.; Ishida, K.; Shin, H.J. Prenylagaramides A and B, new cyclic peptides from two strains of Oscillatoria agardhii. J. Nat. Prod. 1999, 62, 752–755. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.A.; Lin, Z.; Tianero, M.D.B.; Schmidt, E.W. Aestuaramides, a Natural Library of Cyanobactin Cyclic Peptides Resulting from Isoprene-Derived Claisen Rearrangements. ACS Chem. Biol. 2013, 8, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Leikoski, N.; Fewer, D.P.; Jokela, J.; Wahlsten, M.; Rouhiainen, L.; Sivonen, K. Highly Diverse Cyanobactins in Strains of the Genus Anabaena. Appl. Environ. Microbiol. 2010, 76, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Ireland, C.M.; Durso, A.R.; Newman, R.A.; Hacker, M.P. Antineoplastic Cyclic Peptides from the Marine Tunicate Lissoclinum patella. J. Org. Chem. 1982, 47, 1807–1811. [Google Scholar] [CrossRef]
- Wasylyk, J.M.; Biskupiak, J.E.; Costello, C.E.; Ireland, C.M. Cyclic peptide structures from the tunicate Lissoclinum patella by FAB mass spectrometry. J. Org. Chem. 1983, 48, 4445–4449. [Google Scholar] [CrossRef]
- Zabriskie, T.M.; Foster, M.P.; Stout, T.J.; Clardy, J.; Ireland, C.M. Studies on the Solution- and Solid-state Structure of Patellin 2. J. Am. Chem. Soc. 1990, 112, 8080–8084. [Google Scholar] [CrossRef]
- Carroll, A.R.; Coll, J.C.; Bourne, D.J.; MacLeod, J.K.; Mark Zabriskie, T.; Ireland, C.M.; Bowden, B.F. Patellins 1-6 and Trunkamide A: Novel Cyclic Hexa-, Hepta- and Octa-peptides from Colonial Ascidians, Lissoclinurn sp. Aust. J. Chem. 1996, 49, 659–667. [Google Scholar]
- Banker, R.; Carmeli, S. Tenuecyclamides A-D, Cyclic Hexapeptides from the Cyanobacterium Nostoc spongiaeforme var. tenue. J. Nat. Prod. 1998, 61, 1248–1251. [Google Scholar] [CrossRef] [PubMed]
- Lawton, L.; Morris, L.; Jaspars, M. A bioactive modified peptide, aeruginosamide, isolated from the cyanobacterium Microcystis aeruginosa. J. Am. Chem. Soc. 1999, 64, 5329–5332. [Google Scholar]
- Ishida, K.; Nakagawa, H.; Murakami, M. Microcyclamide, a Cytotoxic Cyclic Hexapeptide from the Cyanobacterium Microcystis aeruginosa. J. Nat. Prod. 2000, 63, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Sudek, S.; Haygood, M.G.; Youssef, D.T.A.; Schmidt, E.W. Structure of Trichamide, a Cyclic Peptide from the Bloom-Forming Cyanobacterium Trichodesmium erythraeum, Predicted from the Genome Sequence. Appl. Environ. Microbiol. 2006, 72, 4382–4387. [Google Scholar] [CrossRef] [PubMed]
- Portmann, C.; Blom, J.F.; Gademann, K.; Juttner, F. Aerucyclamides A and B : Isolation and Synthesis of Toxic Ribosomal Heterocyclic Peptides from the Cyanobacterium Microcystis aeruginosa PCC 7806. J. Nat. Prod. 2008, 71, 1193–1196. [Google Scholar] [CrossRef] [PubMed]
- Portmann, C.; Blom, J.F.; Kaiser, M.; Brun, R.; Ju, F.; Gademann, K. Isolation of Aerucyclamides C and D and Structure Revision of Microcyclamide 7806A : Heterocyclic Ribosomal Peptides from Microcystis aeruginosa PCC 7806 and Their Antiparasite Evaluation. J. Nat. Prod. 2008, 71, 1891–1896. [Google Scholar] [CrossRef] [PubMed]
- Ziemert, N.; Ishida, K.; Quillardet, P.; Bouchier, C.; Hertweck, C.; Marsac, N.T.D.; Dittmann, E. Microcyclamide Biosynthesis in Two Strains of Microcystis aeruginosa : From Structure to Genes and Vice Versa. Appl. Environ. Microbiol. 2008, 74, 1791–1797. [Google Scholar] [CrossRef] [PubMed]
