Abstract
Voltage-gated sodium channels (VGSCs) are responsible for the generation of the action potential. Among nine classified VGSC subtypes (Nav1.1–Nav1.9), Nav1.7 is primarily expressed in the sensory neurons, contributing to the nociception transmission. Therefore Nav1.7 becomes a promising target for analgesic drug development. In this study, we compared the influence of an array of VGSC agonists including veratridine, BmK NT1, brevetoxin-2, deltamethrin and antillatoxin (ATX) on membrane depolarization which was detected by Fluorescence Imaging Plate Reader (FLIPR) membrane potential (FMP) blue dye. In HEK-293 cells heterologously expressing hNav1.7 α-subunit, ATX produced a robust membrane depolarization with an EC50 value of 7.8 ± 2.9 nM whereas veratridine, BmK NT1, and deltamethrin produced marginal response. Brevetoxin-2 was without effect on membrane potential change. The ATX response was completely inhibited by tetrodotoxin suggesting that the ATX response was solely derived from hNav1.7 activation, which was consistent with the results where ATX produced a negligible response in null HEK-293 cells. Six VGSC antagonists including lidocaine, lamotrigine, phenytoin, carbamazepine, riluzole, and 2-amino-6-trifluoromethylthiobenzothiazole all concentration-dependently inhibited ATX response with IC50 values comparable to that reported from patch-clamp experiments. Considered together, we demonstrate that ATX is a unique efficacious hNav1.7 activator which offers a useful probe to develop a rapid throughput screening assay to identify hNav1.7 antagonists.
1. Introduction
Voltage-gated sodium channels (VGSCs) are responsible for the rising phase of the action potential in excitable cells such as neurons, cardiac myocytes and skeletal muscle myocytes [1,2]. VGSCs are composed of voltage-sensing and pore-forming elements in one principal α-subunit and one or two auxiliary β-subunits which alter the channel physiological properties and subcellular localization [3]. Based on the amino acid similarity of the α-subunit isoforms, nine VGSC subtypes have been described termed Nav1.1–Nav1.9 [4].
VGSCs represent the molecular targets for a broad range of potent neurotoxins that bind to at least six distinct neurotoxin sites on the sodium channel α-subunit and affect the ion permeation and gating of sodium channels [3]. These toxins include tetrodotoxin (TTX), saxitoxin, and μ-conotoxin (site 1); lipid-soluble alkaloid toxins, including batrachotoxin, veratridine, aconitine, and grayanotoxin (site 2); polypeptide sea anemone and α-scorpion toxins (site 3); β-scorpion toxins (site 4); marine toxins such as brevetoxins (PbTxs) and ciguatoxins (site 5); and δ-conotoxins (site 6) [5]. In addition, pyrethroid insecticides act at a distinctive site on the sodium channel α-subunit to enhance channel activity by shifting activation to more negative membrane potentials as well as by delaying inactivation [6].
The expression of sodium channel α-subunits is tissue-dependent. Nav1.7 is preferentially expressed in the nociceptive neurons such as dorsal root ganglion and trigeminal ganglion as well as the sympathetic ganglion neurons [7] producing “threshold currents” close to resting potential, amplifying small depolarization such as generator potentials [8]. The role of Nav1.7 in the nociception and pain has been well established [9]. Several gain-of-function mutations in SCN9A which encodes Nav1.7 caused primary erythromelalgia, resulting in burning pain and flushing [10,11]. On the contrary, the inflammatory pain responses were reduced or abolished in nociceptor-specific Nav1.7 knock-out mice [12,13]. Deleting SCN9A in both sensory and sympathetic neurons abolished the pain sensations and recapitulated the pain-free phenotype seen in humans with SCN9A loss-of-function mutations [13]. These observations highlighted Nav1.7 as a potentially useful target for the development of novel analgesics.
Patch clamp electrophysiology is the gold standard for characterizing compound activity on the ion channels. While irreplaceable to study the millisecond kinetics of activation and inactivation of VGSCs, the patch clamp technique is laborious and extremely slow, which has greatly limited the utility of this technique to discover novel chemotypes targeting on ion channels. Recently, the automated, multichannel, voltage-clamp instruments provided the possibility for screening larger libraries of compounds. Unfortunately, automated electrophysiology is still quite expensive and is available in few academic laboratories [14,15].
