
Algal Toxin Azaspiracid-1 Induces Early Neuronal Differentiation and Alters Peripherin Isoform Stoichiometry

Abstract
:1. Introduction
2. Results
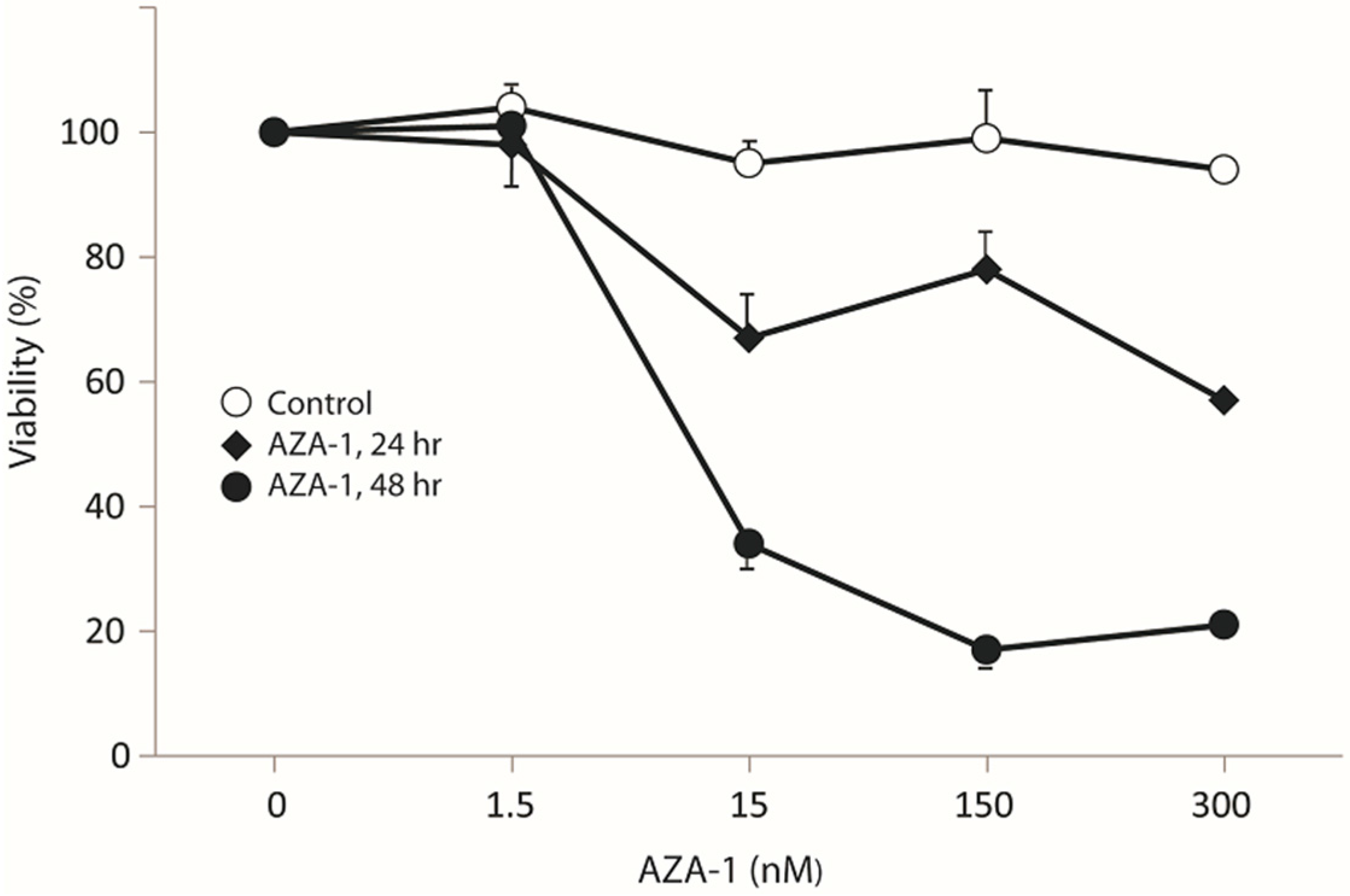
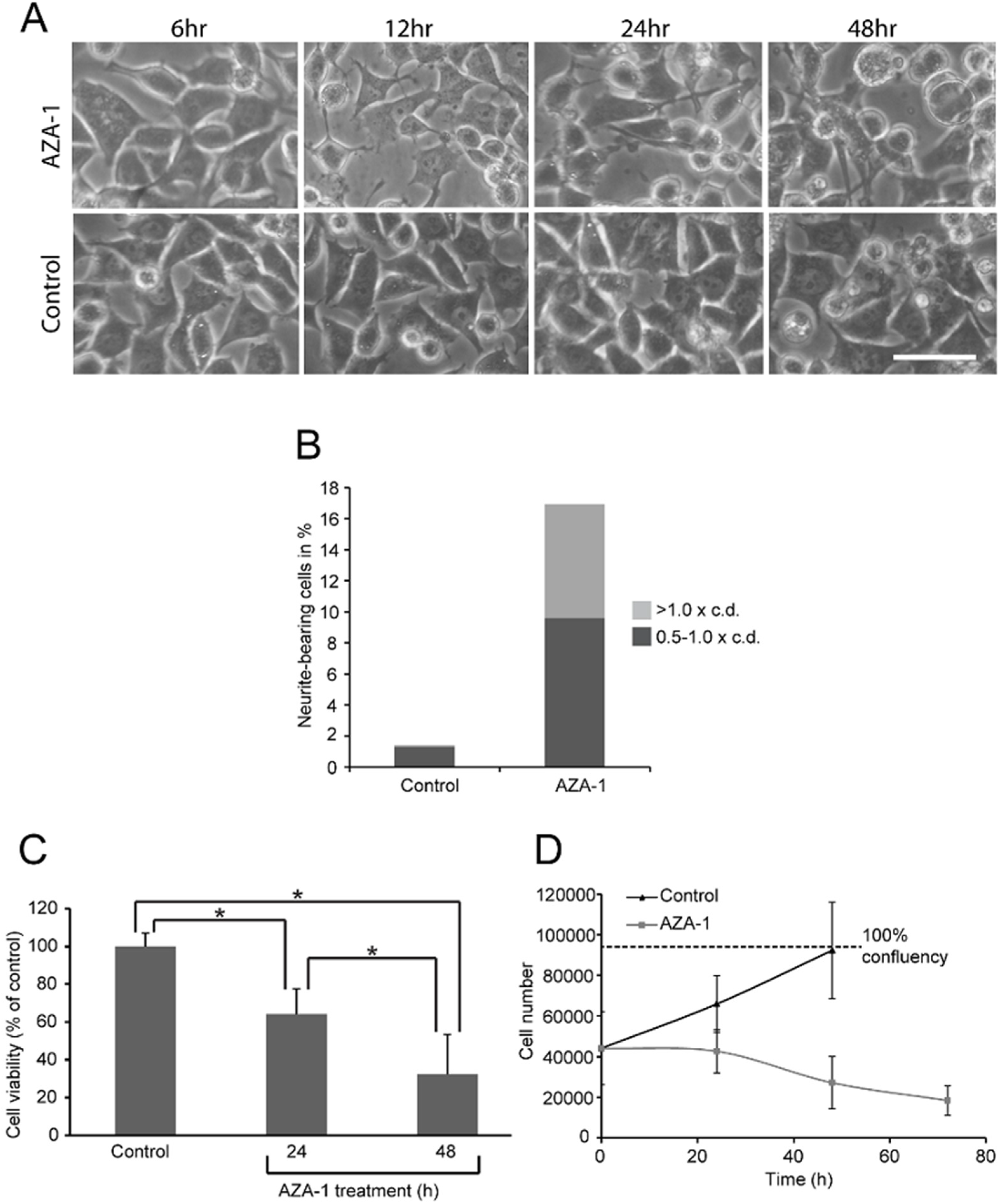
2.1. AZA-1 Induces Morphological Alterations in PC12 Cells


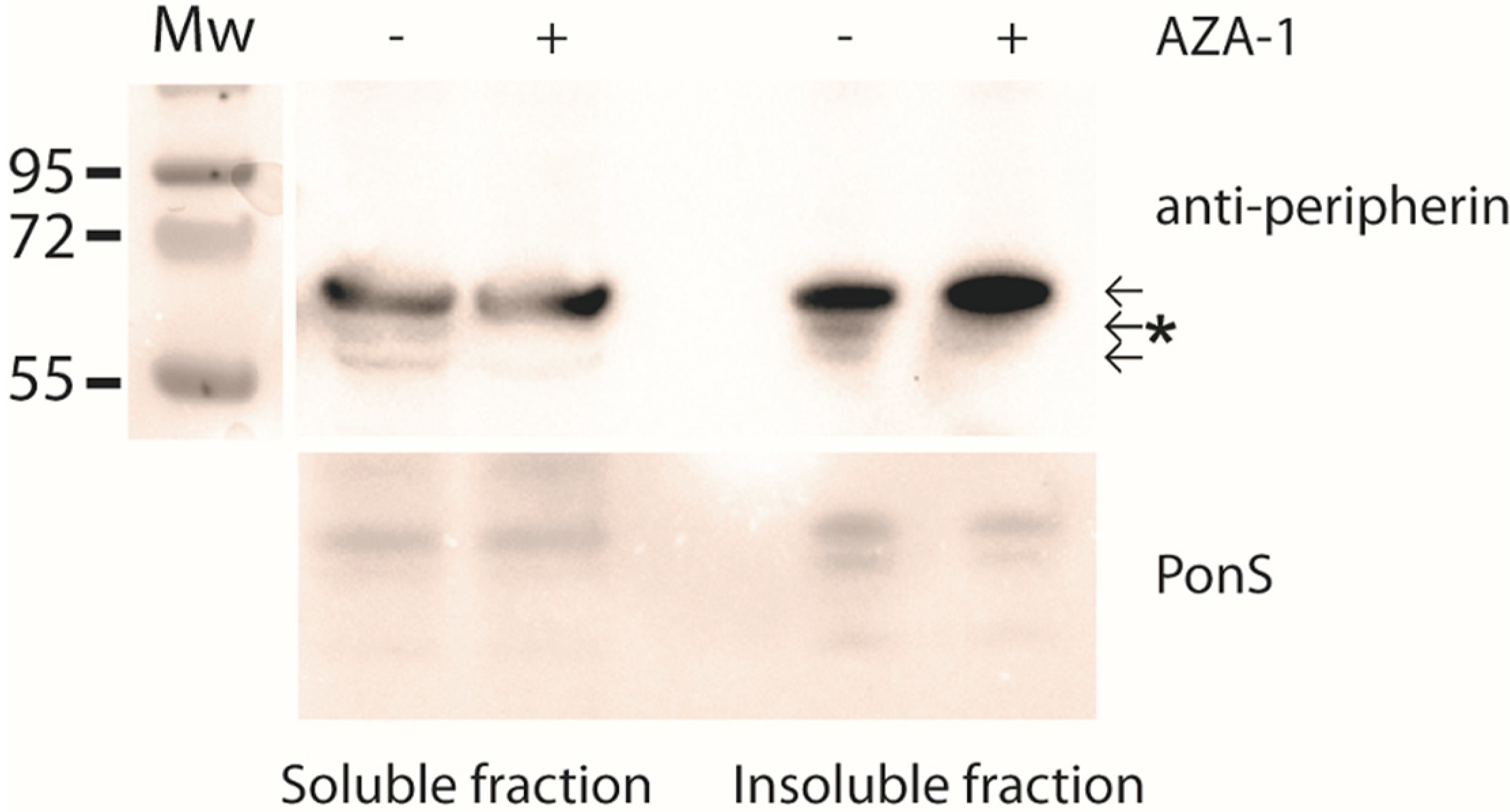
2.2. AZA-1 Down-Regulates a Specific Peripherin Isoform



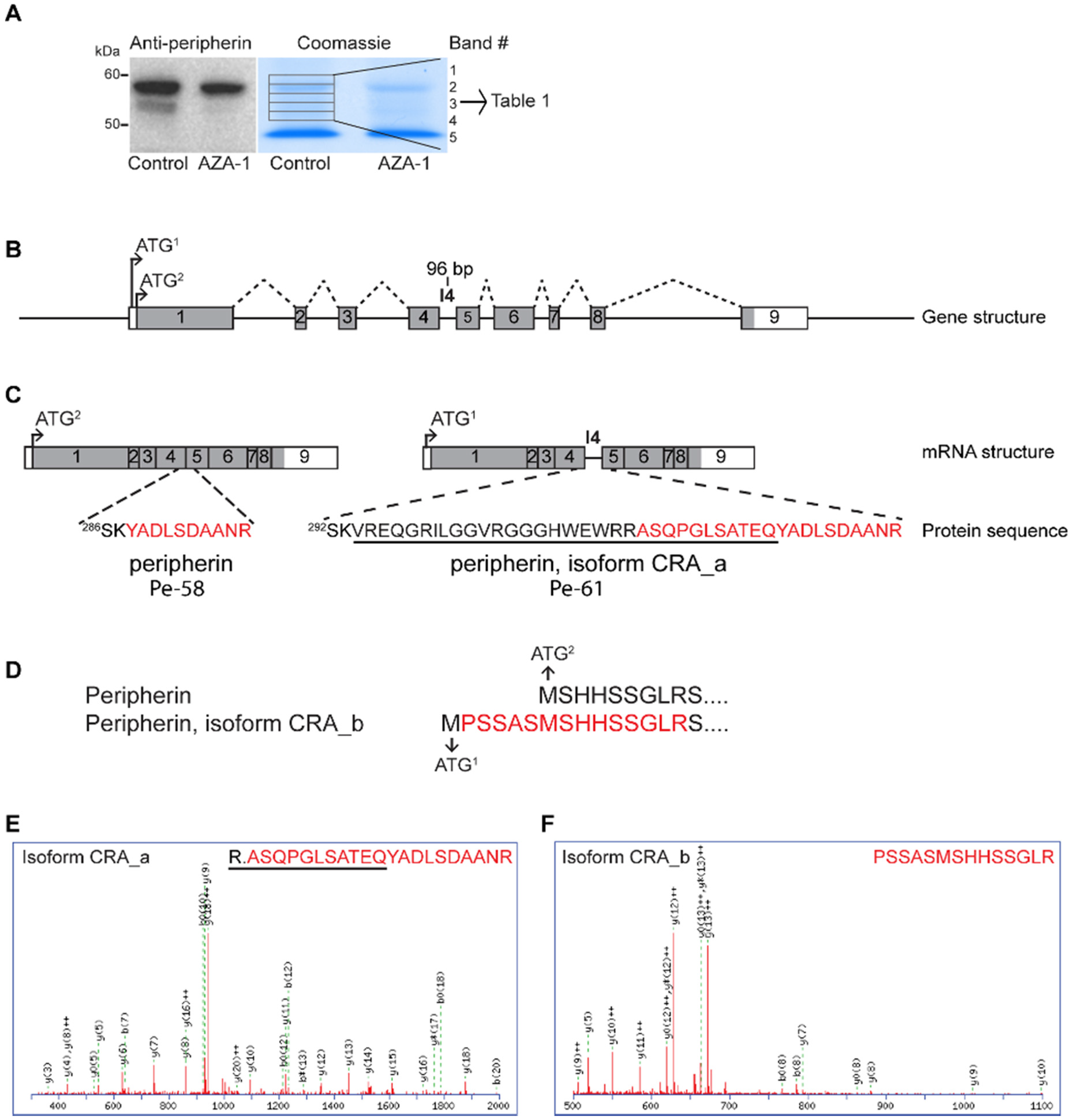
2.3. Mass Spectrometry-Based Verification of Peripherin Isoforms

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GI | Protein Name | No of Amino Acids | Band # | ||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | |||
| 149032092 | Peripherin, isoform CRA_a | 506 | x | ||||
| 129822 | Peripherin | 468 | x | x | x | X | |
| 149032093 | Peripherin, isoform CRA_b | 474 | x | ||||

2.4. Peripherin is Not Down-Regulated at the Transcriptional Level
2.5. Peripherin Down-Regulation Is Not Dependent on the Proteasome, Caspases and Calpains

3. Discussion
4. Experimental Section
4.1. Cell Culturing and Treatment
4.2. Evaluation of Cell Morphology and Viability
4.3. Immunofluorescence
4.4. SDS-PAGE and Western Blotting
4.5. RNA Isolation and Reverse Transcription
4.6. Identification of Peripherin Isoforms
4.7. Data Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ALS | amyotrophic lateral sclerosis |
| AZA-1 | azaspiracid-1 |
| CCK-8 | Cell Counting Kit-8 |
| DSP | diarrhetic shellfish poisoning |
References
- McMahon, T.; Silke, J. Winter toxicity of unknown aetiology in mussels. Harmful Algae News 1996, 14, 2. [Google Scholar]
- Ito, E.; Satake, M.; Ofuji, K.; Kurita, N.; McMahon, T.; James, K.; Yasumoto, T. Multiple organ damage caused by a new toxin azaspiracid, isolated from mussels produced in Ireland. Toxicon 2000, 38, 917–930. [Google Scholar] [CrossRef]