- Milne, B.F.; Long, P.F.; Starcevic, A.; Hranueli, D.; Jaspars, M. Spontaneity in the patellamide biosynthetic pathway. Org. Biomol. Chem. 2006, 4, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Pierce, E.; McIntosh, J.; Schmidt, E.W.; Nair, S.K. Structures of Cyanobactin Maturation Enzymes Define a Family of Transamidating Proteases. Chem. Biol. 2012, 19, 1411–1422. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.S.; Hathaway, B.J.; Sudek, S.; Haygood, M.G.; Rosovitz, M.J.; Ravel, J.; Schmidt, E.W. Natural combinatorial peptide libraries in cyanobacterial symbionts of marine ascidians. Nat. Chem. Biol. 2006, 2, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Houssen, W.E.; Wright, S.H.; Kalverda, A.P.; Thompson, G.S.; Kelly, S.M.; Jaspars, M. Solution Structure of the Leader Sequence of the Patellamide Precursor Peptide, PatE1-34. ChemBioChem 2010, 11, 1867–1873. [Google Scholar] [CrossRef] [PubMed]
- Sardar, D.; Pierce, E.; McIntosh, J.A.; Schmidt, E.W. Recognition Sequences and Substrate Evolution in Cyanobactin Biosynthesis. ACS Synth. Biol. 2015, 4, 167–176. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.A.; Schmidt, E.W. Marine Molecular Machines : Heterocyclization in Cyanobactin Biosynthesis. ChemBioChem 2010, 11, 1413–1421. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.A.; Donia, M.S.; Schmidt, E.W. Insights into heterocyclization from two highly similar enzymes. J. Am. Chem. Soc. 2010, 132, 4089–4091. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.A.; Donia, M.S.; Nair, S.K.; Schmidt, E.W. Enzymatic Basis of Ribosomal Peptide Prenylation in Cyanobacteria. J. Am. Chem. Soc. 2011, 133, 13698–13705. [Google Scholar] [CrossRef] [PubMed]
- Koehnke, J.; Bent, A.; Houssen, W.E.; Zollman, D.; Morawitz, F. The mechanism of patellamide macrocyclization revealed by the characterization of the PatG macrocyclase domain. Nat. Struct. Mol. Biol. 2012, 19, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Cotter, P.D.; Ross, R.P.; Hill, C. Bacteriocins—A viable alternative to antibiotics. Nat. Rev. Microbiol. 2013, 11, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Koehnke, J.; Bent, A.F.; Houssen, W.E.; Mann, G.; Jaspars, M.; Naismith, J.H. The structural biology of patellamide biosynthesis. Curr. Opin. Struct. Biol. 2014, 29, 112–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehnke, J.; Bent, A.F.; Zollman, D.; Smith, K.; Houssen, W.E.; Zhu, X.; Mann, G.; Lebl, T.; Scharff, R.; Shirran, S.; et al. The Cyanobactin Heterocyclase Enzyme : A Processive Adenylase That Operates with a Defined Order of Reaction. Angew. Chem. Int. Ed. 2013, 52, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; McIntosh, J.; Hathaway, B.J.; Schmidt, E.W. Using marine natural products to discover a protease that catalyses peptide macrocyclization of diverse substrates. J. Am. Chem. Soc. 2009, 131, 2122–2124. [Google Scholar] [CrossRef] [PubMed]
- Majmudar, J.; Gibbs, R. Pericyclic prenylation: Peptide modification through a Claisen rearrangement. ChemBioChem 2011, 122723–122726. [Google Scholar] [CrossRef] [PubMed]
- Bent, A.F.; Koehnke, J.; Smith, M.C.M.; Jaspars, M.; Naismith, J.H. Structure of PatF from Prochloron didemni. Acta Crystallogr. 2013, 69, 618–623. [Google Scholar] [CrossRef]
- Melby, J.; Li, X.; Mitchell, D. Orchestration of enzymatic processing by thiazole/oxazole-modified microcin dehydrogenases. Biochemistry (Moscow) 2014, 53, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Houssen, W.E.; Bent, A.F.; MCewan, A.R.; Pieiller, N.; Tabudravu, J.; Koehnke, J.; Mann, G.; Adaba, R.I.; Thomas, L.; Hawas, U.W.; et al. An Efficient Method for the In Vitro Production of Azol(in)e-Based Cyclic Peptides. Angew. Chem. Int. Ed. 2014, 53, 14171–14174. [Google Scholar] [CrossRef] [PubMed]
