1. Introduction
Starfish (
Echinodermata,
Asteroidea) are characterized by a diversity of polar steroids, including polyhydroxylated steroids, structurally related mono-, bi- and triosides, and steroid oligoglycosides (asterosaponins) with carbohydrate chains comprising five or six sugars [
1,
2,
3,
4]. Free polyhydroxysteroids and their glycosides are highly oxygenated steroid compounds, having three to nine hydroxyl groups, and often occur in sulfated forms. Starfish polar steroids have been reported to show a wide spectrum of biological activities, including hemolytic, cytotoxic, antiviral, antibacterial, antifouling, neuritogenic, and antifungal effects [
1,
2,
3,
4]. Recently, some data on the cancer preventive and anticancer activities of starfish polar steroids were obtained. Several asterosaponins and steroid mono- or biosides were found to have anticancer properties showing strong
in vitro cytotoxicity against different tumor cells [
5]. For example, effective cytostatics, asterosaponin 1 and novaeguinoside II from the starfish
Culcita novaeguineae induced apoptosis of human glioblastoma U87MG cells through several signaling transduction pathways [
6,
7]. Moreover, asterosaponin 1 inhibited the proliferation of A549 human lung cancer cells through induction of endoplasmic reticulum stress-associated apoptosis [
8]. Polyhydroxysteroid glycosides isolated from the starfish
Anthenea chinensis exhibited significant activity against promotion of tubulin polymerization
in vitro inhibiting the proliferation of glioblastoma cells [
9]. Leviusculoside G, steroid biglycoside from the starfish
Henricia leviuscila, demonstrated anticarcinogenic action by the induction of p53-dependent apoptosis and inhibition of activator protein 1 (AP-1), nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and extracellular-signal-regulated protein kinases (ERKs) activities in human leukemia HL-60, THP-1, and mouse epidermal JB6 Cl41 cells [
10]. Archasteroside B from the starfish
Archaster typicus induced basal AP-1- and p53-, but not NF-κB-transcriptional activations in JB6 Cl41 cells [
11]. Some asterosaponins and other steroid glycosides from the starfish
Hippasteria kurilensis,
Asteropsis carinifera and
Lethasterias fusca exhibited a significant suppression of the human tumor HT-29, HCT-116, RPMI-7951, and T-47D cell colony formation in a soft agar clonogenic assay [
12,
13,
14]. All these results indicate that further studies of the anticancer properties of polar steroids from starfish are necessary to be conducted.
Recently, we have established structures of six new asterosaponins, leptasteriosides A–F along with one new and one previously known asterogenins from the starfish
Leptasterias ochotensis (order Forcipulatida, family Asteriidae) collected near Shantar Islands in the Sea of Okhotsk. Leptasteriosides A–C demonstrated a substantial suppression of colony formation of human melanoma RPMI-7951 and breast cancer T-47D cells [
15].
Herein, we report the results of the structural elucidation of four new sulfated steroid compounds (1–4) from the fraction of sulfated polyxydroxysteroids and related glycosides from L. ochotensis. Moreover, we discuss the capabilities of 1–4 to inhibit colony formation of cancer RPMI-7951 and T-47D cells ex vivo, the suppressing influence of 1 on the epidermal growth factor (EGF)-induced colony formation of mouse epidermal JB6 Cl41 cells, and the molecular mechanism of cancer preventive effect of 1 implemented through regulation of mitogen-activated protein kinase (MAPK) signaling pathway.
2. Results and Discussion
The concentrated ethanol extract of
L. ochotensis was subjected to sequential separation by chromatography on columns with Polyсhrom-1 and silica gel followed by high-performance liquid chromatography (HPLC) on semipreparative Diasfer-110-C18, Discovery C
18 and analytical Diasfer-110-C18 columns to yield three new sulfated steroid monoglycosides, named as leptaochotensosides A–C (
1–
3), and one new sulfated tetrahydroxylated steroid
4 (
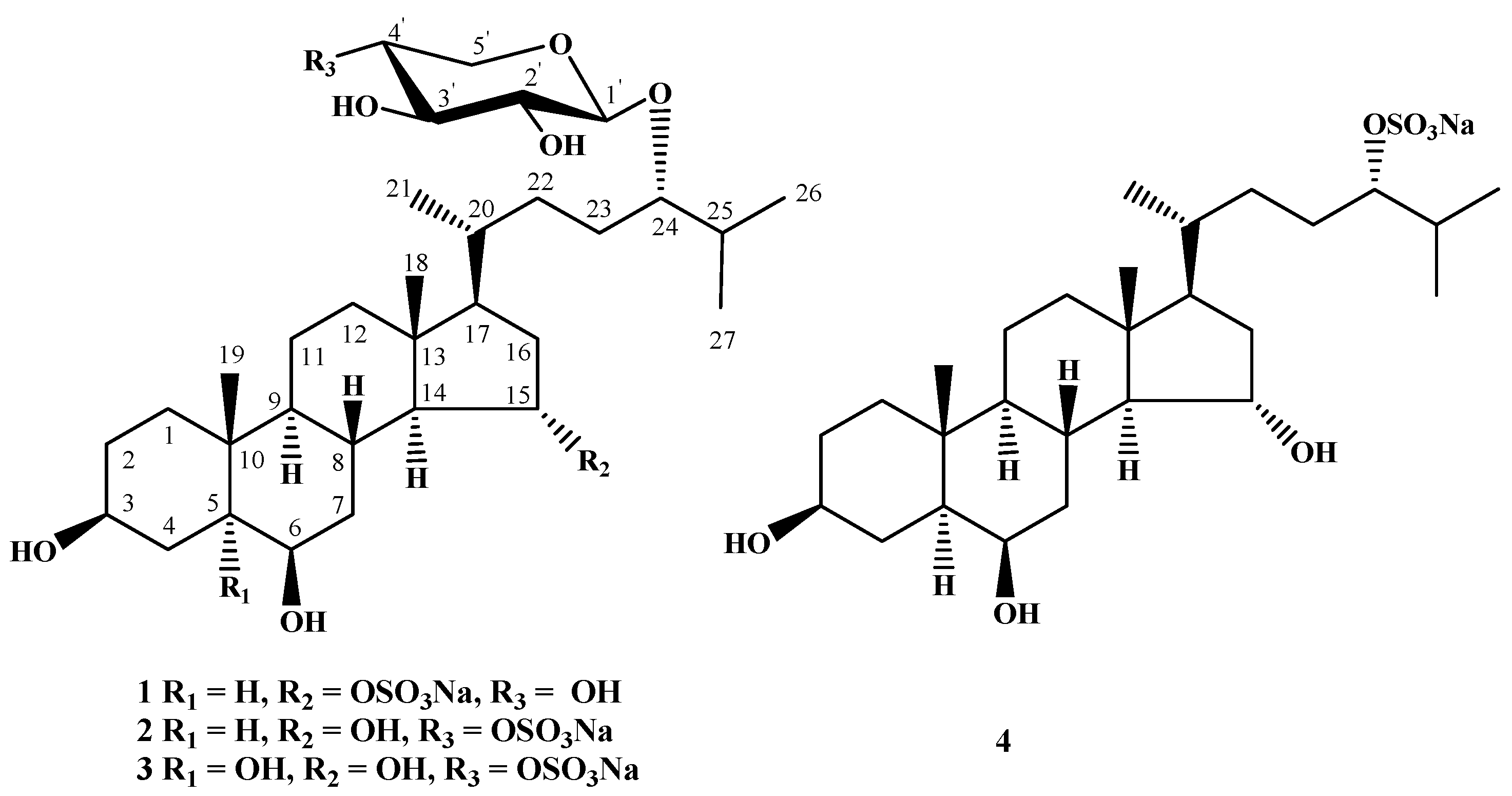
Figure 1).