- Ito, E.; Satake, M.; Ofuji, K.; Higashi, M.; Harigaya, K.; McMahon, T.; Yasumoto, T. Chronic effects in mice caused by oral administration of sublethal doses of azaspiracid, a new marine toxin isolated from mussels. Toxicon 2002, 40, 193–203. [Google Scholar] [CrossRef]
- Satake, M.; Ofuji, K.; James, K.J.; Furey, A.; Yasumoto, T. New toxic event caused by Irish mussels. In Harmful Algae; Reguera, B., Blanco, J., Fernandez, M.L., Wyatt, T., Eds.; Xunta de Galicia and IOC of UNESCO: Santiago de Compostella, Spain, 1998; pp. 468–469. [Google Scholar]
- Twiner, M.J.; Hess, P.; Dechraoui, M.Y.; McMahon, T.; Samons, M.S.; Satake, M.; Yasumoto, T.; Ramsdell, J.S.; Doucette, G.J. Cytotoxic and cytoskeletal effects of azaspiracid-1 on mammalian cell lines. Toxicon 2005, 45, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Ofuji, K.; Satake, M.; McMahon, T.; Silke, J.; James, K.J.; Naoki, H.; Oshima, Y.; Yasumoto, T. Two analogs of azaspiracid isolated from mussels, Mytilus edulis, involved in human intoxication in Ireland. Nat. Toxins 1999, 7, 99–102. [Google Scholar] [CrossRef]
- Vilariño, N.; Nicolaou, K.C.; Frederick, M.O.; Cagide, E.; Alfonso, C.; Alonso, E.; Vieytes, M.R.; Botana, L.M. Azaspiracid Substituent at C1 Is Relevant to in vitro Toxicity. Chem. Res. Toxicol. 2008, 21, 1823–1831. [Google Scholar] [CrossRef] [PubMed]
- Twiner, M.J.; Hanagriff, J.C.; Butler, S.; Madhkoor, A.K.; Doucette, G.J. Induction of Apoptosis Pathways in Several Cell Lines following Exposure to the Marine Algal Toxin Azaspiracid. Chem. Res. Toxicol. 2012, 25, 1493–1501. [Google Scholar] [CrossRef] [PubMed]
- Kellmann, R.; Schaffner, C.A.M.; Grønset, T.A.; Satake, M.; Ziegler, M.; Fladmark, K.E. Proteomic response of human neuroblastoma cells to azaspiracid-1. J. Proteom. 2009, 72, 695–707. [Google Scholar] [CrossRef] [PubMed]