Thus, higher-throughput, less expensive techniques are valuable alternatives to automated electrophysiology. Recently, researchers have developed several fluorescence-based rapid throughput assays for ion channel ligands discovery. The high throughput screening (HTS) thallium (Tl+)-flux assay has been developed to discover modifiers of K+ channels [16,17,18], K+-coupled chloride cotransporters [19], and Na+ and K+-coupled chloride cotransporters [20]. Many efforts have been made to develop the functional HTS assays to identify the VGSC modifiers. These assays including using sodium specific fluorescence dye, sodium-binding benzofuran isophthalate/acetoxymethyl ester (SBFI/AM) in neurons [5,21,22] and in heterologously expressed VGSCs cells [23]. However, the sodium bounded SBFI/AM fluorescence required excitations at two wavelengths (340 and 380 nm) which limited the throughput. In addition, in a heterologously expressed system, the fluorescence signal to noise ratio of the sodium channel agonists at defined recognition sites was minimal [23]. Membrane potential dye such as DiSBAC2(3) was also used to develop the HTS assay for discovering the VGSC antagonists [24,25]. In addition to its two wavelength recording (460 nm and 580 nm), the robust FRET fluorescence signals only can be achieved by co-application of two sodium channel agonists simultaneously. This co-application of two agonists resulted in a low Z′ value (0.15–0.45) which was not suitable for HTS assay [25]. Therefore, an agonist which can efficaciously activate the VGSCs is needed. Antillatoxin (ATX), a structurally unique lipopeptide produced by the marine cyanobacterium, Lyngbya majuscula, is a VGSC agonist [5,23,26,27]. ATX binds to a topological distinct neurotoxin site and allosterically potentiates the VGSC agonists-induced [3H]batrachotoxin binding [27]. More importantly, functional analysis has demonstrated that ATX displays the highest efficacy on the stimulation of sodium influx compared to other VGSC agonists [5,23].