Figure 1.
The structures of compounds 1–4 isolated from L. ochotensis.
Figure 1.
The structures of compounds 1–4 isolated from L. ochotensis.
The molecular formula of compound
1 was determined to be of С
32Н
55О
11SNa from the [M + Na]
+ sodiated adduct ion peak at
m/
z 693.3225 in the positive high resolution electrospray ionization mass spectrometry ((+)HRESIMS) and the [M − Na]
− ion peak at
m/
z 647.3485 in the negative high resolution electrospray ionization mass spectrometry ((−)HRESIMS). The fragment ion peaks at
m/
z 573 [(M + Na) − NaHSO
4]
+, 143 [Na
2HSO
4]
+ in the (+)ESIMS/MS of the ion at
m/
z 693 [M + Na]
+ and
m/
z 97 [HSO
4]
− in the (−)ESIMS/MS of the ion at
m/
z 647 [M − Na]
− showed the presence a sulfate group in
1. The
1H and
13C NMR spectra of the tetracyclic moiety of the aglycon of
1 showed the resonances of protons and carbons of two angular methyls CH
3-18 and CH
3-19 (δ
H 0.77 s, 1.04 s; δ
C 13.8, 16.3), two oxygenated methines CH-3 (δ
H 3.54 m; δ
C 72.4), CH-6 [δ
H 3.74 q (
J = 2.2); δ
C 72.6], and one
O-sulfated methine CH-15 [δ
H 4.49 td (
J = 9.1, 3.2); δ
C 82.2], that were characteristic of a 3β,6β,15α-trihydroxysteroid nucleus sulfated at position C-15 [
16]. The NMR spectra of aglycon side chain indicated the existence of three secondary methyls CH
3-21 [δ
H 0.92 d (
J = 6.7); δ
C 19.1], CH
3-26 [δ
H 0.92 d (
J = 6.7); δ
C 18.4], and CH
3-27 [δ
H 0.92 d (
J = 6.7); δ
C 18.5] and oxygenated CH-24 group (δ
H 3.34 m; δ
C 86.2) bearing an
O-monosaccharide residue. These proton and carbon resonances were similar to those of a 24-
O-glycosylated cholestane chain of fuscaside A earlier isolated from the starfish
Lethasterias fusca [
17]. The
1H NMR spectrum exhibited one resonance in the downfield region due to an anomeric proton of monosaccharide unit at δ
H 4.06 correlated in the HSQC experiment with a carbon signal at δ
C 105.0. The (+)ESIMS/MS of the ion [M + Na]
+ at
m/
z 693 and the (−)ESIMS/MS of the ion [M − Na]
− at
m/z 647 contained the fragment ion peaks corresponding to the loss of a pentose at
m/
z 543 [(M + Na) − С
5H
10O
5]
+ and
m/
z 515 [(M − Na) − С
5H
8O
4]
−, respectively. Thus, the NMR and mass spectral data indicated the presence of a pentose unit and a tetrahydroxylated cholestane aglycon in
1. All the proton and carbon signals of
1 were assigned using 2D NMR experiments, including
1H-
1H correlation spectroscopy (
1H-
1H COSY), heteronuclear single quantum connectivity (HSQC), heteronuclear multiple bond connectivity (HMBC), and nuclear Overhauser effect spectroscopy (NOESY) (
Table 1 and
Table 2,
Supplementary Materials S1–S7). The
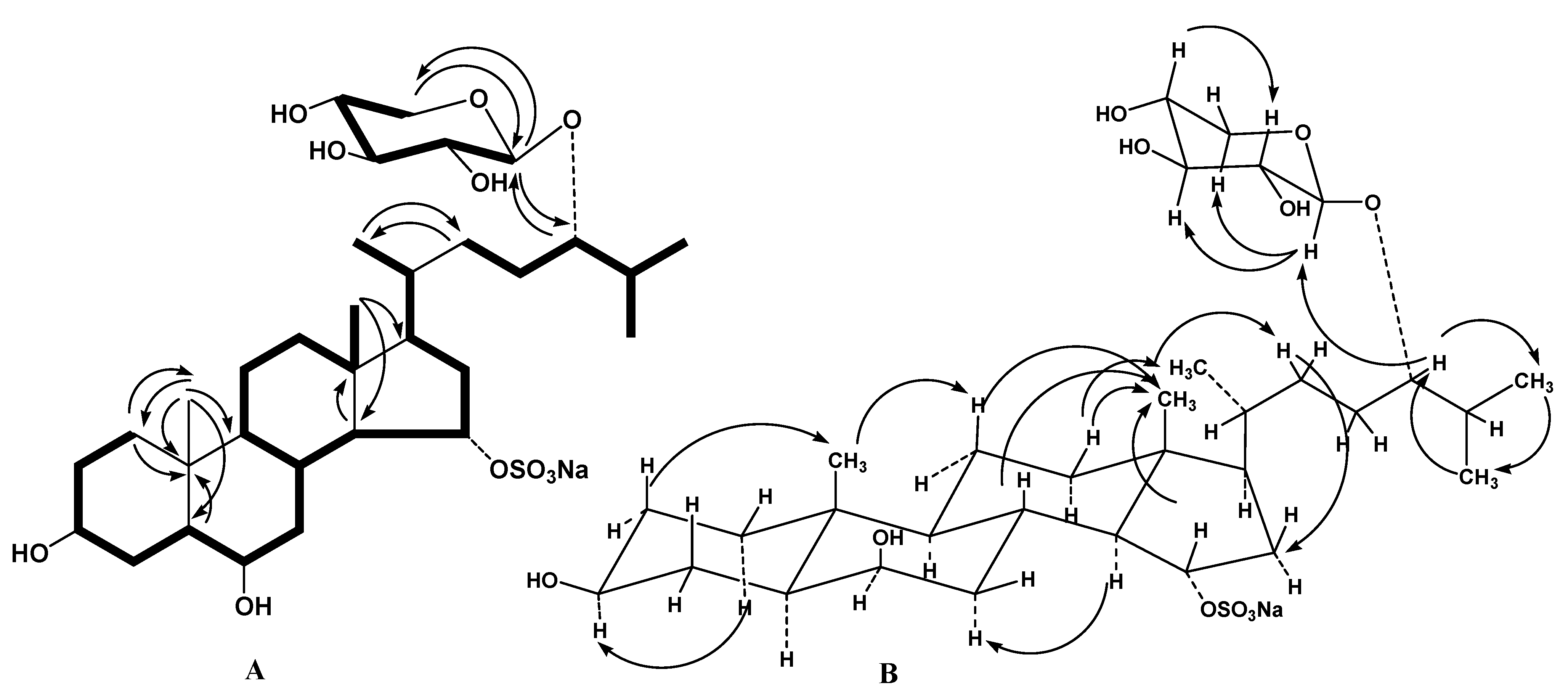
1H-
1H COSY and HSQC correlations confirmed the corresponding sequences of protons at C-1 to C-8, C-8 to C-12 through C-9 and C-11, C-12 to C-18, C-8 to C-14, C-14 to C-17, C-17 to C-21, C-23 and to the end of the side chain (
Figure 2A). The HMBC cross-peaks, such as H-1/C-10, C-19; H-5/C-10; H-14/C-13; H
3-18/C-14, C-17; H
3-19/С-1, С-5, С-9, С-10; H
3-21/C-22; and H
2-22/C-21 supported the overall structure of the steroid moiety of
1 (
Figure 2A). The key NOESY cross-peaks confirmed the common 5α/10β/8β/9α/13β/14α steroid nucleus and the 3β,6β,15α-configurations of oxygenated substituents in
1 (
Figure 2B).