- Vale, C.; Nicolaou, K.C.; Frederick, M.O.; Gómez-Limia, B.; Alfonso, A.; Vieytes, M.R.; Botana, L.M. Effects of Azaspiracid-1, a Potent Cytotoxic Agent, on Primary Neuronal Cultures. A Structure-Activity Relationship Study. J. Med. Chem. 2007, 50, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.Y.; LePage, K.T.; Frederick, M.O.; Nicolaou, K.C.; Murray, T.F. Involvement of Caspase Activation in Azaspiracid-Induced Neurotoxicity in Neocortical Neurons. Toxicol. Sci. 2010, 114, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Vale, C.; Nicolaou, K.C.; Frederick, M.O.; Vieytes, M.R.; Botana, L.M. Cell Volume Decrease as a Link between Azaspiracid-Induced Cytotoxicity and c-Jun-N-Terminal Kinase Activation in Cultured Neurons. Toxicol. Sci. 2010, 113, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Vale, C.; Gómez-Limia, B.; Nicolaou, K.C.; Frederick, M.O.; Vieytes, M.R.; Botana, L.M. The c-Jun-N-Terminal Kinase is Involved in the Neurotoxic Effect of Azaspiracid-1. Cell. Physiol. Biochem. 2007, 20, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Twiner, M.J.; Ryan, J.C.; Morey, J.S.; Smith, K.J.; Hammad, S.M.; van Dolah, F.M.; Hess, P.; McMahon, T.; Satake, M.; Yasumoto, T.; et al. Transcriptional profiling and inhibition of cholesterol biosynthesis in human T lymphocyte cells by the marine toxin azaspiracid. Genomics 2008, 91, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Kulagina, N.V.; Twiner, M.J.; Hess, P.; McMahon, T.; Satake, M.; Yasumoto, T.; Ramsdell, J.S.; Doucette, G.J.; Ma, W.; O’Shaughnessy, T.J. Azaspiracid-1 inhibits bioelectrical activity of spinal cord neuronal networks. Toxicon 2006, 47, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Twiner, M.J.; Doucette, G.J.; Rasky, A.; Huang, X.-P.; Roth, B.L.; Sanguinetti, M.C. Marine Algal Toxin Azaspiracid Is an Open-State Blocker of hERG Potassium Channels. Chem. Res. Toxicol. 2012, 25, 1975–1984. [Google Scholar] [CrossRef] [PubMed]