In this study, we compared the influence of a range of VGSC agonists which bound to VGSC distinct neurotoxin sites using FLIPR membrane potential (FMP) blue dye in a 96-well format in hNav1.7-HEK-293 cells. Among the agonists tested, only ATX produced a potent and efficacious membrane depolarization, providing a good signal/noise ratio. We further demonstrated that six VGSC antagonists including lidocaine, lamotrigine, phenytoin, carbamazepine, riluzole, and 2-amino-6-trifluoromethylthiobenzothiazole (SKA-19) all concentration-dependently inhibited ATX response with IC50 values comparable to that reported from patch-clamp experiments. These data suggested that ATX might represent a useful probe for developing an HTS assay to identify Nav1.7 antagonists.
2. Results
2.1. Influence of VGSC Agonists on Membrane Depolarization in HEK-293 Cells Stably Expressing hNav1.7
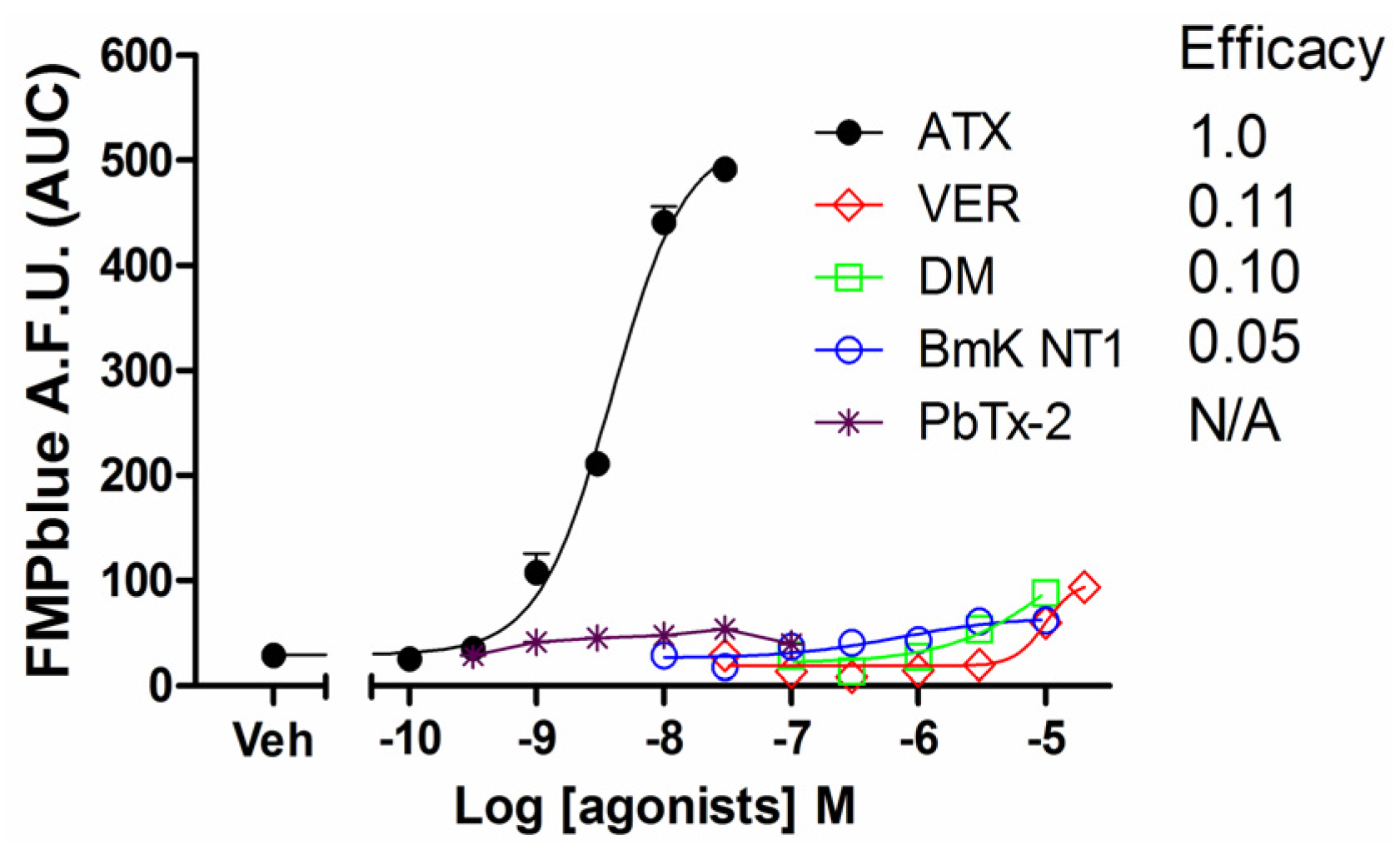
Previous studies have demonstrated that VGSC agonists such as veratridine, brevetoxin-2 (PbTx-2) had minimal effects on the stimulation of sodium influx or membrane depolarization in HEK-293 cells stably expressing VGSCs [23,25]. To identify an efficacious VGSC agonist which can provide a good signal/noise ratio, we examined the response on membrane depolarization in hNav1.7 expressed HEK-293 cells of five VGSC agonists which bound to topologically distinct neurotoxin sites including veratridine (VER, neurotoxin site 2), BmK NT1 (a site 3 α-scorpion toxin) [22], PbTx-2 (neurotoxin site 5) [5], deltamethrin (unrecognized neurotoxin site) [21], and ATX (unrecognized neurotoxin site) [23]. ATX produced a robust membrane depolarization in a concentration-dependent manner (Figure 1A). Veratridine (up to 20 μM), BmK NT1 (up to 10 μM) and deltamethrin (up to 10 μM) produced a marginal response on the membrane depolarization (Figure 1B,C,E). The neurotoxin site 5 agonist PbTx-2 was without effect on the membrane potential change in hNav1.7-HEK-293 cells (Figure 1D). The EC50 value for ATX-stimulated membrane depolarization (area under curve, AUC vs Log (concentration)) was 7.8 ± 2.9 nM with a maximal response of 11.7-fold of vehicle control (Figure 2). Compared to the maximal ATX response (efficacy defined as 1), the maximal responses of veratridine, deltamethrin and BmK NT1 were only 0.11, 0.10 and 0.05, respectively (Figure 2).
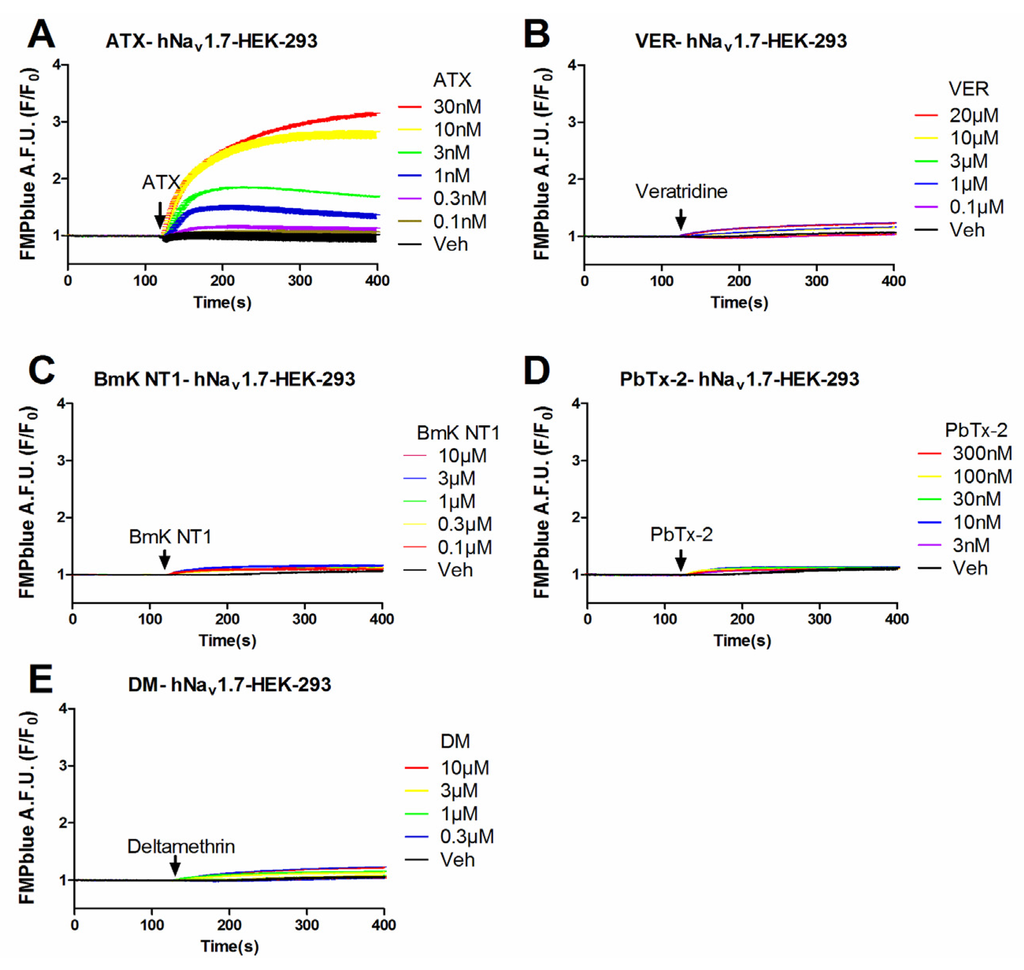
Figure 1.