Table 1.
1H (700.13 MHz) Nuclear magnetic resonance (NMR) chemical shifts of 1–4 in D4-methanol (CD3OD), at 30 °C, δ in ppm, J values in Hz.
Table 1.
1H (700.13 MHz) Nuclear magnetic resonance (NMR) chemical shifts of 1–4 in D4-methanol (CD3OD), at 30 °C, δ in ppm, J values in Hz.
| Position | 1 | 2 | 3 | 4 |
|---|
| 1 | 1.64 m; 0.97 m | 1.71 m; 1.02 m | 1.60 m; 1.32 m | 1.73 m; 1.02 m |
| 2 | 1.73 m; 1.42 m | 1.74 m; 1.39 m | 1.75 m; 1.48 m | 1.74 m; 1.40 m |
| 3 | 3.54 m | 3.47 m | 4.00 m | 3.47 m |
| 4 | 1.74 m
1.55 m | 2.18 m
1.15 m | 2.05 dd (13.0; 11.2)
1.54 m | 2.18 brd (9.8)
1.16 m |
| 5 | 1.12 m | 1.00 m | | 1.00 m |
| 6 | 3.74 q (2.2) | 3.32 m | 3.46 t (2.8) | 3.31 m |
| 7 | 2.27 td (14.5; 3.2)
1.26 m | 2.28 brd (14.9)
1.05 m | 1.85 m | 2.28 td (12.7; 4.3)
1.06 m |
| 8 | 2.00 m | 1.64 m | 1.93 m | 1.65 m |
| 9 | 0.72 m | 0.70 m | 1.40 m | 0.70 m |
| 10 | | | | |
| 11 | 1.54 m; 1.40 m | 1.53 m; 1.31 m | 1.39 m; 1.32 m | 1.54 m; 1.30 m |
| 12 | 1.98 m; 1.23 m | 1.95 brd (12.8)
1.22 m | 1.95 brd (11.8)
1.23 m | 1.95 m
1.22 m |
| 13 | | | | |
| 14 | 1.26 m | 1.04 m | 1.11 m | 1.09 m |
| 15 | 4.49 td (9.1; 3.2) | 3.86 td (11.1; 5.7) | 3.86 td (10.4; 3.3) | 3.84 td (9.5; 3.4) |
| 16 | 2.22 m; 1.92 m | 1.89 m; 1.73 m | 1.88 m; 1.72 m | 1.90 m; 1.74 m |
| 17 | 1.37 m | 1.39 m | 1.39 m | 1.39 m |
| 18 | 0.77 s | 0.71 s | 0.72 s | 0.71 s |
| 19 | 1.04 s | 0.84 s | 1.16 s | 0.84 s |
| 20 | 1.39 m | 1.34 m | 1.36 m | 1.38 m |
| 21 | 0.92 d (6.7) | 0.93 d (7.0) | 0.93 d (6.9) | 0.94 d (7.0) |
| 22 | 1.60 m; 1.04 m | 1.58 m; 0.98 m | 1.59 m; 0.99 m | 1.58 m; 1.07 m |
| 23 | 1.59 m; 1.36 m | 1.56 m; 1.34 m | 1.57 m; 1.33 m | 1.70 m; 1.55 m |
| 24 | 3.34 m | 3.35 m | 3.35 m | 4.11 q (5.9) |
| 25 | 1.84 m | 1.87 m | 1.86 m | 2.00 m |
| 26 | 0.92 d (6.7) | 0.92 d (7.0) | 0.92 d (6.6) | 0.95 d (6.3) |
| 27 | 0.92 d (6.7) | 0.92 d (7.0) | 0.92 d (6.6) | 0.91 d (6.3) |
| 1′ | 4.06 d (6.6) | 4.25 d (7.7) | 4.25 d (7.6) | |
| 2′ | 3.21 dd (9.0; 7.3) | 3.23 dd (9.2; 7.6) | 3.23 dd (9.2; 7.6) | |
| 3′ | 3.29 m | 3.49 t (8.8) | 3.48 t (9.0) | |
| 4′ | 3.54 m | 4.18 m | 4.18 m | |
| 5′ | 3.88 dd (11.5; 5.3)
3.14 dd (11.4; 10.0) | 4.16 dd (10.5; 5.6)
3.28 m | 4.16 dd (10.5; 5.5)
3.28 m | |
The chemical shifts and coupling constants of H-1–H-5 of a pentose unit were determined by the irradiation of anomeric proton in the 1D total correlation spectroscopy (1D TOCSY) experiment. The coupling constant of the anomeric proton of monosaccharide unit (6.6 Hz) corresponded to the β-configuration of the glycosidic bond. The carbon and proton signals and corresponding coupling constants of monosaccharide unit coincided well with those of the terminal β-xylopyranosyl residue [
17]. Acid hydrolysis of glycoside
1 with 2 M trifluoroacetic acid (TFA) followed by obtaining of chiral derivatives were carried out to ascertain an absolute stereochemistry of its monosaccharide unit. Alcoholysis of the monosaccharide by (
R)-(−)-octanol followed by acetylation, gas chromatopgraphy (GC) analysis, and comparison with corresponding derivatives of standard monosaccharides allowed us to establish the
d-configuration of the β-xylose. The attachment position of the β-
d-xylopyranosyl residue to steroid aglycon was confirmed by the HMBC and NOESY spectra, where cross-peaks between H-1′ of Xyl
p and C-24, and, respectively, H-24 of the aglycon were observed. The chemical shift of H
3-21 at δ
H 0.92 and the NOESY cross-peaks H
3-18/H-20, H
3-21/H
β-12, and H-22/H
β-16 were indicative of the (
R)-configuration for the C-20 asymmetric center [
18]. The (24
S)-configuration was proposed by similarity of the
13C NMR data of the side chain of
1 with those of fuscaside A and other steroid 24-
O-β-xylopyranosides isolated from starfish [
17]. Based on the results, the structure of leptaochotensoside A (
1) was determined as (24
S)-24-
O-(β-
d-xylopyranosyl)-5α-cholestane-3β,6β,15α,24-tetraol 15-
O-sulfate. Compound
1 contains the 15-
O-sulfated 3β,6β,15α-trihydroxysteroid nucleus never earlier found in other steroid glycosides from starfish.
The molecular formula of compound
2 was determined to be of С
32Н
55О
11SNa, the same as in the glycoside
1. The fragment ion peaks at
m/
z 573 [(M + Na) − NaHSO
4]
+ in the (+)ESIMS/MS of the ion at
m/
z 693 [M + Na]
+ as well as at
m/
z 97 [HSO
4]
− in the (−)ESIMS/MS of the ion at
m/
z 647 [M − Na]
− indicated the presence of a sulfate group in
2. The thorough comparison of the
1H and
13C NMR data of compound
1 with those of
2 showed that they differed from each other only in position of a sulfate group. The signals H-15 at δ
H 3.86 and C-15 at δ
C 74.2 in the
1H and
13C NMR spectra of
2 were shielded when compared with δ
H 4.49 and δ
C 82.2 in the spectra of
1 that clearly exhibited the lack of a sulfate group at C-15 position. On the other hand, the signals H-4′ at δ
H 4.18 and C-4′ at δ
C 77.7 of the β-
d-xylopyranosyl residue in the
1H and
13C NMR spectra of
2 were deshielded in comparison to δ
H 3.54 and δ
C 71.2 in the spectra
1, respectively, that revealed the presence of a sulfate group at position C-4′ of the β-xylopyranosyl residue in the glycoside
2. The
1H-
1H COSY, HSQC, HMBC, and NOESY experiments led to the assignment of all proton and carbon resonances in the NMR spectra of
2 (
Table 1 and
Table 2,
Supplementary Materials S8 and S9) and confirmed the conclusion about the 3β,6β,15α-trihydroxysubstituted steroid nucleus and 24-
O-glycosylated cholestane side chain bearing the 4-
O-sulfate-β-
d-xylopyranosyl unit [
19]. The attachment of the monosaccharide to the steroid aglycon was also deduced from long-range correlations in the HMBC and NOESY spectra between H-1′ of 4-
O-sulfate-β-Xyl
p and C-24 or H-24, respectively, of the aglycon. The (
S)-configuration at C-24 and the
d-series of 4-
O-sulfate-β-xylose unit were expected by analogy with co-occurring glycoside
1. Thus, the structure of leptaochotensoside B (
2) was established as (24
S)-24-
O-(4-
O-sulfate-β-
d-xylopyranosyl)-5α-cholestane-3β,6β,15α,24-tetraol.