- Chevallier, O.P.; Graham, S.F.; Alonso, E.; Duffy, C.; Silke, J.; Campbell, K.; Botana, L.M.; Elliott, C.T. New insights into the causes of human illness due to consumption of azaspiracid contaminated shellfish. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Aune, T.; Espenes, A.; Aasen, J.A.; Quilliam, M.A.; Hess, P.; Larsen, S. Study of possible combined toxic effects of azaspiracid-1 and okadaic acid in mice via the oral route. Toxicon 2012, 60, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.A.; Tischler, A.S. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA 1976, 73, 2424–2428. [Google Scholar] [CrossRef] [PubMed]
- Satsu, H.; Yokoyama, T.; Ogawa, N.; Fujiwara-Hatano, Y.; Shimizu, M. The changes in the neuronal PC12 and the intestinal epithelial Caco-2 cells during the coculture. The functional analysis using an in vitro coculture system. Cytotechnology 2001, 35, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Brody, B.A.; Ley, C.A.; Parysek, L.M. Selective distribution of the 57 kDa neural intermediate filament protein in the rat CNS. J. Neurosci. 1989, 9, 2391–2401. [Google Scholar] [PubMed]
- Escurat, M.; Djabali, K.; Gumpel, M.; Gros, F.; Portier, M. Differential expression of two neuronal intermediate-filament proteins, peripherin and the low-molecular-mass neurofilament protein (NF-L), during the development of the rat. J. Neurosci. 1990, 10, 764–784. [Google Scholar] [PubMed]
- Leonard, D.G.; Gorham, J.D.; Cole, P.; Greene, L.A.; Ziff, E.B. A nerve growth factor-regulated messenger RNA encodes a new intermediate filament protein. J. Cell Biol. 1988, 106, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Parysek, L.M.; Goldman, R.D. Distribution of a novel 57 kDa intermediate filament (IF) protein in the nervous system. J. Neurosci. 1988, 8, 555–563. [Google Scholar] [PubMed]
