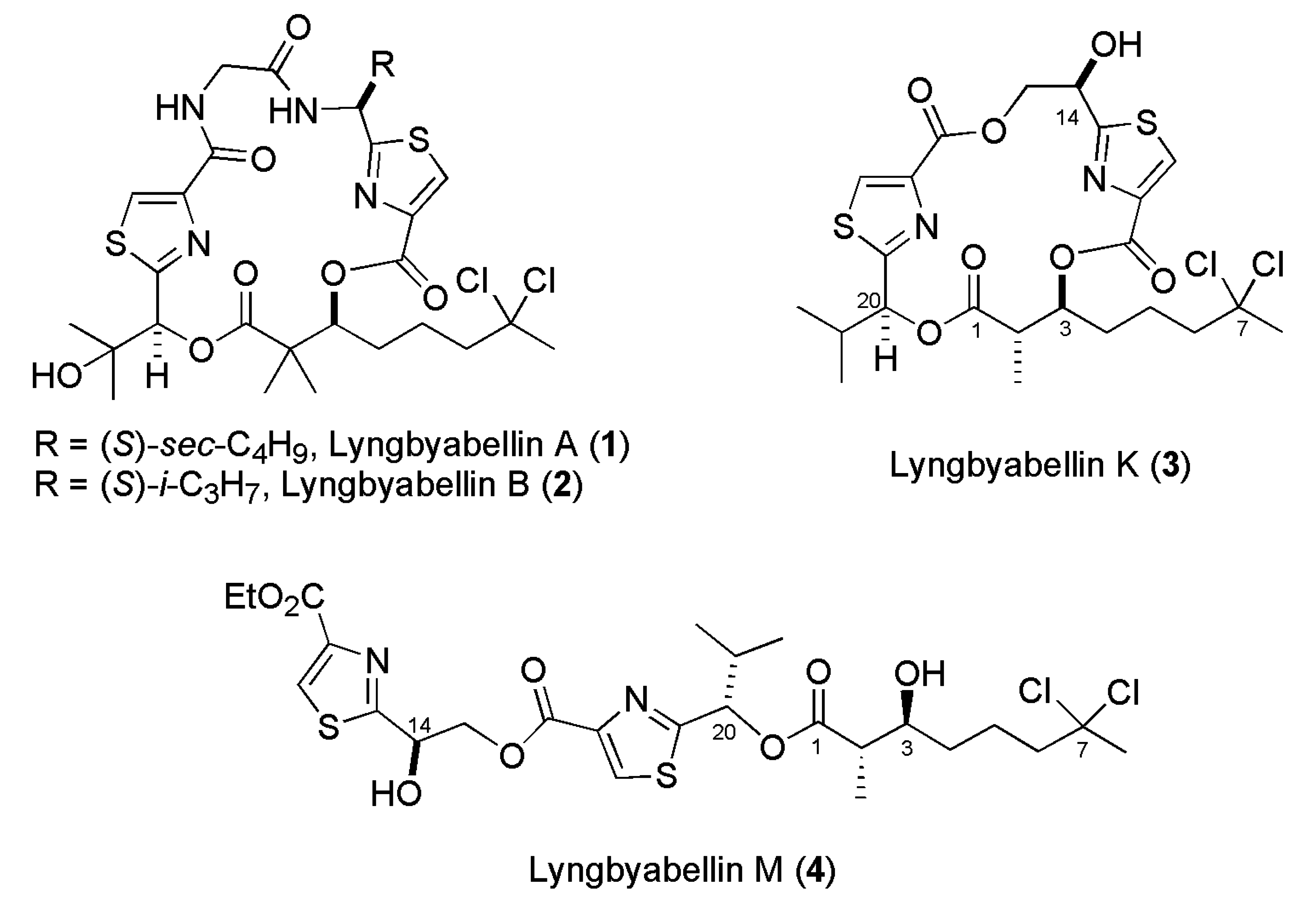
2. Results and Discussion
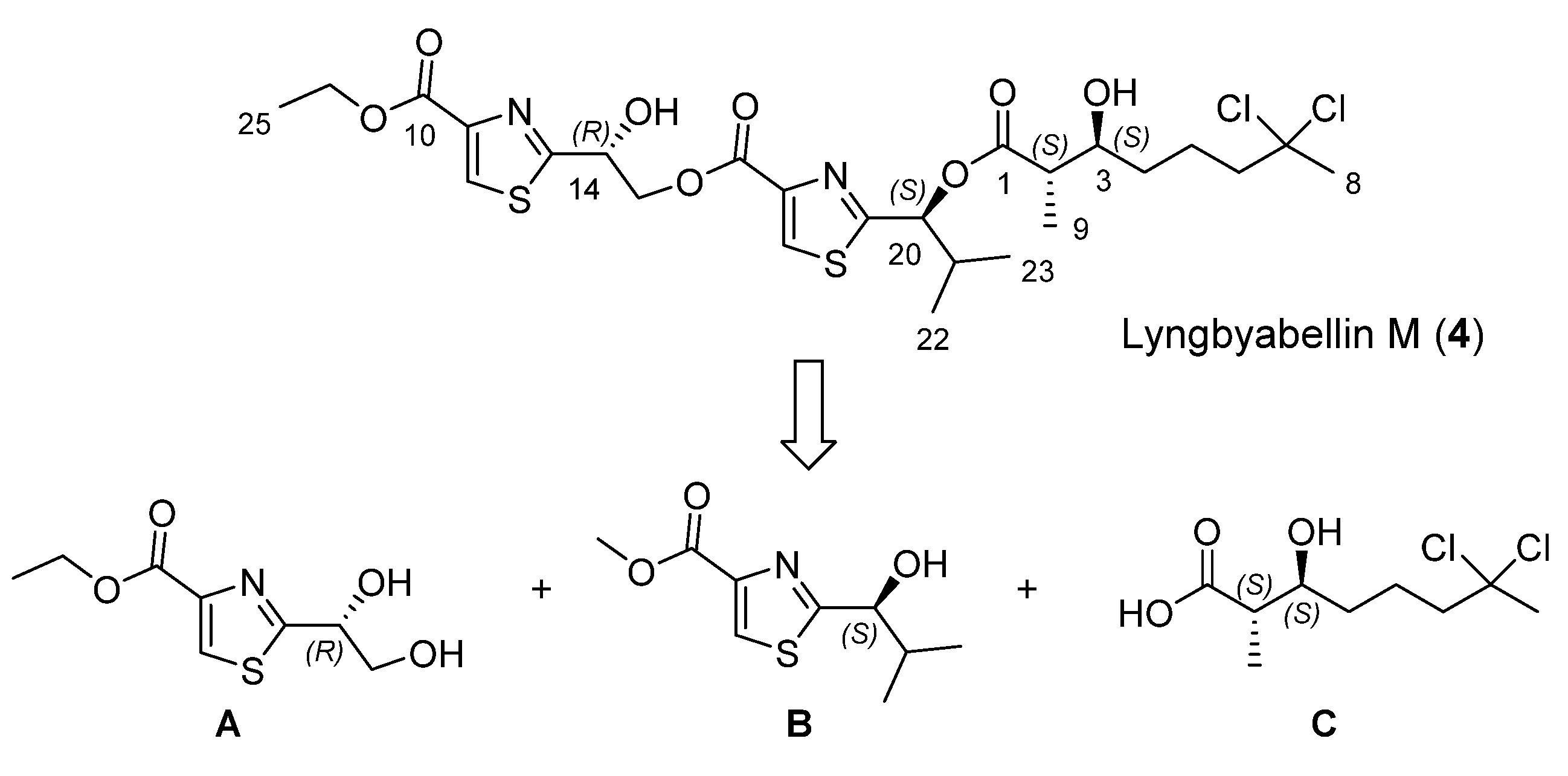
Our approach to lyngbyabellin M (
4) relies on the disconnection at the two ester bonds which simplifies its preparation to the synthesis of three chiral fragments
A–
C (
Scheme 1).