Table 2.
13C (176.04 MHz) nuclear magnetic resonance (NMR) chemical shifts of 1–4 in D4-methanol (CD3OD), at 30 °C, δ in ppm.
Table 2.
13C (176.04 MHz) nuclear magnetic resonance (NMR) chemical shifts of 1–4 in D4-methanol (CD3OD), at 30 °C, δ in ppm.
| Position | 1 | 2 | 3 | 4 |
|---|
| 1 | 39.9, CH2 | 38.7, CH2 | 33.6, CH2 | 38.7, CH2 |
| 2 | 32.2, CH2 | 32.0, CH2 | 31.7, CH2 | 31.9, CH2 |
| 3 | 72.4, CH | 72.0, CH | 68.3, CH | 72.0, CH |
| 4 | 36.4, CH2 | 33.1, CH2 | 41.5, CH2 | 33.0, CH2 |
| 5 | 48.8, CH | 52.9, CH | 76.6, C | 52.8, CH |
| 6 | 72.6, CH | 70.3, CH | 76.4, CH | 70.3, CH |
| 7 | 40.2, CH2 | 43.0, CH2 | 35.3, CH2 | 42.9, CH2 |
| 8 | 31.3, CH | 35.4, CH | 31.3, CH | 35.4, CH |
| 9 | 55.6, CH | 55.5, CH | 46.6, CH | 55.4, CH |
| 10 | 36.6, C | 37.4, C | 39.3, C | 37.3, C |
| 11 | 22.0, CH2 | 22.3, CH2 | 22.1, CH2 | 22.2, CH2 |
| 12 | 41.4, CH2 | 41.6, CH2 | 41.7, CH2 | 41.4, CH2 |
| 13 | 44.0, C | 44.7, C | 44.9, C | 44.7, C |
| 14 | 61.4, CH | 63.8, CH | 63.5, CH | 63.8, CH |
| 15 | 82.2, CH | 74.2, CH | 74.3, CH | 74.1, CH |
| 16 | 38.7, CH2 | 41.8, CH2 | 41.8, CH2 | 41.7, CH2 |
| 17 | 54.9, CH | 55.3, CH | 55.1, CH | 54.8, CH |
| 18 | 13.8, CH3 | 14.0, CH3 | 13.8, CH3 | 13.7, CH3 |
| 19 | 16.3, CH3 | 14.1, CH3 | 17.4, CH3 | 13.9, CH3 |
| 20 | 36.7, CH | 36.8, CH | 36.8, CH | 36.7, CH |
| 21 | 19.1, CH3 | 18.3, CH3 | 18.3, CH3 | 19.1, CH3 |
| 22 | 32.4, CH2 | 33.0, CH2 | 33.0, CH2 | 32.4, CH2 |
| 23 | 28.6, CH2 | 28.8, CH2 | 28.6, CH2 | 28.1, CH2 |
| 24 | 86.2, CH | 86.2, CH | 86.1, CH | 86.0, CH |
| 25 | 31.9, CH | 32.2, CH | 32.0, CH | 31.7, CH |
| 26 | 18.4, CH3 | 18.2, CH3 | 18.2, CH3 | 18.6, CH3 |
| 27 | 18.5, CH3 | 19.1, CH3 | 19.1, CH3 | 17.8, CH3 |
| 1′ | 105.0, CH | 104.6, CH | 104.6, CH | |
| 2′ | 75.3, CH | 75.3, CH | 75.3, CH | |
| 3′ | 77.9, CH | 76.2, CH | 76.1, CH | |
| 4′ | 71.2, CH | 77.8, CH | 77.7, CH | |
| 5′ | 66.7, CH2 | 64.8, CH2 | 64.6, CH2 | |
The molecular formula of
3 was determined to be of С
32Н
55О
12SNa from the [M + Na]
+ sodiated adduct ion peak at
m/
z 709.3182 in the (+)HRESIMS and the [M − Na]
− ion peak at
m/
z 663.3427 in the (–)HRESIMS. The fragment ion peaks at
m/
z 589 [(M + Na) − NaHSO
4]
+ in the (+)ESIMS/MS of the ion at
m/
z 709 [M + Na]
+and
m/
z 97 [HSO
4]
− in the (−)ESIMS/MS of the ion at
m/
z 663 [M − Na]
− revealed the presence of a sulfate group in
3. The molecular mass difference of 16 amu between
3 and
2 assumed the existence of an additional hydroxyl group in
3 compared to glycoside
2. The comparison of the NMR data of
3 with those of
2 clearly showed that glycoside
3 contained the same 24-
O-glycosylated cholestane side chain bearing the 4-
O-sulfate-β-
d-xylopyranosyl unit. Most of the signals in the NMR spectra of
3 attributable to steroid nucleus were similar to those of
2 with exception of some resonances belonging to steroid A and B-rings. The signals of H-3α (m) and H
3-19 (s) in
3 were downfield shifted from δ
H 3.47 to 4.00 and from δ
H 0.84 to 1.16, respectively, in comparison with those of
2. The multiplet of H-4β (δ
H 2.18) in
2 was replaced by the doublet doublets (δ
H 2.05,
J = 13.0, 11.2 Hz) in the spectrum of
3. Along with the
13C NMR spectrum, these data indicated the presence an additional hydroxyl group at C-5 (δ
C 76.6) in
3. The detailed analysis of the carbon and proton signals and the corresponding coupling constants in the NMR spectra of the aglycon part of
3 testified that compound
3 contained the 3β,5,6β,15α-tetrahydroxysubstituted steroid nucleus (
Table 1 and
Table 2,
Supplementary Materials S10 and S11) [
20]. Accordingly, the structure of leptaochotensoside C (
3) was determined as (24
S)-24-
O-(4-
O-sulfate-β-
d-xylopyranosyl)-5α-cholestane-3β,5,6β,15α,24-pentaol.
Figure 2.
(A) 1H-1H Correlation spectroscopy (1H-1H COSY) and key heteronuclear multiple bond connectivity (HMBC) correlations for compound 1; (B) Key nuclear Overhauser effect spectroscopy (NOESY) correlations for compound 1.
Figure 2.
(A) 1H-1H Correlation spectroscopy (1H-1H COSY) and key heteronuclear multiple bond connectivity (HMBC) correlations for compound 1; (B) Key nuclear Overhauser effect spectroscopy (NOESY) correlations for compound 1.