Time-response relationships for ATX (A); veratridine (VER) (B); BmK NT1 (C); PbTx-2 (D); and deltamethrin (DM) (E) on membrane depolarization in hNav1.7-HEK-293 cells. This experiment was performed in three independent cultures, each in triplicate.
Figure 1.
Time-response relationships for ATX (A); veratridine (VER) (B); BmK NT1 (C); PbTx-2 (D); and deltamethrin (DM) (E) on membrane depolarization in hNav1.7-HEK-293 cells. This experiment was performed in three independent cultures, each in triplicate.
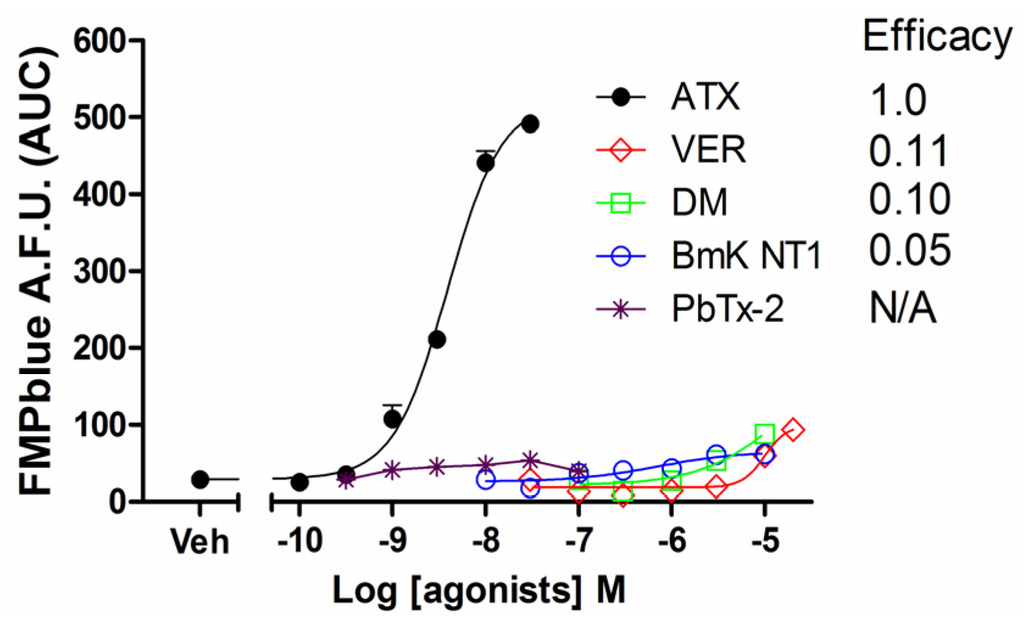
Figure 2.
Concentration-response relationship curves for ATX, veratridine, deltamethrin, BmK NT1 and PbTx-2 induced membrane potential changes. Each data point represents the mean ± SEM from two experiments, each in triplicates. ATX produced an efficacious response in the membrane depolarization while veratridine, deltamethrin and BmK NT1 only produced marginal response with efficacies of 0.11, 0.10, and 0.05 respectively. PbTx-2 was without effect on the membrane depolarization.
Figure 2.
Concentration-response relationship curves for ATX, veratridine, deltamethrin, BmK NT1 and PbTx-2 induced membrane potential changes. Each data point represents the mean ± SEM from two experiments, each in triplicates. ATX produced an efficacious response in the membrane depolarization while veratridine, deltamethrin and BmK NT1 only produced marginal response with efficacies of 0.11, 0.10, and 0.05 respectively. PbTx-2 was without effect on the membrane depolarization.
2.2. ATX-Induced Membrane Depolarization Was Dependent on the Activation of hNav1.7
Given the efficacious response on the membrane potential change in hNav1.7-HEK-293 cells, we examined whether this membrane depolarization was from the activation of hNav1.7. Pre-treatment of TTX, a pore blocker of VGSC, concentration-dependently suppressed the ATX (10 nM)-induced membrane depolarization with an IC50 value of 49.8 nM (31.0–80.4 nM, 95% CI) (Figure 3A,B). In null HEK-293 cells, ATX produced minimal response on the membrane depolarization which, compared to the ATX-induced response in hNav1.7-HEK-293 cells, was marginal (Figure 3C–E).