Scheme 1.
Retrosynthetic analysis for lyngbyabellin M (4).
Scheme 1.
Retrosynthetic analysis for lyngbyabellin M (4).
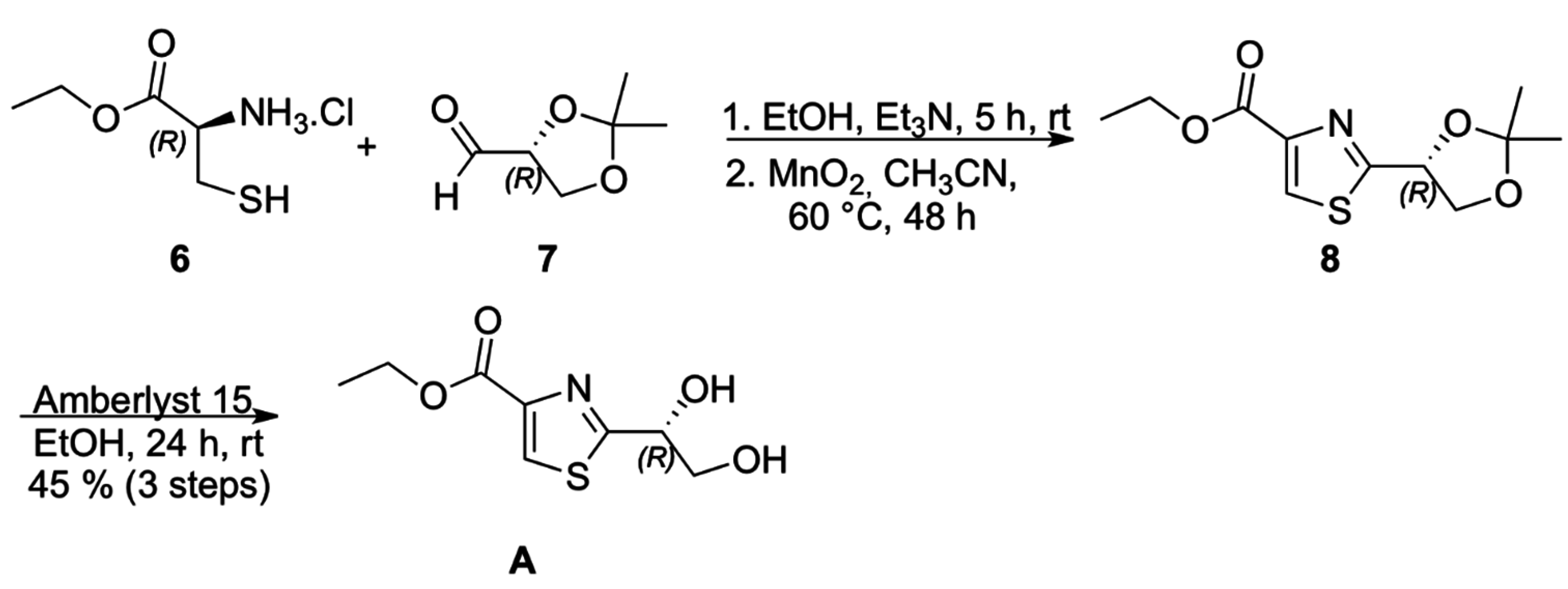
Chiral thiazole
A (
Scheme 2) was prepared from condensation of (
S)-cysteine ethyl ester hydrochloride (
6) with (
R)-isopropylidene glyceraldehyde (
7), followed by MnO
2 oxidation of the intermediate thiazolidine, according to the methodology described by Iwakawa and coworkers for the corresponding methyl ester, to afford 3,4-disubstituted thiazole
8 which underwent acetonide deprotection to provide thiazole
A in 36% overall yield and specific optical rotation [α]
D20 = +59 (MeOH, 1.15) [
11,
12,
13].
Scheme 2.
Synthesis of thiazole A.
Scheme 2.
Synthesis of thiazole A.
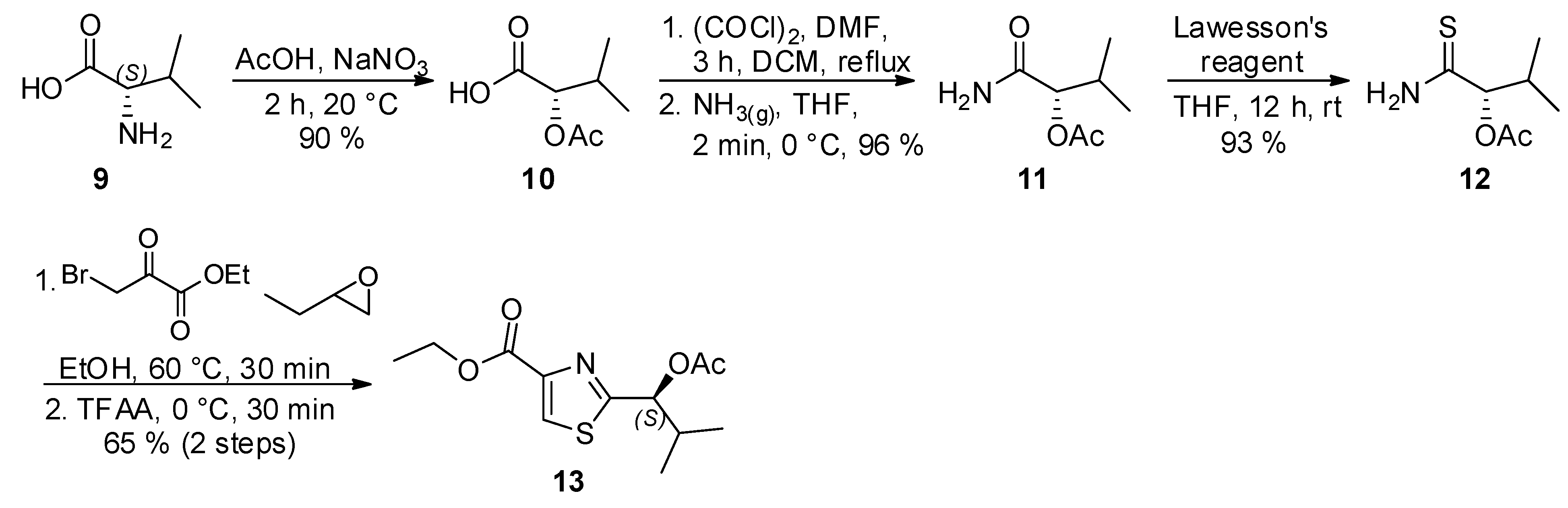
As to the preparation of thiazole
B, a modification of the Hantzch synthesis was employed to prepare
13, following the procedure described by Schmidt and coworkers (
Scheme 3). Accordingly, the reaction of thioamide
12, readily obtained from (
S)-valine (
9), with ethyl bromopyruvate, followed by dehydration of the intermediate upon treatment with trifluoroacetic anhydride (TFAA) at 0 °C afforded thiazole
13 as white solid ([α]
D25 = −37.0 (
c 0.90, CHCl
3); [α]
D25 = −38.6 (
c 1.09, CHCl
3)) [
14].
Scheme 3.
Synthesis of thiazole 13.
Scheme 3.
Synthesis of thiazole 13.
Methanolysis of thiazole
13 required a detailed investigation of the reaction conditions in order to avoid extensive racemization (
Table 1). Initially, we carried out the hydrolysis/transesterification step according to the e procedure in the literature which provided the desired methyl ester in 70% yield but its optical rotation [α]
D25 = −25.0 (
c 0.65, CHCl
3) was slightly lower than the one previously reported ([α]
D25 = −31.2 (
c 0.61,CHCl
3)) [
11,
12,
13]. Other hydrolytic conditions were tested and, in our hands, the best one proved to be the use of dibutyltin oxide in refluxing methanol [
15] which afforded the desired fragment
B in 71% yield and [α]
D25 = −27.0 (
c 0.70, CHCl
3 ). Despite the less than optimal optical purity for intermediate
B, we decided to move on in our synthetic plan as fragment
B would be coupled with fragment
C which was expected to be prepared in high enantiomeric ratio. In the event, the undesired diastereoisomer obtained after the coupling of fragments
B and
C would be removed by chromatography, affording the desired ester
25 in pure form.