The molecular formula of compound
4 was determined to be of С
27Н
47О
7SNa from the [M + Na]
+ sodiated adduct ion peak at
m/
z 561.2864 in the (+)HRESIMS and the [M − Na]
− ion peak at
m/
z 515.3043 in the (−)HRESIMS. The fragment ion peak at
m/
z 97 [HSO
4]
− in the (−)ESIMS/MS of the ion at
m/
z 515 [M − Na]
− indicated the existence of a sulfate group in
4. Analysis of the
1H and
13C NMR spectra of
4 and
2 clearly revealed that compound
4 had the same 3β,6β,15α-trihydroxysubstituted steroid nucleus. The protons sequence from H-17 to H-27, correlated with the corresponding carbon atoms of the side chain of
4, was assigned using the
1H-
1H COSY and HSQC experiments (
Table 1 and
Table 2,
Supplementary Materials S12 and S13). The HMBC correlations H
3-21/C-17, C-20, C-22; H
3-26/C-24, C-25, C-27; H
3-27/C-24, C-25, C-26, and chemical shifts of CH-24 [δ
H 4.11 q (
J = 5.9), δ
C 86.0] supported the total structure of the side chain with oxygen-containing function at C-24. Comparison of the carbon and proton resonances and corresponding coupling constants of the side chain with those data reported in literature [
20] showed that compound
4 contained the 24-
O-sulfated cholestane side chain. Thereby, the steroid
4 was proved to be 24-
O-sulfated aglycon of
2 and its structure was established as (24
S)-5α-cholestane-3β,6β,15α,24-tetraol 24-
O-sulfate.
2.1. Cytotoxic Activity of Leptaochotensosides A–C (1–3) and Sulfated Polyhydroxysteroid 4 ex Vivo
Ex vivo cytotoxicity tests [
21] are necessary to define the concentration range for further and more detailed
ex vivo experiments and to provide meaningful information on signal transduction pathway and molecular targets of compounds.
In the studies on cytotoxic properties of
1–
4, human melanoma RPMI-7951 and human breast cancer T-47D cells were treated with various concentrations of
1–
4 (0–200 µM) for 24 h and then cell viability was assessed by the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2
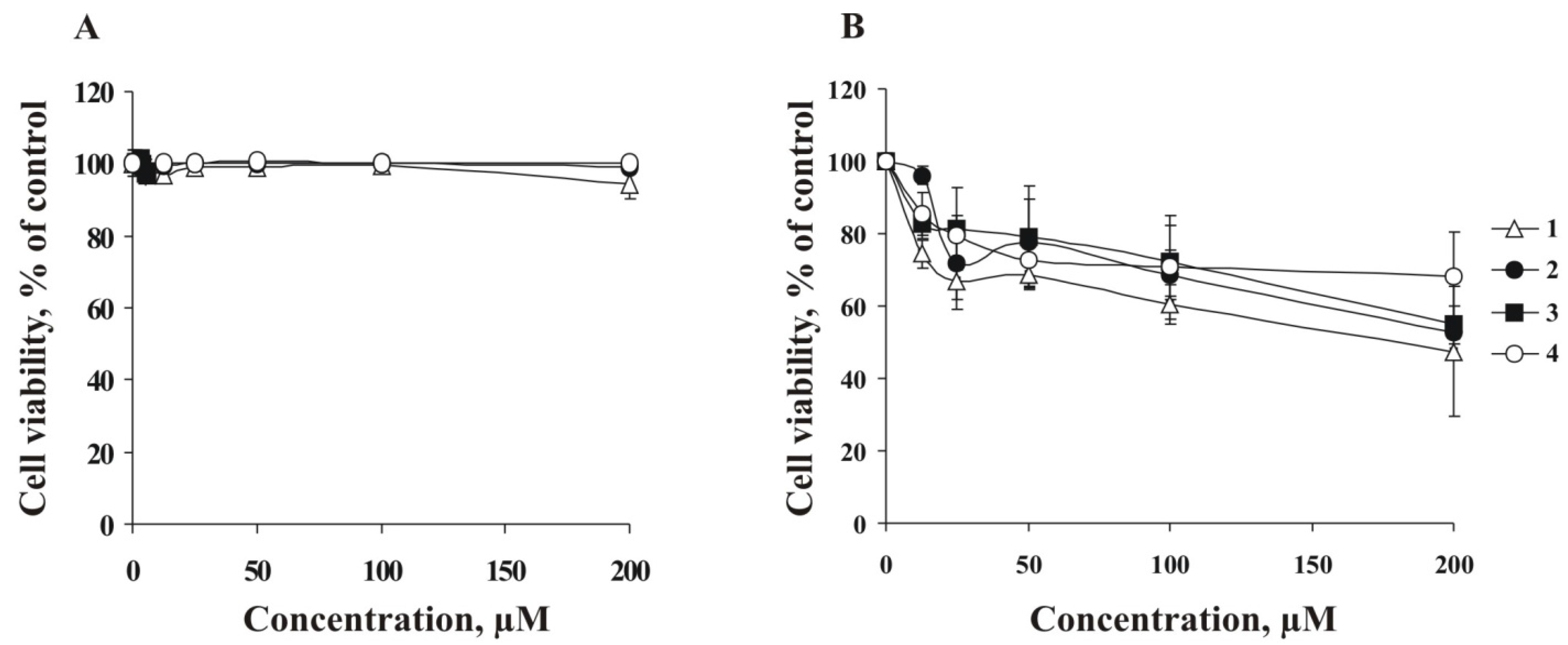
H-tetrazolium (MTS) assay. The results showed that
1–
4 did not decrease cell viability of RPMI-7951 cells within the concentration range of 0–200 µM (
Figure 3A). At the same time, the compounds
1–
4 possess only slight cytotoxic activity against T-47D cells at the highest of the used concentrations (200 µM) (
Figure 3B). For further experiments, RPMI-7951 and T-47D cells were treated with investigated compounds at non-cytotoxic concentrations (lower than 100 μM).
Figure 3.
Ex vivo cytotoxicities of leptaochotensosides A–C (1–3) and sulfated polyhydroxysteroid 4. (A) Human melanoma RPMI-7951 cells (8 × 103/well) and (B) breast cancer T-47D cells (8 × 103/well) were incubated with compounds 1–4 (0–200 µM) for 24 h at 37 °C in an 5% CO2 incubator. Compounds cytotoxicities were estimated using MTS assay. Data are represented as the mean ± SD as determined from triplicate experiments.
Figure 3.
Ex vivo cytotoxicities of leptaochotensosides A–C (1–3) and sulfated polyhydroxysteroid 4. (A) Human melanoma RPMI-7951 cells (8 × 103/well) and (B) breast cancer T-47D cells (8 × 103/well) were incubated with compounds 1–4 (0–200 µM) for 24 h at 37 °C in an 5% CO2 incubator. Compounds cytotoxicities were estimated using MTS assay. Data are represented as the mean ± SD as determined from triplicate experiments.
2.3. The Leptaochotensoside A (1) Inhibits the EGF-induced Colony Formation of JB6 Cl41 Cells and Signal Transduction in JB6 Cl41 Cells
The carcinogenesis is multistage process, including initiation (cell transformation), promotion (colony formation), and progression (metastasis) [
22]. One of the perspective approaches for cancer therapy is the search and development of nontoxic compounds, which are effective in preventing cancer initiation. The mouse epidermal JB6 Cl41 cells are known to respond irreversibly to tumor promoters such as epidermal growth factor (EGF) by induction of anchorage independent increase in a number of cell colonies in soft agar [
23]. That is why this well-established culture system was used to identify the effect of leptaochotensoside A (
1) on EGF-induced cell transformation.