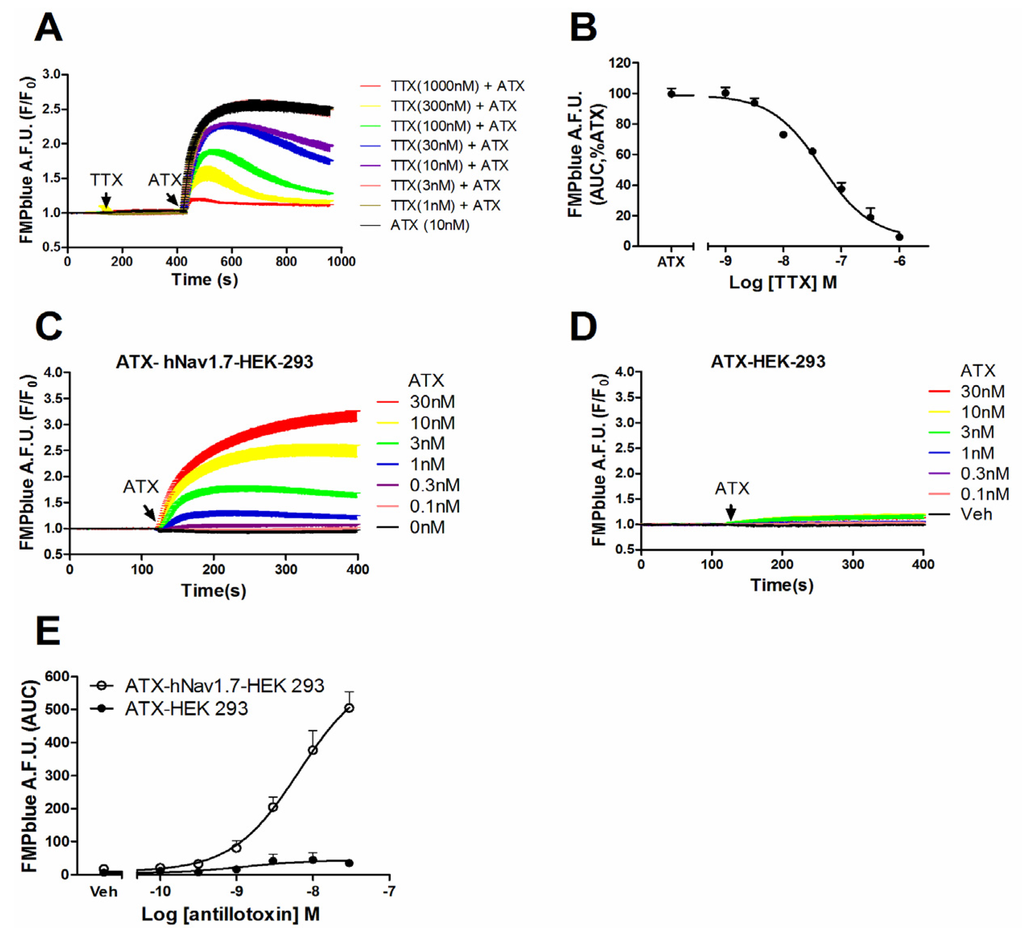
Figure 3.
ATX-produced membrane depolarization was dependent on the activation of hNav1.7. (A) TTX antagonized ATX-induced membrane depolarization in hNav1.7 HEK-293 cells as a function of time; (B) Concentration-response curve for TTX suppressed ATX-induced depolarization in hNav1.7 HEK 293 cells. Each data point represents the mean ± SEM from two independent cultures performed in triplicate; (C) and (D) ATX response on membrane depolarization in hNav1.7-HEK-293 cells and null HEK-293 cells as a function of time, respectively; (E) Concentration-response relationships of ATX response on membrane depolarization in hNav1.7-HEK-293 cells and null HEK-293 cells. This experiment was performed in two independent cultures, each in triplicate with similar results.
Figure 3.
ATX-produced membrane depolarization was dependent on the activation of hNav1.7. (A) TTX antagonized ATX-induced membrane depolarization in hNav1.7 HEK-293 cells as a function of time; (B) Concentration-response curve for TTX suppressed ATX-induced depolarization in hNav1.7 HEK 293 cells. Each data point represents the mean ± SEM from two independent cultures performed in triplicate; (C) and (D) ATX response on membrane depolarization in hNav1.7-HEK-293 cells and null HEK-293 cells as a function of time, respectively; (E) Concentration-response relationships of ATX response on membrane depolarization in hNav1.7-HEK-293 cells and null HEK-293 cells. This experiment was performed in two independent cultures, each in triplicate with similar results.