- Mann, G.; Koehnke, J.; Bent, A.F.; Graham, R.; Houssen, W.; Jaspars, M.; Schwarz-Linek, U.; Naismith, J.H. The structure of the cyanobactin domain of unknown function from PatG in the patellamide gene cluster. Acta Crystallogr. 2014, F70, 1597–1603. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, K.L.; Kishore, V.; Bible, K.C.; Sakai, P.; Sullins, D.W.; Li, K.M. Didemnins and Tunichlorin: Novel Natural Products from the Marine Tunicate Trididemnum solidum. J. Nat. Prod. 1988, 51, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, Y.; Endo, M.; Nakagawa, H.; Nakanishi, T.; Mizukawa, J. A new cyclic peptide, ascidiacyclamide, isolated from ascidian. J. Chem. Soc. Chem. Commun. 1983, 323. [Google Scholar] [CrossRef]
- Hamada, Y.; Shibata, M.; Shioiri, T. New methods and reagents in organic synthesis. 58. : A synthesis of patellamide a, a cytotoxic cyclic peptide from a tunicate. Revision of its proposed structure. Tetrahedron Lett. 1985, 26, 5155–5158. [Google Scholar] [CrossRef]
- Ishida, T.; Inoue, M.; Hamada, Y.; Kato, S.; Shioiri, T. X-Ray crystal structure of ascidiacyclamide, a cytotoxic cyclic peptide from ascidian. J. Chem. Soc. Chem. Commun. 1987, 370–371. [Google Scholar] [CrossRef]
- Degnan, B.M.; Hawkins, C.J.; Lavin, M.F.; Mccaffrey, E.J.; Parry, D.L.; Brenk, A.L.V.D.; Watterst, D.J. New Cyclic Peptides with Cytotoxic Activity from the Ascidian Lissoclinum patella. J. Med. Chem. 1989, 32, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, F.J.; Ksebati, M.B.; Chang, J.S.; Wang, J.L.; Hossain, B.M.; van de Helm, D. Cyclic Peptides from the Ascidian Lissoclinum patella: Conformational Analysis of patellamide D by X-ray Analysis and Molecular, modeling. J. Org. Chem. 1989, 54, 3463–3472. [Google Scholar] [CrossRef]
- Biskupiak, J.E.; Ireland, C.M. Absolute configuration of thiazole amino acids in peptides. J. Org. Chem. 1983, 48, 2302–2304. [Google Scholar] [CrossRef]
- Williams, D.E.; Moore, R.E. The Structure of Ulithiacyclamide B. Antitumor Evaluation of Cyclic Peptides and Macrolides from Lissoclinum Patella. J. Nat. Prod. 1989, 52, 732–739. [Google Scholar] [CrossRef] [PubMed]
- McDonald, L.A.; Ireland, C. Patellamide E: A new cyclic peptide from the ascidian Lissoclinum patella. J. Nat. Prod. 1992, 55, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.A.; Gustafson, K.R.; Cardellina, J.H.; Boyd, M.R. Patellamide F, a new cytotoxic cyclic peptide from the colonial ascidian Lissoclinum patella. J. Nat. Prod. 1995, 58, 594–597. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Do, T.; Schmitz, F.J.; Andrusevich, V.; Engel, M.H. New Cyclic Peptides from the Ascidian Lissoclinum patella. J. Nat. Prod. 1998, 61, 1547–1551. [Google Scholar] [CrossRef] [PubMed]
- Wipf, P.; Uto, Y. Total Synthesis and Revision of Stereochemistry of the Marine Metabolite Trunkamide A. J. Org. Chem. 2000, 65, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Caba, J.M.; Rodriguez, I.M.; Manzanares, I.; Giralt, E.; Albericio, F. Solid-Phase Total Synthesis of Trunkamide A1. J. Org. Chem. 2001, 66, 7568–7574. [Google Scholar] [CrossRef] [PubMed]
- McKeever, B.; Pattenden, G. Total synthesis of the prenylated cyclopeptide trunkamide A, a cytotoxic metabolite from Lissoclinum sp. Tetrahedron Lett. 2001, 42, 2573–2577. [Google Scholar] [CrossRef]
- Salvatella, X.; Caba, J.M.; Albericio, F.; Giralt, E. Solution Structure of the Antitumor Candidate Trunkamide A by 2D NMR and Restrained Simulated Annealing Methods. J. Org. Chem. 2003, 68, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Shioiri, T.; Hamada, Y.; Kato, S.; Shibata, M.; Kondo, Y. Cytotoxic activity of cyclic peptides of marine origin and their derivates: Importance of oxazoline functions. Biochem. Pharmacol. 1987, 3, 4181–4185. [Google Scholar] [CrossRef]