Table 1.
Hydrolysis conditions for the preparation of fragment
B.
![Marinedrugs 13 03309 i001]()
Table 1.
Hydrolysis conditions for the preparation of fragment B. ![Marinedrugs 13 03309 i001]()
| Entry | Conditions | Yield (%) | [α]D25 (c, CHCl3) |
|---|
| 1 | MeONa/MeOH, rt, 4 h | 85 | −25 (0.78) |
| 2 | K2CO3, MeOH, reflux, 2 h | 70 | −25 (0.65) |
| 3 | (i) LiOH.H2O, MeOH, rt, 1 h
(ii) TMSCH2N2, THF, rt, 20 min | 34 | −20 (0.66) |
| 4 | Bu2SnO, MeOH, reflux, 12 h | 71 | −27 (0.78) |
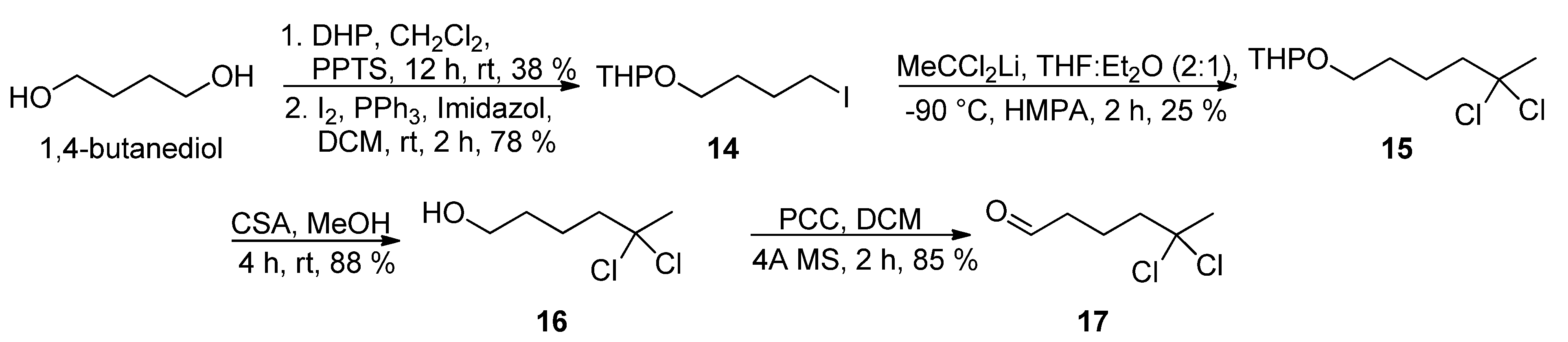
The preparation of
gem-dichloro aldehyde
17 was initially investigated via the nucleophilic substitution reaction of iodide
14 [
16,
17] with the lithium anion derived from 1,1-dichloroethane to afford
15, followed by removal of the tetrahydropyranyl group and oxidation of the primary alcohol in
16 (
Scheme 4). However, this was not a practical solution as the
gem-dichloro pyranyl ether
15 was obtained in 25% yield at most.
Scheme 4.
Synthesis of aldehyde 17.
Scheme 4.
Synthesis of aldehyde 17.
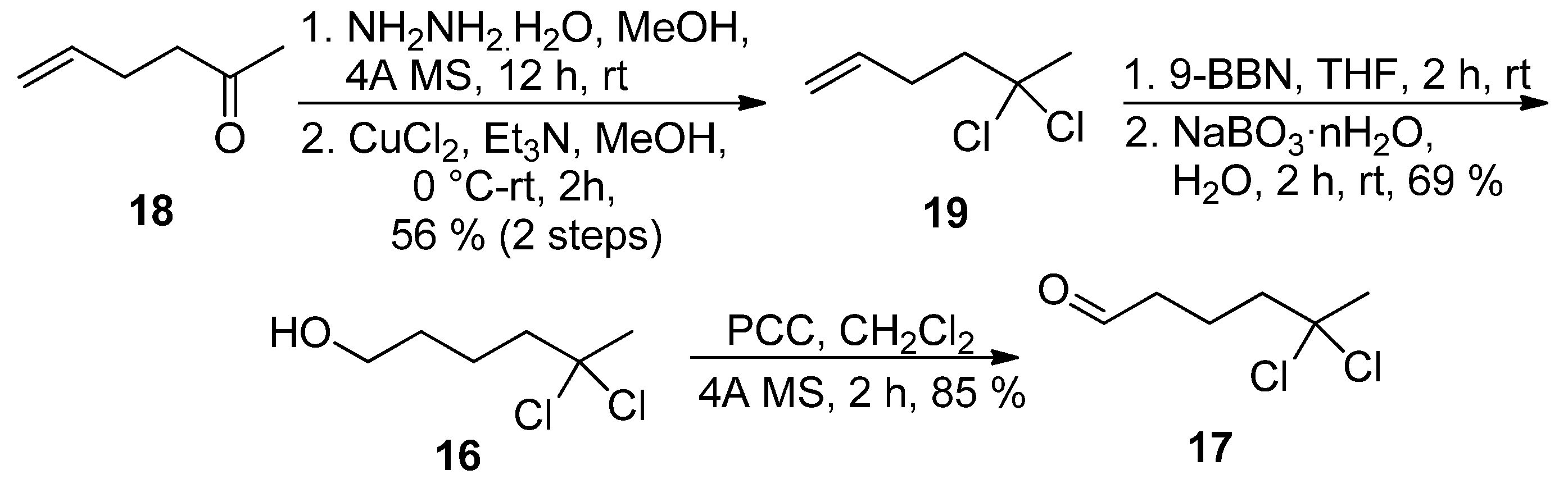
The best solution came with the preparation of
gem-dichloro alkene
19 in 56% yield (two steps) from ketone
18 using the oxidation of the corresponding hydrazone by CuCl
2 as described by Takeda and coworkers [
18]. Hydroboration-oxidation sequence, followed by oxidation of primary alcohol
16 secured preparation of
gem-dichloro aldehyde
17 in 32% overall yield (
Scheme 5).
Scheme 5.
Alternative synthesis of aldehyde 17.
Scheme 5.
Alternative synthesis of aldehyde 17.
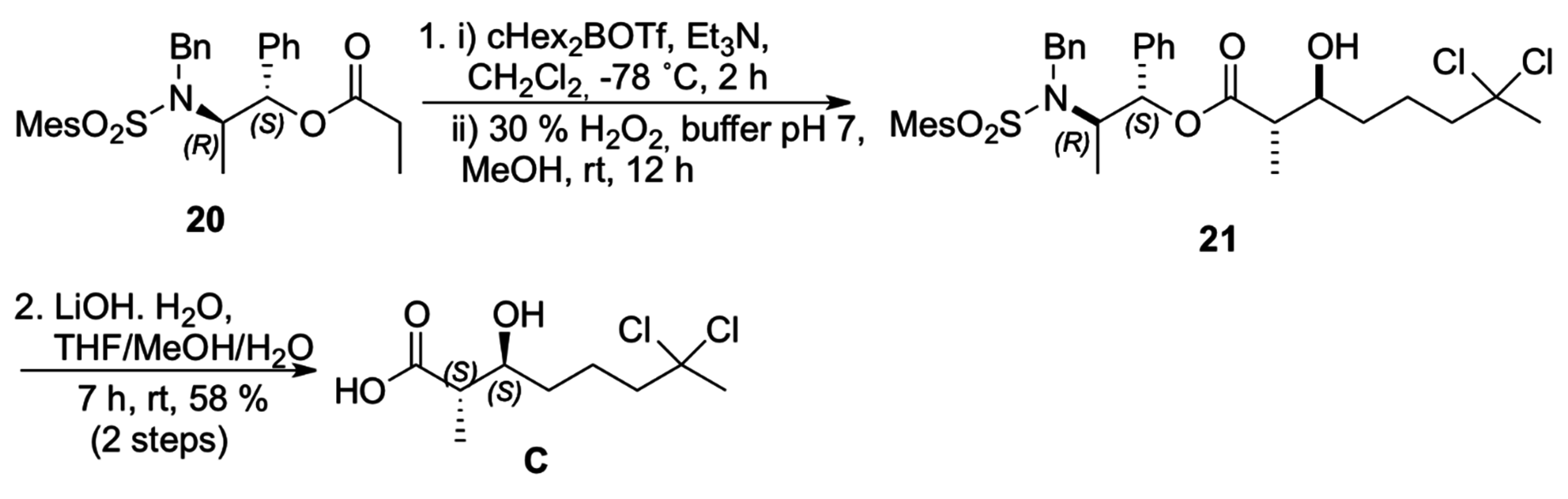
The construction of carboxylic acid
C was then accomplished using the
anti-selective boron-mediated asymmetric aldol reaction with propionate
20 prepared from Masamune’s chiral auxiliary [
19].