- Portier, M.M.; de Nechaud, B.; Gros, F. Peripherin, a new member of the intermediate filament protein family. Dev. Neurosci. 1983, 6, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Aletta, J.M.; Angeletti, R.; Liem, R.K.H.; Purcell, C.; Shelanski, M.L.; Greene, L.A. Relationship Between the Nerve Growth Factor-Regulated Clone 73 Gene Product and the 58-Kilodalton Neuronal Intermediate Filament Protein (Peripherin). J. Neurochem. 1988, 51, 1317–1320. [Google Scholar] [CrossRef] [PubMed]
- Helfand, B.T.; Mendez, M.G.; Pugh, J.; Delsert, C.; Goldman, R.D. A Role for Intermediate Filaments in Determining and Maintaining the Shape of Nerve Cells. Mol. Biol. Cell 2003, 14, 5069–5081. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.; Doroudchi, M.M.; Nguyen, M.D.; Durham, H.D.; Strong, M.J.; Shaw, G.; Julien, J.-P.; Mushynski, W.E. A neurotoxic peripherin splice variant in a mouse model of ALS. J. Cell Biol. 2003, 160, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Maioli, M.; Rinaldi, S.; Migheli, R.; Pigliaru, G.; Rocchitta, G.; Santaniello, S.; Basoli, V.; Castagna, A.; Fontani, V.; Ventura, C.; et al. Neurological morphofunctional differentiation induced by REAC technology in PC12. A neuro protective model for Parkinson’s disease. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Shea, T.B.; Beermann, M.L. Respective roles of neurofilaments, microtubules, MAP1B, and tau in neurite outgrowth and stabilization. Mol. Biol. Cell 1994, 5, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Landon, F.; Wolff, A.; de Nechaud, B. Mouse peripherin isoforms. Biol. Cell 2000, 92, 397–407. [Google Scholar] [CrossRef]
- Landon, F.; Lemonnier, M.; Benarous, R.; Huc, C.; Fiszman, M.; Gros, F.; Portier, M.M. Multiple mRNAs encode peripherin, a neuronal intermediate filament protein. EMBO J. 1989, 8, 1719–1726. [Google Scholar] [PubMed]