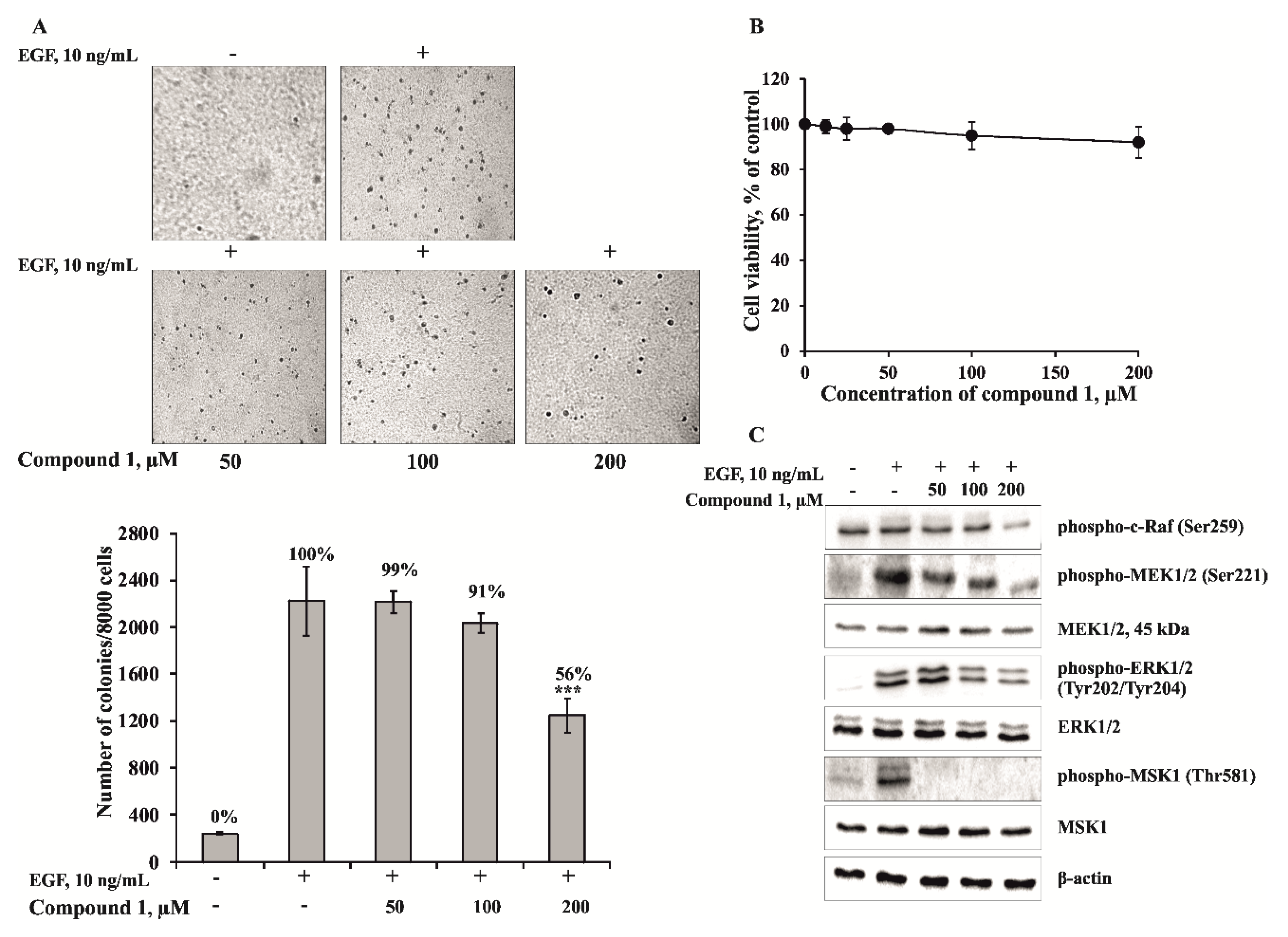
To estimate the effect of
1, JB6 Cl41 cells were treated with EGF (10 ng/mL) in the absence or presence of
1 (50, 100, 200 µM) in a soft agar matrix as described in Experimental Section. Our results revealed that JB6 Cl41 cells treated with compound
1 at concentration 200 µM formed less colonies compared with control cells treated with EGF only. It decreased EGF-induced colony number on 44% of control (
Figure 5A). The inhibition of by the compound
1 was not due to its cytotoxicity because the effective concentration range for suppressing cell transformation did not affect cell viability of JB6 Cl41 cells (
Figure 5B). Since the anchorage-independent growth ability is an
ex vivo indicator and a key characteristic of the transformed cell phenotype [
24], these results suggest that
1 can reduce the malignant potential of JB6 Cl41 cells induced by EGF.
To elucidate cancer prevention molecular mechanism, we tested the effect of leptaochotensoside A (
1) on activation of mitogen-activated protein kinases (MAPKs). MAPK pathways comprise three kinase modules, in which MAPK is activated upon phosphorylation by a mitogen-activated protein kinase kinase (MAPKK), which in turn is activated, when phosphorylated by MAPKKK [
25]. The most thoroughly characterized subgroups of the MAP kinase family include ERKs, c-Jun
N-terminal kinases (JNKs)/stress-activated protein kinases, and p38 kinases [
26]. MAPK pathways are evolutionarily conserved kinase modules that link extracellular signals to the machinery that controls fundamental cellular processes such as growth, proliferation, differentiation, migration and apoptosis [
27]. Herein, we describe the effects of compound
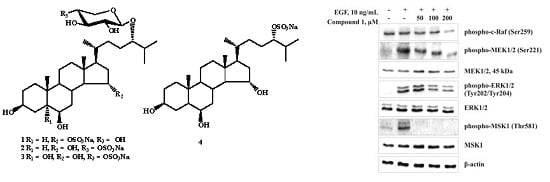
1 on the phosphorylation of proto-oncogene serine/threonine-protein (c-Raf), mitogen-activated protein kinase 1/2 (MEK1/2), extracellular signal-regulated 1/2 (ERK1/2), and mitogen- and stress-activated protein (MSK-1) kinases in JB6 Cl41 cells. Compound
1 was shown to inhibit effectively the EGF-induced phosphorylation of ERK1/2 and MSK-1 kinases (
Figure 5C). These data suggest that the anticlonogenic effects of compound
1 might be mediated by regulating the activity of MAP kinases, and ERK1/2 and MSK-1 kinases play a key role in its cancer preventive action.
It is well known that steroids such as steroid hormones act through intracellular receptors on gene transcription and protein synthesis (genomic mechanism of action). On the other hand, the regulatory cascades,
i.e., MAP kinases, the phosphatidylinositol 3-OH kinase (PI3K) and tyrosine kinases are modulated through non-transcriptional mechanisms by steroid hormones [
28,
29]. Therefore, we can assume that the action of starfish steroid glycosides on MAPK signal pathway is realized through non-genomic mechanism. The identification of target molecule of leptaochotensoside A (
1) and elucidation of its binding peculiarity with a target protein or cellular receptor is an aim of further research.
Figure 5.
The effect of leptaochotensoside A (1) on the epidermal growth factor (EGF)-induced colony formation of JB6 Cl41 cells and molecular mechanism in JB6 Cl41 cells. (A) Leptaochotensoside A (1) inhibits EGF-induced anchorage-independent growth of mouse epidermal JB6 Cl41 cells. JB6 Cl41 cells (8 × 103) were exposed to EGF (10 ng/mL) and treated with 1 (0–200 µM) in 1 mL of 0.3% Basal Medium Eagle (BME) agar containing 10% fetal bovine serum, 2 mM l-glutamine, and 25 µg/mL gentamicin. The cultures were maintained at 37 °C in an 5% CO2 incubator for 14 days, and the cell colonies were scored using a microscope Motic AE 20 (Motic) and the Motic Image Plus computer program. Data are represented as the mean ± SD as determined from triplicate experiments and the asterisks indicate a significant (*** p < 0.001) decrease of the colony formation of the cells treated with leptaochotensoside A (1) compared with the phosphate buffered saline (PBS)-treated group; (B) The absence of cytotoxic effect of leptaochotensoside A (1) on JB6 Cl41 cells. An MTS assay was used after treatment of cells with 1 for 24 h. All the experiments were performed in triplicate, and the mean absorbance values were calculated. Data are represented as the mean ± SD as determined from triplicate experiments; (C) Leptaochotensoside A (1) inhibits MAPK signaling pathway in JB6 Cl41 cells. After cells (6 × 105) were cultured in a 10-cm dish overnight, they were treated with compound 1 (0–200 µM) for 24 h. Then, the cells were starved in serum-free medium for another 12 h and treated with EGF (10 ng/mL) for 15 min. Cells were harvested and protein levels were determined by Western blot analysis.