anti-Aldol
21 was prepared via enolization of ester with dicyclohexylboron triflate and triethylamine at −78 °C (
Scheme 6) [
20]. The absolute configuration at the two newly formed stereogenic centers was assigned after conversion of the aldol adduct
21 to the corresponding carboxylic acid
C (58% yield for two steps) [
21].
Scheme 6.
Synthesis of carboxilyc acid C.
Scheme 6.
Synthesis of carboxilyc acid C.
Despite the previous results on the aldol reaction with propionate derived from Masamune’s auxiliary described in the literature [
19], we decided to confirm the
anti relative configuration via conversion of the carboxylic acid obtained from
ent-
21 to the corresponding acetonide
22, after reduction with LiAlH
4 and treatment with 2,2-dimethoxypropane under acid catalysis (40% overall yield). The large coupling constant (
3J = 11.6 Hz) observed between Ha and Hb in
22 revealed their
trans disposition which translates to the expected
anti configuration in the corresponding carboxylic acid (
Scheme 7).
Scheme 7.
Conversion of ent-21 to acetonide 22.
Scheme 7.
Conversion of ent-21 to acetonide 22.
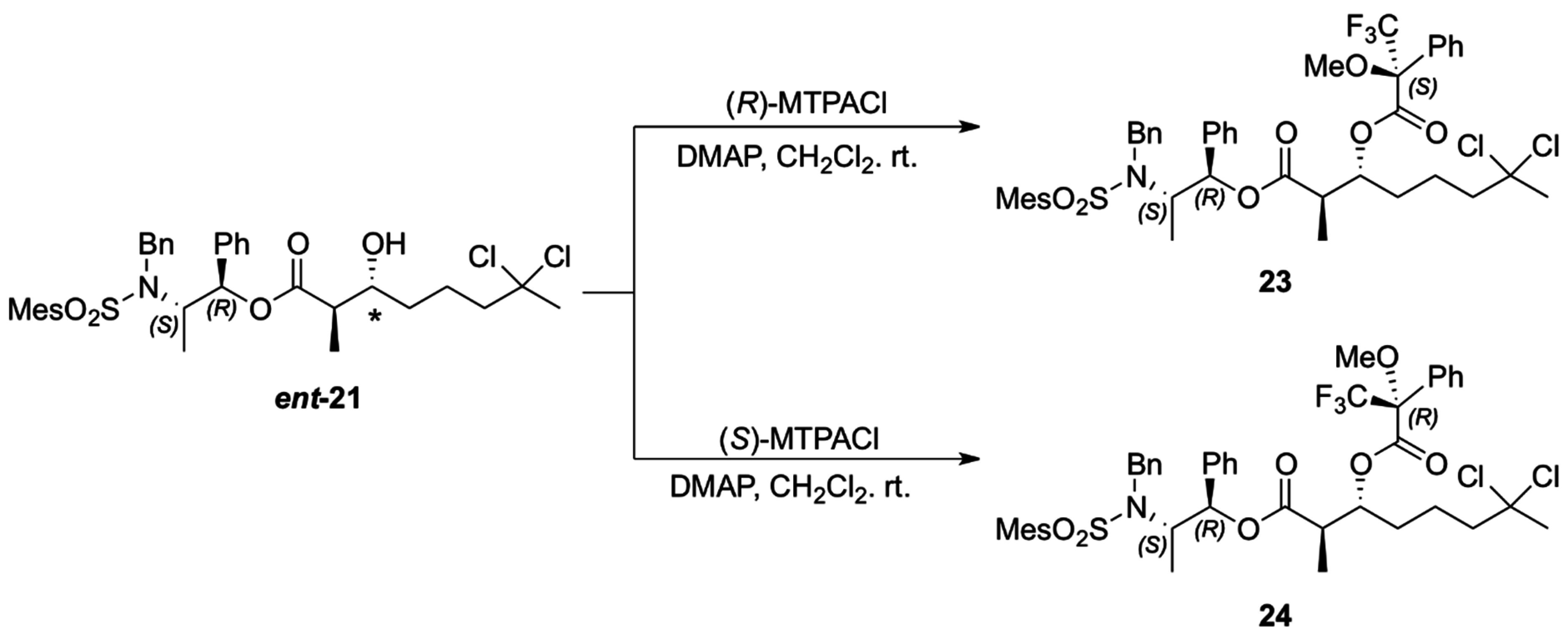
The absolute configuration of the two newly generated stereogenic centers in
ent-21 was assigned via the corresponding Mosher esters
23 and
24, prepared from (
R)- and (
S)-α-methoxy-α- trifluoromethylphenylacetyl chloride (MTPACl), respectively (
Scheme 8). Analyses of the
1H-NMR spectra for Mosher esters
23 and
24 revealed Δδ
S − R < 0 for H14, H15, H16 and H19 (up to −0.12 ppm) and positive values for H5 (+0.05 ppm) which translate to
R configuration for both the carbinolic carbon and the methine carbon α to the carbonyl group in
ent-
21 (
Table 2) [
22,
23,
24].
Scheme 8.
Preparation of the MTPA esters of ent-21.
Scheme 8.
Preparation of the MTPA esters of ent-21.
Table 2.
Chemical shifts and Δδ
S − R values for MTPA esters derived from
ent-
21.
![Marinedrugs 13 03309 i002]()
Table 2.