- Kim, C.; Yun, N.; Lee, Y.M.; Jeong, J.Y.; Baek, J.Y.; Song, H.Y.; Ju, C.; Youdim, M.B.H.; Jin, B.K.; Kim, W.K.; et al. Gel-based Protease Proteomics for Identifying the Novel Calpain Substrates in Dopaminergic Neuronal Cell. J. Biol. Chem. 2013, 288, 36717–36732. [Google Scholar] [CrossRef] [PubMed]
- Goll, D.E.; Thompson, V.F.; Li, H.; Wei, W.; Cong, J. The calpain system. Phys. Rev. 2003, 83, 731–801. [Google Scholar] [CrossRef] [PubMed]
- Parysek, L.M.; Goldman, R.D. Characterization of intermediate filaments in PC12 cells. J. Neurosci. 1987, 7, 781–791. [Google Scholar] [PubMed]
- Corbo, M.; Hays, A.P. Peripherin and Neurofilament Protein Coexist in Spinal Spheroids of Motor Neuron Disease. J. Neuropathol. Exp. Neurol. 1992, 51, 531–537. [Google Scholar] [CrossRef] [PubMed]
- McLean, J.; Liu, H.N.; Miletic, D.; Weng, Y.C.; Rogaeva, E.; Zinman, L.; Kriz, J.; Robertson, J. Distinct biochemical signatures characterize peripherin isoform expression in both traumatic neuronal injury and motor neuron disease. J. Neurochem. 2010, 114, 1177–1192. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Tjostheim, S.; Sanelli, T.; McLean, J.R.; Horne, P.; Fan, Y.; Ravits, J.; Strong, M.J.; Robertson, J. An Aggregate-Inducing Peripherin Isoform Generated through Intron Retention Is Upregulated in Amyotrophic Lateral Sclerosis and Associated with Disease Pathology. J. Neurosci. 2008, 28, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Beaulieu, J.-M.; Nguyen, M.D.; Julien, J.-P. Late Onset Death of Motor Neurons in Mice Overexpressing Wild-Type Peripherin. J. Cell Biol. 1999, 147, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Oblinger, M.; Wong, J.; Parysek, L. Axotomy-induced changes in the expression of a type III neuronal intermediate filament gene. J. Neurosci. 1989, 9, 3766–3775. [Google Scholar] [PubMed]