Figure 5.
The effect of leptaochotensoside A (1) on the epidermal growth factor (EGF)-induced colony formation of JB6 Cl41 cells and molecular mechanism in JB6 Cl41 cells. (A) Leptaochotensoside A (1) inhibits EGF-induced anchorage-independent growth of mouse epidermal JB6 Cl41 cells. JB6 Cl41 cells (8 × 103) were exposed to EGF (10 ng/mL) and treated with 1 (0–200 µM) in 1 mL of 0.3% Basal Medium Eagle (BME) agar containing 10% fetal bovine serum, 2 mM l-glutamine, and 25 µg/mL gentamicin. The cultures were maintained at 37 °C in an 5% CO2 incubator for 14 days, and the cell colonies were scored using a microscope Motic AE 20 (Motic) and the Motic Image Plus computer program. Data are represented as the mean ± SD as determined from triplicate experiments and the asterisks indicate a significant (*** p < 0.001) decrease of the colony formation of the cells treated with leptaochotensoside A (1) compared with the phosphate buffered saline (PBS)-treated group; (B) The absence of cytotoxic effect of leptaochotensoside A (1) on JB6 Cl41 cells. An MTS assay was used after treatment of cells with 1 for 24 h. All the experiments were performed in triplicate, and the mean absorbance values were calculated. Data are represented as the mean ± SD as determined from triplicate experiments; (C) Leptaochotensoside A (1) inhibits MAPK signaling pathway in JB6 Cl41 cells. After cells (6 × 105) were cultured in a 10-cm dish overnight, they were treated with compound 1 (0–200 µM) for 24 h. Then, the cells were starved in serum-free medium for another 12 h and treated with EGF (10 ng/mL) for 15 min. Cells were harvested and protein levels were determined by Western blot analysis.
![Marinedrugs 13 04418 g005]()
3. Experimental Section
3.1. General Methods
Optical rotations were determined on a PerkinElmer 343 polarimeter (Perkin Elmer, Waltham, MA, USA). The 1H and 13C NMR spectra were recorded on Bruker Avance III 700 spectrometers at 700.13 and 176.04 MHz, respectively, with tetramethylsilane used as an internal standard. The HRESI mass spectra were recorded on an Agilent 6510 quadrupole-time of flight liquid chromatography/mass spectrometry (Q-TOF LC/MS) mass spectrometer (Agilent Technologies, Santa Clara, CA, USA); the samples were dissolved in MeOH (c 0.001 mg/mL). GC analysis was performed on an Agilent 6850 Series chromatograph (Agilent Technologies), equipped with a capillary column HP-5 MS (30 m × 0.25 mm) over the temperature range 100–270 °C at 5 °C/min with the carrier gas He (1.7 mL/min); the temperatures of the injector and the detector were 250 and 270 °C, respectively. HPLC separations were carried out on an Agilent 1100 Series chromatograph that was equipped with a differential refractometer; the Diasfer-110-C18 (10 μm, 250 mm × 15 mm), Discovery C18 (5 μm, 250 mm × 10 mm), and Diasfer-110-C18 (5 μm, 250 mm × 4.6 mm) columns were used. Low-pressure column liquid chromatography was performed with the Polychrom 1 (powdered Teflon, Biolar, Latvia), silica gel KSK (50–160 μm, Sorbpolimer, Krasnodar, Russia). Sorbfil silica gel plates (4.5 × 6.0 cm, 5–17 μm, Sorbpolimer) were used for thin-layer chromatography.
MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay kit was purchased from Promega (Madison, WI, USA).
Antibodies against phospho-c-Raf (Ser259), phospho-MEK 1/2 (Ser221), phospho-ERK 1/2 (Tyr202/Tyr204), phospho-MSK 1 (Thr581), MEK 1/2, ERK 1/2, MSK 1 were obtained from Cell Signaling Technology (Danvers, MA, USA), β-actin and horseradish peroxidase (HRP) conjugated secondary antibody from rabbit and mouse were purchased from Santa Cruz Biotechnology (Dallas, TX, USA) and Proteintech Group (Chicago, IL, USA), respectively.
The chemiluminescence’s detection kit ECL Plus was from Amersham (Pittsburgh, PA, USA). The Basal Medium Eagle (BME), Minimum Essential Medium (MEM), Roswell Park Memorial Institute medium (RPMI 1640), phosphate buffered saline (PBS), l-glutamine, gentamicin solution, trypsin, fetal bovine serum (FBS), sodium hydrocarbonate (NaHCO3), and agar were purchased from Sigma and Gibco (Carlsbad, CA, USA). All other common chemicals, solvents and reagents were of highest grade available from various commercial sources.
3.2. Animal Materials
Specimens of Leptasterias ochotensis Brandt, 1851 (order Forcipulatida, family Asteriidae) were collected at a depth of 20–40 m in the Sea of Okhotsk near the Island of Bolshoy Shantar during the research vessel Akademik Oparin 29th scientific cruise in August 2003. Species identification was carried out by A.V. Smirnov (Zoological Institute of the Russian Academy of Sciences, St. Petersburg, Russia). A voucher specimen (no. 029-052) is on deposit at the marine specimen collection of the G.B. Elyakov Pacific Institute of Bioorganic Chemistry of the FEB RAS, Vladivostok, Russia.
3.3. Extraction and Isolation
The fresh animals (0.35 kg) were chopped and extracted twice with EtOH at 20 °C. The H2O/EtOH layer was evaporated, and the residue was dissolved in H2O (0.5 L). The H2O-soluble materials were passed through a Polychrom 1 column (6.5 cm × 21 cm), eluted with distilled H2O until a negative chloride ion reaction was obtained, and then eluted with EtOH. The combined EtOH eluate was evaporated to give a brownish residue (4.8 g). This material was chromatographed over a silica gel column (4 cm × 20 cm) using CHCl3/EtOH (stepwise gradient, 8:1 to 1:2) to yield six fractions, 1–6, that were then analyzed by TLC on silica gel plates in the eluent system BuOH/EtOH/H2O (4:1:2). Fractions 1, 2, 4, and 5 mainly contained the sulfated polyhydroxysteroids and related glycosides, fraction 3 mainly contained the free sulfated asterogenins, and fraction 6 mainly contained the asterosaponins. HPLC separation of fraction 5 on a Diasfer-110-C18 column (10 μm, 250 mm × 15 mm, 2.5 mL/min) with EtOH/H2O (65:35) as an eluent system followed by the further separation on a Discovery C18 column (5 μm, 250 mm × 10 mm, 2.5 mL/min) with MeOH/H2O (70:30) as an eluent system yielded pure 1 (38.6 mg, tR 18.7 min) and several additional subfractions, containing compounds 2–4. Further HPLC separation of these subfractions on a Diasfer-110-C18 column (5 μm, 250 mm × 4.6 mm, 1.0 mL/min) with MeOH/H2O (75:25) as an eluent system yielded pure 2 (1.5 mg, tR 7.4 min), 3 (7.5 mg, tR 6.8 min), and 4 (1.7 mg, tR 11.2 min).