2.3. Z′ Factor Determination
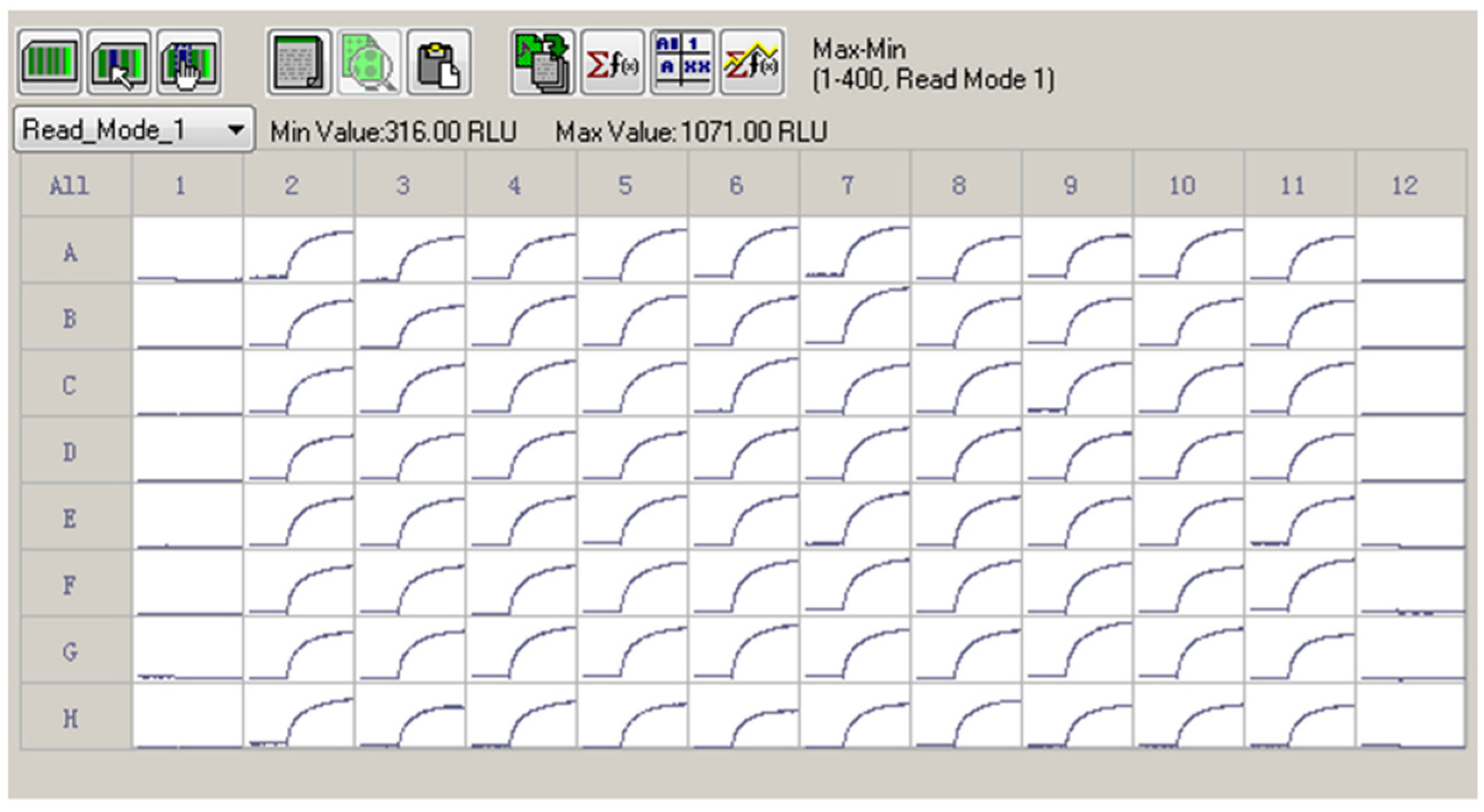
Given the efficacious ATX response on the membrane depolarization which was solely dependent on the hNav1.7 activation, we determined Z′ factor to test the suitability to use ATX and membrane potential dye for HTS assay. As shown in Figure 4, 30 nM ATX produced a robust, yet consistent response on the membrane depolarization. The calculated Z′ value was 0.7598.
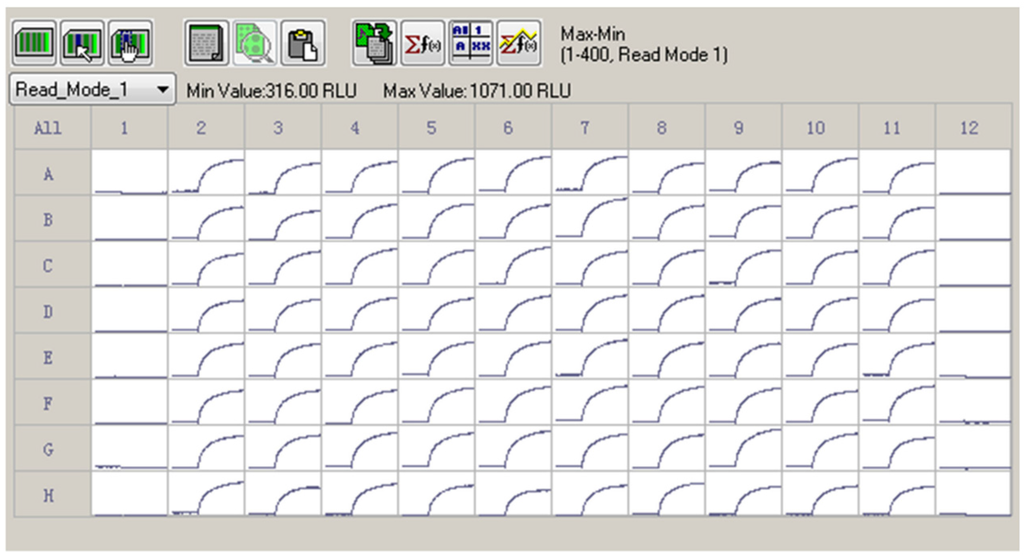
Figure 4.