Chemical shifts and ΔδS − R values for MTPA esters derived from ent-21. ![Marinedrugs 13 03309 i002]()
| H | (S)-MTPA Ester (23) | (R)-MTPA Ester (24) | Δ (δS − δR) |
|---|
| δH (ppm) | Multiplicity (J) | δH (ppm) | Multiplicity (J) |
|---|
| H-5 | 1.98 | s | 1.93 | s | +0.053 |
| H-14 | 0.90 | d (3H, J = 7.4) | 1.02 | d (3H, J = 7.2) | −0.12 |
| H-15 | 5.73 | d (1H, J = 5.1) | 5.76 | d (1H, J = 5.1) | −0.034 |
| H-16 | 4.04 | m (1H) | 4.06 | m (1H) | −0.02 |
| H-19 | 1.07 | d (3H, J = 6.9) | 1.10 | d (3H, J = 6.8) | −0.036 |
| H-37/38 | 2.37 | s (6H) | 2.37 | s (6H) | −0.025 |
| H-39 | 2.20 | s (3H) | 2.21 | s (3H) | −0.005 |
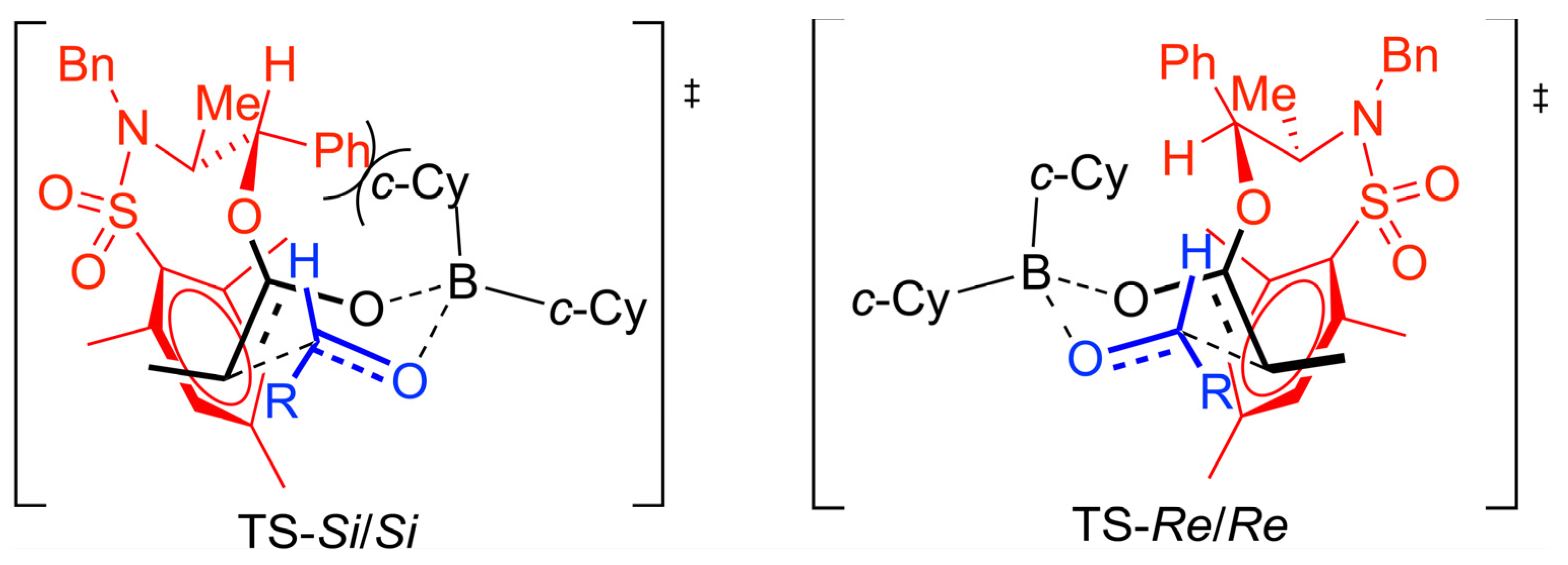
The preference for the
anti stereochemistry observed for the major aldol product requires a kinetically controlled approach of the
E-boron enolate
Re face to the
Re face of the aldehyde according to a 6-membered Zimmerman-Traxler transition state model. The stabilizing interaction involving the electron-deficient
N-sulfonylmesityl group and the electron-rich double bond of the boron enolate can be accommodated in the transition state corresponding to the
Re/
Re topology while for the
Si/
Si topology which would lead to the diastereoisomeric
anti aldol product, a destabilizing interaction develops between the pseudoaxial cyclohexyl group and the
N-mesityl group of the
N-mesitylsulfonyl norephedrine chiral auxiliary (
Scheme 9).
Scheme 9.
Proposed transition state model for the aldol reaction.
Scheme 9.
Proposed transition state model for the aldol reaction.
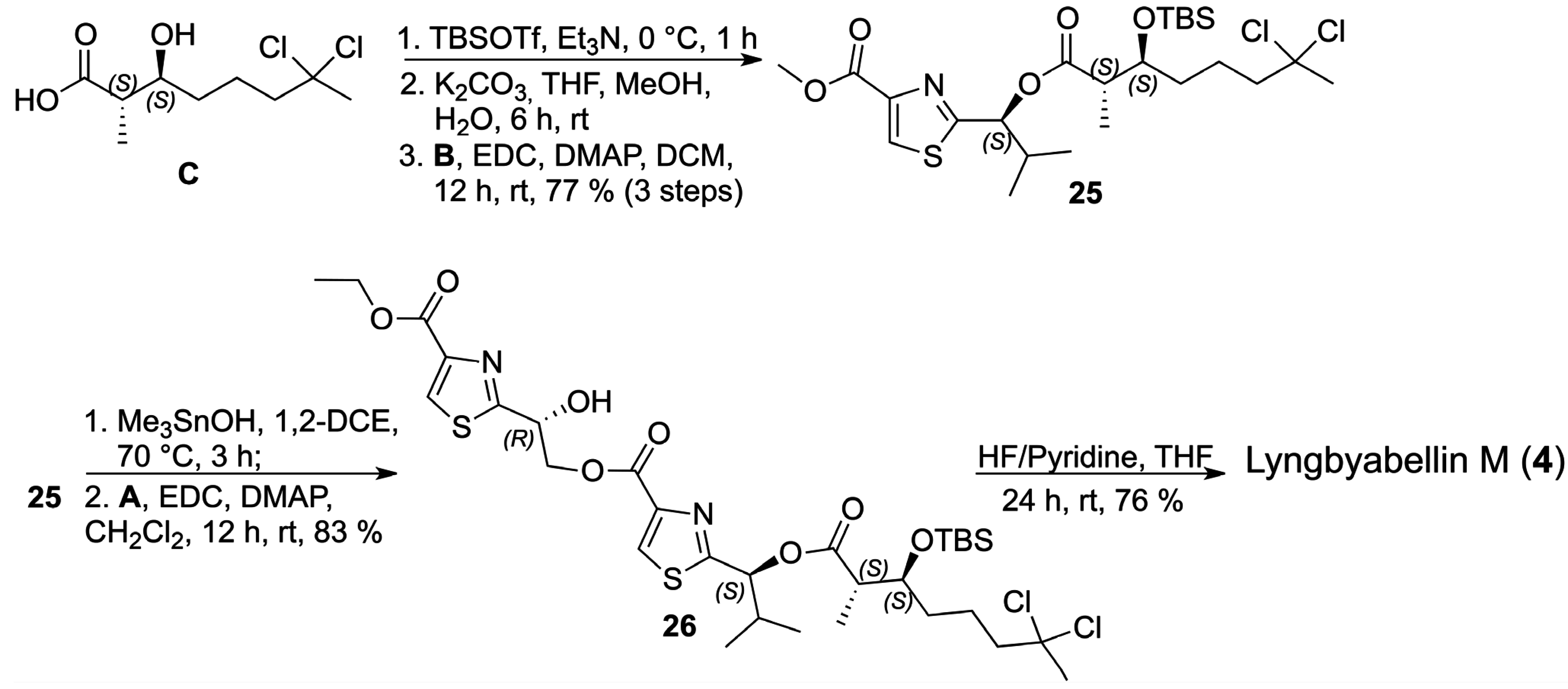
With the three key fragments in hands, we initiated the assembly of the structure assigned for lyngbyabellin M (
4) by coupling the carboxylic acid
C with thiazole
B which required the protection of the secondary alcohol in
C as the corresponding
terc-butyldimethylsilyl (TBS) ether, hydrolysis to the carboxylic acid, followed by coupling with thiazole
B. After purification, the corresponding ester
25 was obtained in 77% yield as a single diastereoisomer over 3 steps, as indicated by the analysis of the
1H- and
13C-NMR spectra. In order to install thiazole
A, a chemoselective hydrolysis of the methyl ester in
25 was required which was accomplished using hydroxytrimethyltin in 1,2-dichloroethane without any indication of epimerization by
1H- and
13C-NMR analyses [
25,
26,
27]. The corresponding carboxylic acid was prepared from
25 and it was coupled to thiazole
A via the EDC/DMAP protocol to afford
26 in 83% yield over two steps (
Scheme 10).
Scheme 10.
Final steps in the total synthesis of lyngbyabellin M (4).