- McLean, J.R.; Smith, G.A.; Rocha, E.M.; Osborn, T.M.; Dib, S.; Hayes, M.A.; Beagan, J.A.; Brown, T.B.; Lawson, T.F.S.; Hallett, P.J.; et al. ALS-associated peripherin spliced transcripts form distinct protein inclusions that are neuroprotective against oxidative stress. Exp. Neurol. 2014, 261, 217–229. [Google Scholar] [CrossRef] [PubMed]
- McLean, J.R.; Robertson, J. Isoform-specific expression and ratio changes accompany oxidant-induced peripherin aggregation in a neuroblastoma cell line. Brain Res. 2011, 1422, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Bruening, W.; Durham, H.D.; Mushynski, W.E. Activation of Stress-Activated Protein Kinases Correlates with Neurite Outgrowth Induced by Protease Inhibition in PC12 Cells. J. Neurochem. 1999, 72, 1081–1087. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Zhang, H.; Zhang, Y.; Zhang, Y.; Bai, L.; Chen, Q.; Wu, J.; Zhang, L. Differential protein profile of PC12 cells exposed to proteasomal inhibitor lactacystin. Neurosci. Lett. 2014, 575, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Masaki, R.; Saito, T.; Yamada, K.; Ohtani-Kaneko, R. Accumulation of phosphorylated neurofilaments and increase in apoptosis-specific protein and phosphorylated c-Jun induced by proteasome inhibitors. J. Neurosci. Res. 2000, 62, 75–83. [Google Scholar] [CrossRef]
- Song, E.J.; Hong, H.-M.; Yoo, Y.S. Proteasome inhibition induces neurite outgrowth through posttranslational modification of TrkA receptor. Int. J. Biochem. Cell Biol. 2009, 41, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Tarjányi, O.; Berta, G.; Harci, A.; Bacsa, E.B.; Stark, B.; Pap, M.; Szeberényi, J.; Sétáló, G., Jr. The role of Src protein in the process formation of PC12 cells induced by the proteasome inhibitor MG-132. Neurochem. Int. 2013, 63, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J.; Leystra-Lantz, C.; Ge, W.