- Kohda, K.; Ohta, Y.; Yokoyama, Y.; Kawazoe, Y.; Kato, T.; Suzumura, Y.; Hamada, Y.; Shioiri, T. Mechanistic aspects of the cytocidal action of ulithiacyclamide on mouse leukemia L1210 cells in vitro. Biochem. Pharmacol. 1990, 38, 4497–4500. [Google Scholar]
- Kohda, K.; Ohta, Y.; Kawazoe, Y.; Kato, T.; Suzumura, Y.; Hamada, Y.; Shioiri, T. Ulicyclamide is cytotoxic against L1210 cells in vitro and inhibits both DNA and RNA syntheses. Biochem. Pharmacol. 1989, 38, 4500–4502. [Google Scholar] [PubMed]
- Houssen, W.E.; Jaspars, M. Azole-Based Cyclic Peptides from the Sea Squirt Lissoclinum Patella : Old Scaffolds, New Avenues. ChemBioChem 2010, 11, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Bowden, B.F.; Gravalos, D.G. Cyclic Hepta-Peptide Derivate from Colonial Ascidians Lissoclinum sp. U.S. Patent 6,025,466, 15 February 2000. [Google Scholar]
- Zafrir-ilan, E.; Carmeli, S. Two new microcyclamides from a water bloom of the cyanobacterium. Tetrahedron Lett. 2010, 51, 6602–6604. [Google Scholar] [CrossRef]
- Raveh, A.; Carmeli, S. Aeruginazole A, a novel thiazole-containing cyclopeptide from the cyanobacterium Microcystis sp. Org. Lett. 2010, 12, 3536–3539. [Google Scholar] [CrossRef] [PubMed]
- Adiv, S.; Pierce, E.; McIntosh, J.; Schmidt, E.W.; Nair, S.K. New aeruginazoles. A group of thiazole containing cyclic peptides from Microcystis aeruginosa. Chem. Biol. 2012, 19, 1411–1422. [Google Scholar] [CrossRef]
- Ishida, K.; Matsuda, H.; Murakami, M.; Yamaguchi, K. Kawaguchipeptin B, an antibacterial cyclic undecapeptide from the cyanobacterium Microcystis aeruginosa. J. Nat. Prod. 1997, 60, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Todorova, A.K.; Juttner, F.; Linden, A.; Pluss, T.; von Philipsborn, W. Nostocyclamide: A new macrocyclic, thiazole-containing allelochemical from Nostoc sp. 31. J. Org. Chem. 1995, 60, 7891–7895. [Google Scholar] [CrossRef]
- Juttner, F.; Todorova, A.K.; Walch, N.; von Philipsborn, W. Nostocyclamide M: A cyanobacterial cyclic peptide with allelopathic activity from Nostoc 31. Phytochemistry 2001, 57, 613–619. [Google Scholar] [CrossRef]
- Bertram, A.; Pattenden, G. Marine metabolites: Metal binding and metal complexes of azole-based cyclic peptides of marine origin. Nat. Prod. Rep. 2007, 1, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Comba, P. Strains and stresses in coordination compounds. Coord. Chem. Rev. 1999, 182, 343–371. [Google Scholar] [CrossRef]
- Comba, P.; Dovalil, N.; Gahan, L.R.; Hanson, G.R.; Westphal, M. Cyclic peptide marine metabolites and CuII. R. Soc. Chem. 2014, 43, 1935–1956. [Google Scholar] [CrossRef] [PubMed]
- Comba, P.; Dovalil, N.; Hanson, G.R.; Linti, G. Synthesis and Cu II Coordination Chemistry of a Patellamide Derivative : Consequences of the Change from the Natural Thiazole/Oxazoline to the Artificial Imidazole Heterocycles. Inorg. Chem. 2011, 50, 5165–5174. [Google Scholar] [CrossRef] [PubMed]
- Van den Brenk, A.L.; Byriel, K.A.; Fairlie, D.P.; Gahan, L.R.; Hanson, G.R.; Hawkins, C.J.; Jones, A.; Kennard, H.L.; Moubaraki, B.; Murray, K.S. Crystal Structure, Electrospray Ionization Mass Spectrometry, Electron Paramagnetic Resonance, and Magnetic Susceptibility of Cu2(ascidH2)(1,2-u-CO3)(H2O)2).2H2O, the Bis(copper(II)) Complex of Ascidiacyclamide (ascidH4), a Cyclic Peptide Isolated from the Ascidian lissoclinum patella. Inorg. Chem. 1994, 33, 3549–3557. [Google Scholar] [CrossRef]
- Van den Brenk, A.L.; Fairlie, D.P.; Hanson, G.R.; Gahan, L.R.; Hawkins, C.J.; Jones, A. Binding of copper(II) to the Cyclic Octapeptide Patellamide D. Inorg. Chem. 1994, 33, 2280–2289. [Google Scholar] [CrossRef]