Scheme 10.
Final steps in the total synthesis of lyngbyabellin M (4).
Comparison of the NMR data of the synthetic sample of lyngbyabellin M (
4) prepared after deprotection of the TBS group in
26 with HF/pyridine with those available in the literature for lyngbyabellin M (
4) revealed almost complete match except for C-13, C-14, C-15, C-16, C-20 and C-21. Personal information provided by Gerwick and coworkers after completion of our work provided revised
13C-NMR data for natural lyngbyabelin M (
4) which nicely fit those obtained for the synthetic sample thus confirming the 2
S,3
S,14
R,20
S configuration proposed for the natural product by spectroscopic analysis and chemical correlation (
Table 3).
Table 3.
Comparison of the 13C-NMR data for natural (CDCl3, 125 MHz) and synthetic (CDCl3, 125 MHz) lyngbyabellin M.
Table 3.
Comparison of the 13C-NMR data for natural (CDCl3, 125 MHz) and synthetic (CDCl3, 125 MHz) lyngbyabellin M.
| Carbon | Natural Lyngbyabellin M *δ (ppm) | Synthetic Lyngbyabellin M δ (ppm) |
|---|
| 1 | 174.4 | 174.3 |
| 2 | 44.4 | 44.4 |
| 3 | 75.4 | 75.3 |
| 4 | 34.4 | 34.4 |
| 5 | 21.5 | 21.5 |
| 6 | 50.0 | 49.9 |
| 7 | 90.8 | 90.8 |
| 8 | 37.7 | 37.6 |
| 9 | 15.0 | 14.9 |
| 10 | 161.8 | 161.7 |
| 11 | 147.6 | 147.5 |
| 12 | 128.2 | 128.2 |
| 13 | 171.2 (172.1) ** | 172.1 |
| 14 | 70.30 (70.05) ** | 70.0 |
| 15 | 70.26 (70.15) ** | 70.2 |
| 16 | 161.7 (160.6) * | 160.5 |
| 17 | 145.9 | 145.7 |
| 18 | 129.2 | 129.1 |
| 19 | 170.5 | 170.4 |
| 20 | 77.5 (77.1) ** | 77.2 |
| 21 | 34.4 (34.2) ** | 34.3 |
| 22 | 19.5 | 19.4 |
| 23 | 16.8 | 16.7 |
| 24 | 61.8 | 61.7 |
| 25 | 14.7 | 14.6 |
Although synthetic lyngbyabellin M (4) displayed the same absolute configuration as that originally proposed by Gerwick and coworkers for the natural product, the synthetic sample was shown to be dextrorotatory ([α]D25 = +12 (c = 0.5, MeOH)) while natural lyngbyabellin M was reported as levorotatory ([α]D25 = −4.5 (c =0.5, MeOH)). We are presently not able to explain this discrepancy since a sample of the natural product was not available for us to compare natural and synthetic lyngbyabellins M by chiral HPLC or other chiroptical techniques.
3. Experimental Section
Reagents and solvents were commercial grade and were used as supplied except when specified in the experimental procedure. Triethylamine and dichloromethane were distilled from calcium hydride and tetrahydrofuran was distilled from Na/benzophenone. Reactions were monitored by thin layer chromatography analysis using Merck Silica Gel 60 F-254 thin layer plates. Flash column chromatography was performed on Acros silica gel 60, 0.040–0.063 mm. 1H NMR and 13C NMR data were recorded on a Varian Inova (500 MHz for 1H and 125 MHz for 13C NMR) or Bruker Avance (250 MHz for 1H and 62.5 MHz for 13C NMR) spectrometers using as internal standard the residual nondeuterated solvent (CHCl3) or TMS (1H NMR). High resolution mass spectra (HRMS) for novel compounds were measured on a Waters XEVO Quadrupole-Time of Flight (Q-TOF) spectrometer (ESI) or in a Waters Premier (EI). Infrared spectra (IR) were obtained on iS5 spectrometer and absorptions are reported in reciprocal centimeters. Melting points were recorded on an Electrothermal 9100 melting point apparatus and were uncorrected.
(
R)-Ethyl 2-(2,2-dimethyl-1,3-dioxolan-4-yl)thiazole-4-carboxylate (
8). To a stirred solution of
l-cysteine ethyl ester hydrochloride (4.28 g, 23 mmol) and triethylamine (3.37 mL, 1.1 equivalent) in ethanol (66 mL), a solution of freshly prepared (
R)-(+)-glyceraldehyde acetonide [
29,
30] (2.86 g, 22 mmol) in ethanol (10 mL) was added. After stirring for 5 h at room temperature, the solvent was evaporated under reduced pressure. The crude product was dissolved in ether and washed with water (20 mL) and brine (15 mL). The organic phase was dried over MgSO
4 and the solvent evaporated under reduced pressure. A colorless oil was obtained which was stirred in CH
3CN (130 mL) in the presence of MnO
2 (30.2 g, 20 equiv.) at 60 °C. The reaction progression was followed by TLC and after 48 h the crude mixture was filtered over Celite. Solvent evaporation under reduced pressure was followed by purification by column chromatography on silica gel, with hexanes–ethyl acetate (7:3) mixture as the eluent. An orange oil was obtained (2.951 g, 11.44 mmol) in 52% yield. IR (cm
−1, ATR): 2986, 1719, 1372, 1208;
1H NMR (250 MHz, CDCl
3): δ 8.19 (s, 1H), 5,43 (dd, 1H,
J = 6.7 and 5.1 Hz), 4.52-4.36 (m, 3H), 4.10 (dd, 1H,
J = 8.8 and 5.1 Hz), 1.58 (s, 3H), 1.46 (s, 3H), 1.40 (t, 3H,
J = 7.1 Hz);
13C NMR (62.5 MHz, CDCl
3): δ 172.8, 160.5, 146.7, 126.9, 110.4, 74.7, 69.6, 60.6, 25.7, 24.4, 13.7. HRMS (ESI/+,
m/
z): Calcd for: C
11H
15NO
4S, 258.0800; found: 258.0804; [α]
D25 = +46 (CH
2Cl
2, 1.07).
(R)-Ethyl 2-(1,2-dihydroxyethyl)thiazole-4-carboxylate (A). To a stirred solution of ester 8 (2.01 g, 7.8 mmol) in ethanol (30 mL), Amberlist 15 (10% m/m, 0.20 g) was added. The reaction progression was followed by TLC and after 3 h, the mixture was filtered and the solvent was evaporated under reduced pressure. The crude product was recrystallized from hexanes–ethyl acetate (2:1) and a white solid (1.19 g, 5.46 mmol) was obtained in 70% yield. MP 152–153 °C; IR (cm−1, ATR): 3293, 3114, 1716, 1507, 1234, 1052; 1H NMR (250 MHz, MeOD): δ 8.32 (s, 1H), 4.96 (dd, 1H, J = 6.0 and 3.8 Hz), 4.37 (q, 2H, J = 7.1 Hz), 3.93 (dd, 1H, J = 11.5 and 3.8 Hz), 3.76 (dd, 1H, J = 11.5 and 6.0 Hz), 1.38 (t, 3H, J = 7.1 Hz); 13C NMR (62.5 MHz, MeOD): δ 177.2, 162.8, 147.7, 129.2, 73.6, 67.2, 62.4, 14.6. HRMS (ESI/+, m/z): Calcd: C8H11NO4S, 218.0487; Found: 218.0475; [α]D25 = +59 (MeOH, 1.15).