A representative 96-well plate for ATX (30 nM) response on the membrane depolarization. The cells in Columns 2 to 11 were exposed to 30 nM of ATX. The Columns 1 and 12 were negative controls (0.1% DMSO). ATX produced a robust, yet consistent membrane depolarization. The Z′ factor was calculated to be 0.7589. This experiment was performed in two independent cultures.
Figure 4.
A representative 96-well plate for ATX (30 nM) response on the membrane depolarization. The cells in Columns 2 to 11 were exposed to 30 nM of ATX. The Columns 1 and 12 were negative controls (0.1% DMSO). ATX produced a robust, yet consistent membrane depolarization. The Z′ factor was calculated to be 0.7589. This experiment was performed in two independent cultures.
2.4. Influence of an Array of VGSC Antagonists on ATX-Induced Membrane Depolarization
We next tested the influence of six VGSC antagonists on ATX (10 nM)-induced membrane depolarization. All the VGSC antagonists tested including lidocaine, lamotrigine, phenytoin, carbamazepine, riluzole, and SKA-19 produced concentration-dependent inhibition of ATX (10 nM)-induced membrane depolarization (Figure 5). The IC50 values for SKA-19, riluzole, phenytoin, lamotrigine, carbamazepine, and lidocaine were 2.02 (1.49–2.74 μM, 95% CI), 3.58 (2.67–4.80 μM, 95% CI), 18.7 (11.8–29.7 μM, 95% CI), 66.3 (40.7–108.1 μM, 95% CI), 77.7 (49.9–121.0 μM, 95% CI) and 150.6 (92.9–244.0 μM, 95% CI), respectively (Table 1). The IC50 values generated here are consistent to that generated from patch clamp (Table 1). It should be noted that riluzole, SKA-19, carbamazepine, and lamotrigine all produced nearly complete inhibition on ATX-induced membrane depolarization. However, the maximal inhibition of lidocaine and phenytoin on ATX-induced depolarization was somewhat smaller representing a maximal suppressing of 80.2% ± 5.8% and 78.8% ± 5.5%, respectively.
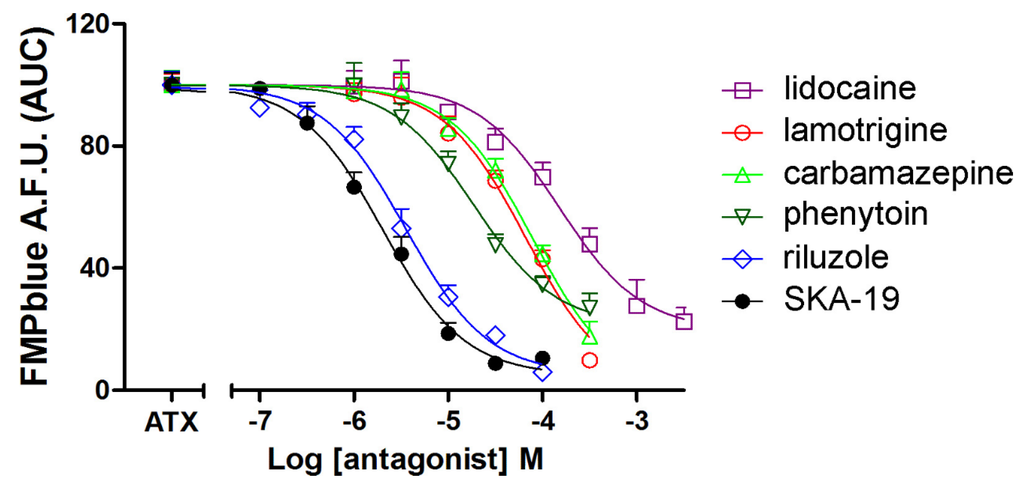
Figure 5.
Influence of VGSC antagonists including riluzole, SKA-19, phenytoin, lidocaine, carbamazepine, and lamotrigine on ATX (10 nM)-induced membrane depolarization. Data are presented as percentage of 10 nM ATX-induced fluorescence change. Each data point represents the mean ± SEM from two experiments, each in triplicates.
Figure 5.
Influence of VGSC antagonists including riluzole, SKA-19, phenytoin, lidocaine, carbamazepine, and lamotrigine on ATX (10 nM)-induced membrane depolarization. Data are presented as percentage of 10 nM ATX-induced fluorescence change. Each data point represents the mean ± SEM from two experiments, each in triplicates.