- Freeman, D.J.; Pattenden, G.; Drake, A.F.; Siligardi, G. Marine metabolites and metal ion chelation. Circular dichroism studies of metal binding to Lissoclinum cyclopeptides. J. Chem. Soc. Perkin Trans. 1998, 2, 129–135. [Google Scholar] [CrossRef]
- Morris, L.A.; Jaspars, M.; Kettenes-van den Bosh, J.J.; Versuis, K.; Heck, A.J.R.; Kelly, S.M.; Prince, N.C. Metal Binding of Lissoclinum patella metabolites. Part 1: Patellamides A, C and ulithiacyclamide A. Tetrahedron 2001, 57, 3185–3197. [Google Scholar] [CrossRef]
- Morris, L.A.; Milne, B.F.; Jaspars, M.; Kettenes-van den Bosh, J.J.; Versuis, K.; Heck, A.J.R.; Kelly, S.M.; Prince, N.C. Metal Binding of Lissoclinum patella metabolites. Part 2: Lissoclinamides 9 and 10. Tetrahedron 2001, 57, 3199–3207. [Google Scholar] [CrossRef]
- Comba, P.; Gahan, L.R.; Hanson, G.R.; Maeder, M.; Westphal, M. Carbonic anhydrase activity of dinuclear CuII complexes with patellamide model ligands. R. Soc. Chem. 2014, 43, 3144–3152. [Google Scholar]
- Wipf, P. Synthetic Studies of Biologically Active Marine Cyclopeptides. Chem. Rev. 1995, 95, 2115–2134. [Google Scholar] [CrossRef]
- Driggers, E.M.; Hale, J.; Lee, J.; Terrent, N.K. The exploration of macrocycles for drug discovery—An underexploited structural class. Nat. Rev. Drug Discov. 2008, 7, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Tianero, M.D.B.; Donia, M.S.; Young, T.S.; Schultz, P.G.; Schmidt, E.W. Ribosomal Route to Small-Molecule Diversity. J. Am. Chem. Soc. 2012, 134, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Ruffner, D.E.; Schmidt, E.W.; Heemstra, J.R. Assessing the combinatorial potential of the RiPP cyanobactin tru pathway. ACS Synth. Biol. 2015, 4, 482–492. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.A.; Robertson, C.R.; Agarwal, V.; Nair, S.K.; Bulaj, G.W.; Schmidt, E.W. Circular Logic: Nonribosomal Peptide-like Macrocyclization with a Ribosomal Peptide Catalyst. J. Am. Chem. Soc. 2010, 132, 15499–15501. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Ito, Y.; Kato, Y.; Tsunoda, S.; Suga, H. One-Pot Synthesis of Azoline-Containing Peptides in a Cell-free Translation System Integrated with a Posttranslational Cyclodehydratase. Chem. Biol. 2014, 21, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Koehnke, J.; Mann, G.; Bent, A.F.; Ludewig, H.; Shirran, S.; Botting, C.; Lebl, T.; Houssen, W.E.; Jaspars, M.; Naismith, H. Structural analysis of leader peptide binding enables leader-free cyanobactin process. Nat. Chem. Biol. 2015, 11, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Sardar, D.; Lin, Z.; Schmidt, E.W. Modularity of RiPP Enzymes Enables Designed Synthesis of Decorated Peptides. Chem. Biol. 2015, 22, 907–916. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, J.; Vasconcelos, V. Cyanobactins from Cyanobacteria: Current Genetic and Chemical State of Knowledge. Mar. Drugs 2015, 13, 6910-6946. https://doi.org/10.3390/md13116910
Martins J, Vasconcelos V. Cyanobactins from Cyanobacteria: Current Genetic and Chemical State of Knowledge. Marine Drugs. 2015; 13(11):6910-6946. https://doi.org/10.3390/md13116910
Chicago/Turabian StyleMartins, Joana, and Vitor Vasconcelos. 2015. "Cyanobactins from Cyanobacteria: Current Genetic and Chemical State of Knowledge" Marine Drugs 13, no. 11: 6910-6946. https://doi.org/10.3390/md13116910
APA StyleMartins, J., & Vasconcelos, V. (2015). Cyanobactins from Cyanobacteria: Current Genetic and Chemical State of Knowledge. Marine Drugs, 13(11), 6910-6946. https://doi.org/10.3390/md13116910