3.4. Spectral Data of New Compounds
Leptaochotensoside A (
1): С
32Н
55О
11SNa, amorphous powder;
+12.3 (
с 0.45, MeOH); the
1H and
13C NMR data are listed in
Table 1 and
Table 2; (+)HRESIMS
m/
z 693.3225 [M + Na]
+ (calculated for C
32H
55O
11SNa
2, 693.3255). (+)ESIMS/MS of the ion [M + Na]
+ at
m/
z 693: 573 [(M + Na) − NaHSO
4]
+, 543 [(M + Na) − С
5H
8O
4 − H
2O]
+, 143 [Na
2HSO
4]
+. (−)HRESIMS
m/
z 647.3485 [M − Na]
− (calculated for C
32H
55O
11S, 647.3471). (–)ESIMS/MS of the ion [M − Na]
− at
m/
z 647: 629 [(M − Na) − H
2O]
−, 515 [(M − Na) − С
5H
8O
4]
−, 497 [(M − Na) − С
5H
8O
4 − H
2O]
−, 97 [HSO
4]
−.
Leptaochotensoside B (
2): C
32H
55O
11SNa, amorphous powder;
+3.5 (
с 0.1, MeOH); the
1H and
13C NMR data are listed in
Table 1 and
Table 2; (+)HRESIMS
m/
z 693.3238 [M + Na]
+ (calculated for C
32H
55O
11SNa
2, 693.3255). (+)ESIMS/MS of the ion [M + Na]
+ at
m/z 693: 573 [(M + Na) − NaHSO
4]
+. (−)HRESIMS
m/
z 647.3480 [M − Na]
− (calculated for C
32H
55O
11S, 647.3471). (−)ESIMS/MS of the ion [M − Na]
− at
m/
z 647: 97 [HSO
4]
−.
Leptaochotensoside C (
3): C
32H
55O
12SNa, amorphous powder;
+3.5 (
с 0.1, MeOH); the
1H and
13C NMR data are listed in
Table 1 and
Table 2; (+)HRESIMS
m/
z 709.3182 [M + Na]
+ (calculated for C
32H
55O
12SNa
2, 709.3204). (+)ESIMS/MS of the ion [M + Na]
+ at
m/
z 709: 589 [(M + Na) − NaHSO
4]
+. (−)HRESIMS
m/
z 663.3427 [M − Na]
− (calculated for C
32H
55O
12S, 663.3420). (−)ESIMS/MS of the ion [M − Na]
− at
m/
z 663: 97 [HSO
4]
−.
(24
S)-5α-Cholestane-3β,6β,15α,24-tetraol 24-
O-sulfate (
4): C
27H
47O
7SNa, amorphous powder;
+7.0 (
с 0.1, MeOH); the
1H and
13C NMR data are listed in
Table 1 and
Table 2; (+)HRESIMS
m/
z 561.2864 [M + Na]
+ (calculated for C
27H
47O
7SNa
2, 561.2838). (−)HRESIMS
m/
z 515.3043 [M − Na]
− (calculated for C
27H
47O
7S, 515.3042). (−)ESIMS/MS of the ion [M − Na]
− at
m/
z 515: 97 [HSO
4]
−.
3.5. Acid Hydrolysis and Determination of Absolute Configuration of Monosaccharide
The acid hydrolysis of 1 (2.0 mg) was carried out in a solution of 2 M TFA (1 mL) in a sealed vial on a H2O bath at 100 °C for 2 h. The H2O layer was washed with CHCl3 (3 × 1.0 mL) and concentrated in vacuo. One drop of concentrated TFA and 0.5 mL of R-(−)-2-octanol (Aldrich) were added to the sugar mixture, and the sealed vial was heated on a glycerol bath at 130 °C for 6 h. The solution was evaporated in vacuo and treated with a mixture of pyridine/acetic anhydride (1:1, 0.6 mL) for 24 h at room temperature. The acetylated 2-octylglycosides were analyzed by GC using the corresponding authentic samples prepared by the same procedure. The following peaks of monosaccharide unit were detected in the hydrolysate of 1: d-xylose (tR 24.32, 24.55, and 24.80 min). The retention times of the authentic samples were as follows: d-xylose (tR 24.28, 24.57, and 24.78 min) and l-xylose (tR 24.07, 24.15, 24.71, and 24.92 min).
3.6. Bioactivity Assay
3.6.1. Cell Lines and Culture Conditions
Mouse epidermal JB6 Cl41 cells (АТСС # CRL-2010™), human malignant melanoma RPMI-7951 cells (ATCC # HTB-66™), and human breast cancer cells T-47D (ATCC # HTB-133™) were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA).
The JB6 Cl41, RPMI-7951, and T-47D cells were cultured in MEM/5% FBS, MEM/10% FBS, and RPMI-1640/10% FBS media, respectively. The cell cultures were maintained at 37 °C in an 5% CO2 incubator (MCO-18AIC, Sanyo, Moriguchi, Osaka, Japan). The cells were grown for 3–4 days and after reaching 90% of confluence were harvested by exposure to 0.25% trypsin-ethylenediaminetetraacetic acid (trypsin-EDTA) solution and then passed into new T-75 tissue culture flasks.
3.6.2. MTS Assay
To estimate cell viability, JB6 Cl41, RPMI-7951, and T-47D cells (8 × 103/well) were seeded in 96-well plates for 24 h at 37 °C in 5% CO2 incubator. The attached cells were fed with fresh medium containing various concentrations of compounds 1–4 from L. ochotensis (0–200 µM) for additional 24 h. After that, the cytotoxicity of 1–4 was measured using an MTS assay kit according to the manufacturer’s instructions. All the experiments were performed in triplicate, and the mean absorbance values were calculated. The results are expressed as the percentage of inhibition that produced a reduction in absorbance after treatment with polar steroids compared to the non-treated cells (control).
3.6.4. Western Blot Analysis
After JB6 Cl41 cells (6 × 105) were cultured in a 10-cm dish overnight, they were treated with 1 (50, 100, 200 µM) for 24 h. Then, the cells were starved in serum-free medium for another 12 h and treated with EGF (10 ng/mL) for 15 min. The harvested cells were lysed with lysis buffer (50 mM Tris-HCl (pH 7.4), 150 mM·NaCl, 1 mM EDTA, 1 mM ethylene glycol tetraacetic acid (EGTA), 10 mg/mL aprotinin, 10 mg/mL leupeptin, 5 mM phenylmethanesulfonyl fluoride (PMSF), 1 mM dithiolthreitol (DTT) containing 1% Triton X-100). Insoluble debris was removed by centrifugation at 12,000 rpm for 15 min, and protein’s content was determined using Bradford reagent (Bio-Rad, Hercules, CA, USA). Lysate protein (20–40 µg) was subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrophoretically transferred to a polyvinylidene difluoride membranes (PVDF) (Millipore, Billerica, MA, USA). The membranes were blocked with 5% non-fat milk for 1 h and then incubated with the respective specific primary antibody at 4 °C overnight. Protein bands were visualized using an enhanced chemiluminescence reagent (ECL Plus, GE Healthcare, Marlborough, MA, USA) after hybridization with a HRP-conjugated secondary antibody. Band density was quantified using the ImageJ software program (National Institutes of Health, Bethesda, MD, USA).
3.6.5. Statistical Analysis
All assays were performed at least in triplicate. The results are expressed as the mean ± standard deviation (SD). A Student’s t-test was used to evaluate the data with the following significance levels: * p < 0.05, ** p < 0.01, *** p < 0.001.




 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