(S)-Methyl 2-(1-hydroxy-2-methylpropyl)thiazole-4-carboxylate (Thiazole B). To a solution of thiazol 1314 (0.503 g, 1.85 mmol) in anhydrous methanol (25 mL) was added dibutyltinoxide (2.30 g, 9.38 mmol). The suspension was stirred 12 h under reflux and the reaction course was monitored by TLC. The solids were removed by filtration and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography in silica gel using an isocratic mixture of hexanes-ethyl acetate (7:3, V/V). A colorless solid was isolated in 71% yield (0.283 g, 1.31 mmol). MP 50–52 °C; IR (cm−1, ATR): 3279, 2968, 1735, 1714, 1212. 1H NMR (250 MHz, CDCl3): δ 8.11 (s, 1H), 4.86 (d, J = 4.7 Hz, 1H), 3.90 (s, 3H), 3.36 (br s, 1H), 2.29–2.15 (m, 1H), 0.98 (d, J = 6.8 Hz, 3H), 0.90 (d, J = 6.8 Hz, 3H). 13C NMR (62.5 MHz, CDCl3): δ 176.2, 161.9, 146.2, 127.5, 76.5, 52.4, 34.9, 18.8, 16.2. HRMS (ESI/+, m/z): Calcd: C9H14NO3S, 216.0694; Found: 216.0703. [α]D25 = −27 (CHCl3, 0.78).
5,5-Dichlorohexan-1-ol (16). To a flask containing 4Å molecular sieves (15 g) was added methanol (70 mL) and hydrazine monohydrate (5 mL, 5 equiv.) were added successively with stirring. After 20 min, 5-hexen-2-one (18, 2.4 mL, 20.4 mmol) was added dropwise to the reaction mixture and the mixture was stirred overnight. Molecular sieves were filtered off and washed with ethanol. The combined filtrate was concentrated under reduced pressure and the excess hydrazine was further removed by azeotropic distillation with ethanol under reduced pressure. The crude product was dissolved in methanol (60 mL). A solution of anhydrous copper(II) chloride (12.1 g, 6 equiv.) and triethylamine (8.5 mL, 3 equiv.) in methanol (120 mL) was prepared after stirring for 10 min. at 20 °C and cooled to 0 °C. To this solution was added dropwise the above hydrazone solution over 15 min. The cooling bath was removed and stirring was continued for 2 h. The reaction mixture was quenched by the addition of aqueous ammonia solution (3.5% v/v, 10 mL) and the mixture was extracted with cold pentane (3 × 150 mL), washed with brine, dried (Na2SO4), and carefully concentrated under reduced pressure. A volatile colorless oil (1.74 g, 11.4 mmol) was obtained in 56% crude yield. 1H NMR (250 MHz, CDCl3): δ 5.94–5.74 (m, 1H), 5.16–4.95 (m, 2H), 2.50-2.36 (m, 2H), 2.35–2.25 (m, 2H), 2.17 (s, 3H); 13C NMR (62.5 MHz, CDCl3): δ 136.4, 115.6, 90.1, 48.8, 37.4, 29.9. A round bottomed flask equipped with a nitrogen inlet containing the colorless oil above, 9-borabicylcononane solution (0.5 M in THF, 24 mL, 11.6 mmol) was added via syringe. After stirring 2 h at room temperature, water (10 mL) was added, followed by sodium perborate (4.3 g, 3 equiv.). The mixture was vigorously stirred for 2 h and extracted with diethyl ether. The combined organic phase was washed with saturated NaCl solution (10 mL) and dried (MgSO4), and the product was purified by column chromatography on silica gel using hexanes–ethyl acetate (6:4) mixture as the eluent. A colorless oil (1.325 g, 7.75 mmol) was obtained in 38% overall yield from 18. IR (cm−1, ATR): 3345, 2936, 2873, 1440, 1380, 1169, 1071, 1050; 1H NMR (250 MHz, CDCl3): δ 3.60 (t, 2H, J = 6.3 Hz), 2.82 (br s, 1H), 2.23–2.13 (m, 2H), 2.10 (s, 3H), 1.75–1.49 (m, 4H); 13C NMR (62.5 MHz, CDCl3): δ 90.7, 62.2, 49.5, 37.3, 31.8, 22.1.
5,5-Dichlorohexanal (17). To a mixture of pyridinium chlorochromate (0.97 g, 2 equiv.) and 4Å molecular sieves (1 g) in DCM (40 mL) was added a solution of the alcohol 16 (0.38 g, 2.2 mmol) in DCM (5 mL). The mixture was then stirred at room temperature for 1 h and the residue was filtered through a short pad of Florisil and silica gel, washed with DCM and the solvent was evaporated under reduced pressure. A volatile colorless oil (0.82 g, 85% crude yield) was obtained and used without purification in the next step. IR (cm−1, ATR): 2932, 2726, 1723, 1448, 1381, 1171; 1H NMR (250 MHz, CDCl3): δ 9.78 (t, 1H, J = 1.3 Hz), 2.54 (td, 2H, J = 7.1 and 1.3 Hz), 2.26–2.18 (m, 2H), 2.15 (s, 3H), 2.07–1.93 (m, 2H); 13C NMR (62.5 MHz, CDCl3): δ 201.3, 90.0, 48.7, 42.8, 37.3, 18.3.
(2S,3S)-7,7-Dichloro-3-hydroxy-2-methyloctanoic acid (C). To an oven-dried round-bottomed flask, a stirred solution of (1S,2R)-20 (0.500 g, 1.04 mmol) and triethylamine (0.35 mL, 2.4 equiv.) in DCM (5 mL) was added and cooled to −78 °C. A solution of freshly prepared c-Hex2BOTf (1.0 M in hexane, 2.3 mL, 2.2 equiv.) was added dropwise via syringe over 15 min. The resulting solution was stirred at −78 °C for 30 min. and a solution of aldehyde 17 (0.21 g, 1.2 equiv.) in DCM (2 mL) was added dropwise. The reaction mixture was stirred at −78 °C for 2 h and allowed to warm to room temperature over 1 h. Then, the reaction mixture was quenched by the addition of phosphate buffer solution (pH 7, 5 mL), followed by MeOH (15 mL) and 30% aqueous H2O2 (2 mL, careful addition). The mixture was vigorously stirred overnight and then concentrated under reduced pressure. The residue was partitioned between water (15 mL) and DCM (30 mL). The aqueous layer was extracted with DCM and the combined organic layer was concentrated. The aldol product was isolated by chromatography on silica gel using hexanes-ethyl acetate (8:2) as eluent. A viscous colorless oil containing a small quantity of cyclohexanol was dissolved in a water-MeOH-THF solution (1:1:1, 9 mL) and LiOH·H2O (126 mg, 5 equiv.) was added. The mixture was stirred overnight at room temperature and poured into water (10 mL). After extraction with ether to recover the chiral auxiliary, the aqueous layer was acidified with 1M HCl to pH 2 and extracted with DCM. The organic layer was washed with brine and dried with Na2SO4. After solvent concentration under reduced pressure, the product was purified by chromatography on a short pad of silica gel with gradient. The nonpolar products were removed with hexanes-ethyl acetate (7:3) and the acid C was removed with ethyl acetate to give a viscous colorless oil (0.145 g, 0.60 mmol) in 58% yield over two steps. IR (cm−1, ATR): 3344, 2984, 2964, 1694, 1208; 1H NMR (250 MHz, CDCl3): δ 3.65–3.85 (m, 1H), 2.57 (qt, 1H, J = 7.1), 2.28–2.18 (m, 2H), 2.14 (s, 3H), 1.96–1.48 (m, 4H), 1.24 (d, 3H, J = 7.1); 13C NMR (62.5 MHz, CDCl3): δ 180.8, 90.5, 73.0, 49.5, 45.3, 37.3, 33.6, 21.9, 14.1. HRMS (ESI/−, m/z): Calcd: C9H16Cl2O3, 241.0398; Found: 241.0439. [α]D25 = −6 (MeOH, 1.00).