-W. Intermediate filament steady-state mRNA levels in amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2004, 316, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Palokangas, H.; Ying, M.; Väänänen, K.; Saraste, J. Retrograde Transport from the Pre-Golgi Intermediate Compartment and the Golgi Complex Is Affected by the Vacuolar H+-ATPase Inhibitor Bafilomycin A1. Mol. Biol. Cell 1998, 9, 3561–3578. [Google Scholar] [CrossRef] [PubMed]
- Aasebo, E.; Opsahl, J.A.; Bjorlykke, Y.; Myhr, K.M.; Kroksveen, A.C.; Berven, F.S. Effects of blood contamination and the rostro-caudal gradient on the human cerebrospinal fluid proteome. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hjørnevik, L.V.; Frøyset, A.K.; Grønset, T.A.; Rungruangsak-Torrissen, K.; Fladmark, K.E. Algal Toxin Azaspiracid-1 Induces Early Neuronal Differentiation and Alters Peripherin Isoform Stoichiometry. Mar. Drugs 2015, 13, 7390-7402. https://doi.org/10.3390/md13127072
Hjørnevik LV, Frøyset AK, Grønset TA, Rungruangsak-Torrissen K, Fladmark KE. Algal Toxin Azaspiracid-1 Induces Early Neuronal Differentiation and Alters Peripherin Isoform Stoichiometry. Marine Drugs. 2015; 13(12):7390-7402. https://doi.org/10.3390/md13127072
Chicago/Turabian StyleHjørnevik, Linda V., Ann K. Frøyset, Toril A. Grønset, Krisna Rungruangsak-Torrissen, and Kari E. Fladmark. 2015. "Algal Toxin Azaspiracid-1 Induces Early Neuronal Differentiation and Alters Peripherin Isoform Stoichiometry" Marine Drugs 13, no. 12: 7390-7402. https://doi.org/10.3390/md13127072
APA StyleHjørnevik, L. V., Frøyset, A. K., Grønset, T. A., Rungruangsak-Torrissen, K., & Fladmark, K. E. (2015). Algal Toxin Azaspiracid-1 Induces Early Neuronal Differentiation and Alters Peripherin Isoform Stoichiometry. Marine Drugs, 13(12), 7390-7402. https://doi.org/10.3390/md13127072