Methyl-2-((S)-1-(((2S,3S)-3-((tert-butyldimethylsilyl)oxy)-7,7-dichloro-2-methyloctanoyl)oxy)-2-methylpropyl)thiazole-4-carboxylate (25). Carboxylic acid C (0.14 g, 0.60 mmol) was dissolved in DCM (3 mL) and cooled to 0 °C. To this solution, triethylamine (0.5 mL, 6 equiv.) and tert-butyldimethylsilyl triflate (0.36 mL, 2.6 equiv.) were added. The mixture was stirred for 2 h at 0 °C. Then potassium carbonate (0.41 g, 5.85 mmol), water (4 mL), MeOH (4 mL), and THF (4 mL) were added, and the mixture was stirred for 6 h at room temperature. To this solution ethyl acetate (20 mL) and brine (5 mL) were added and the mixture was acidified with 1M HCl (pH 2). The organic layer was separated, and the aqueous layer was extracted with ethyl acetate (2 × 20 mL). The combined organic layers were dried (Na2SO4), and concentrated under reduced pressure. The remaining oil (0.21 g) was dissolved in DCM (3 mL) and EDC (0.13 g, 1.2 equiv.), thiazole B (0.14 g, 1.1 equiv.) and catalytic quantity of DMAP were added. The mixture was stirred overnight at room temperature and quenched by addition of brine (5 mL), followed by extraction with ethyl acetate (3 × 20 mL). The organic layer was dried with Na2SO4 and after solvent evaporation under reduced pressure, the crude product was purified by chromatography on silica gel using hexanes-ethyl acetate (7:3). A colorless oil (0.256 g, 0.462 mmol) was obtained in 77% yield for two steps. IR (ATR, cm−1): 2956, 2931, 2856, 1744, 1244, 1107; 1H NMR (250 MHz, CDCl3): δ 8.12 (s, 1H), 6.03 (d, 1H, J = 5.3 Hz), 4.13–4.03 (m, 1H), 3.92 (s, 3H), 2.86–2.71 (m, 1H), 2.48–2.33 (m, 1H), 2.10 (s, 3H), 1.84–1.12 (m, 9H), 0.95 (s, 3H), 0.93 (s, 3H), 0.83 (s, 9H), 0.07 (s, 3H), 0.01 (s, 3H); 13C NMR (62.5 MHz, CDCl3): δ 172.9, 170.5, 161.7, 146.9, 127.2, 90.5, 77.1, 72.6, 52.4, 49.7, 45.5, 37.3, 33.5, 32.1, 25.7, 21.7, 18.6, 17.9, 17.0, 10.9, −4.6, −4.7. HRMS (ESI/+, m/z): Calcd: C24H42Cl2NO5SSi, 554.1930; Found: 554.1943. [α]D25 = −10 (CH2Cl2, 0.8).
(R)-2-(4-(Ethoxycarbonyl)thiazol-2-yl)-2-hydroxyethyl-2-((S)-1-(((2S,3S)-3-((tert-butyldimethylsilyl)oxy)-7,7-dichloro-2-methyloctanoyl)oxy)-2-methylpropyl)thiazole-4-carboxylate (26). Carboxylic ester 25 (88.7 mg, 0.16 mmol) was dissolved in 1,2-dichloroethane (3 mL) and after addition of trimethyltin hydroxide (0.18 g, 6 equiv.), the mixture was heated at 70 °C until the reaction was complete by TLC analysis. After 2 h, the mixture was concentrated in vacuo, and the residue was taken up in ethyl acetate (15 mL). The organic layer was washed successively with HCl (5% v/v, 5 mL) and brine (5 mL) and dried over sodium sulfate. Removal of the solvent in vacuo afforded the corresponding carboxylic acid as colorless oil (73 mg) which was dissolved in DCM (2 mL). To this solution EDC (17 mg, 1.1 equiv.), compound A (17 mg, 1.1 equiv.) and catalytic quantity of DMAP were added. The mixture was stirred overnight at room temperature and quenched by addition of brine (5 mL), followed by extraction with ethyl acetate (3 × 20 mL). The organic layer was dried with Na2SO4 and after solvent concentration, the product was purified by preparative thin layer chromatography on silica gel using chloroform-ethyl acetate (7:3). A colorless oil (0.0982 g, 0.133 mmol) was obtained in 83% yield. IR (ATR, cm−1): 2956, 2931, 2856, 1744, 1244, 1107; 1H NMR (250 MHz, CDCl3): δ 8.17 (s, 2H), 5.98 (d, 1H, J = 5.6 Hz), 5.47 (dd, 1H, J = 6.9 and 3.2 Hz), 4.88 (dd, 1H, J = 11.5 and 3.2 Hz), 4.62 (dd, 1H, J = 11.5 and 7.4 Hz), 4,41 (q, 2H, J = 7.1 Hz), 4.12–4.04 (m, 1H), 2.83–2.76 (m, 1H), 2.43–2.34 (m, 1H), 2.13 (s, 3H), 1.83–1.49 (m, 4H), 1.40 (t, 3H, J = 7.2 Hz), 1.18 (d, 3H, J = 7.0 Hz), 0.96 (dd, 6H, J = 6.2 Hz), 0.84 (s, 9H), 0.08 (s, 3H), 0.01 (s, 3H); 13C NMR (62.5 MHz, CDCl3): δ 173.0, 171.9, 170.9, 161.3, 161.1, 147.2, 146.0, 128.3, 127.9, 90.6, 77.0, 72.7, 70.5, 68.9, 61.5, 49.7, 45.4, 37.4, 33.5, 32.2, 29.6, 25.7, 21.6, 18.6, 17.9, 17.2, 14.3, 11.0, −4.6, −4.7. HRMS (ESI/+. m/z): Calcd: C31H49Cl2N2O8S2Si. 739.2076; Found: 739.2107; [α]D25 = +23 (CHCl3, 0.65).
(R)-2-(4-(Ethoxycarbonyl)thiazol-2-yl)-2-hydroxyethyl 2-((S)-1-(((2S,3S)-7,7-dichloro-3-hydroxy-2-methyloctanoyl)oxy)-2-methylpropyl)thiazole-4-carboxylate (4). A teflon test tube was charged with a solution of compound 26 (0.0210 g, 0.0284 mmol) in THF (1 mL) and a solution of pyridine-HF (70%, 0.15 mL, 250 equiv.) was added. The solution was stirred for 24 h at room temperature and the reaction mixture was dissolved in ethyl acetate (20 mL), washed with water (5 mL), brine (5 mL) and dried over Na2SO4. After solvent concentration under reduced pressure, the product was purified by preparative thin layer chromatography on silica gel using chloroform–ethyl acetate (6:4). A colorless oil (0.0135 g, 0.0216 mmol) was obtained in 76% yield. IR (ATR, cm−1): 3337, 3118, 2967, 2935, 1731, 1485, 1325, 1210, 1170, 1099; 1H NMR (500 MHz, CDCl3): δ 8.30 (s, 1H), 8.18 (s, 1H), 5.99 (d, 1H, J = 3.7 Hz), 5.42 (dd, 1H, J = 9.1 and 2.5 Hz), 4.95 (dd, 1H, J = 11.3 and 2.5 Hz), 4.43 (q, 2H, J = 7.1 Hz), 4.31 (dd, 1H, J = 11.3 and 9.0 Hz), 3.81 (q, 2H, J = 5.1 Hz), 2.89–2.81 (m, 1H), 2.39–2.19 (m, 4H), 2.16 (s, 3H), 1.96–1.83 (m, 4H), 1.41 (t, 3H, J = 7.1 Hz), 1.26 (d, 4H, J = 7.1 Hz), 1.15 (d, 3H, J = 6.7 Hz), 1.00 (d, 3H, J = 7.1 Hz); 13C NMR (125 MHz, CDCl3): δ 174.4, 172.1, 170.5, 161.7, 160.5, 147.5, 145.7, 129.1, 128.2, 90.8, 75.4, 70.2, 70.2, 61.7, 49.9, 37.7, 34.5, 34.3, 21.5, 19.4, 16.7, 15.0, 14.6. HRMS (ESI/+. m/z): Calcd: C25H35Cl2N2O8S2: 625.1212, Found: 625.1185, [α]D25 = +12 (MeOH, 0.5).















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