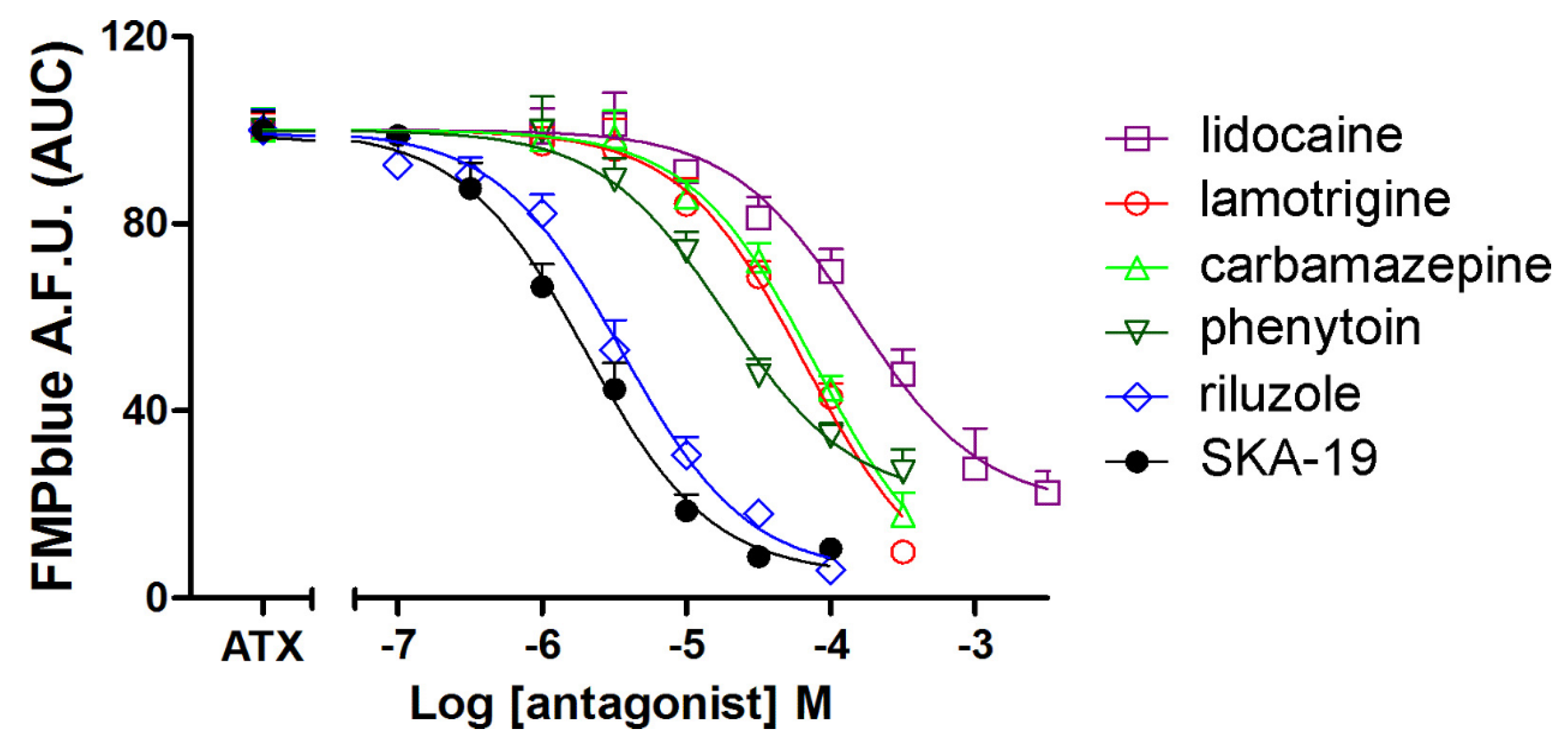

Table 1.
Comparison of the IC50 values generated from this study with that from patch clamp.
| Compounds | IC50 (μM) (95% CI) | Patch-Clamp IC50 (μM) | Reference |
|---|---|---|---|
| SKA-19 | 2.02 (1.49–4.74) | 5.8 | [28] |
| Riluzole | 3.58 (2.67–4.80) | 2 | [29] |
| Phenytoin | 18.7 (11.8–29.7) | 31.6 | [24] |
| Lamotrigine | 66.3 (40.6–108.1) | 79 | [30] |
| Carbamazepine | 77.7 (49.9–121.0) | 101 | [30] |
| Lidocaine | 150.6 (92.9–244.0) | 110 | [31] |
3. Discussion
In primary cultured neuronal preparation, the VGSC agonists ATX, veratridine, BmK NT1, PbTx-2 and deltamethrin which bound to topologically distinct neurotoxin sites all produced robust and significant sodium influx with distinct efficacies [5,22,23]. However, several studies have pointed out that in heterologous expression systems, these VGSC agonists produced a minimal response on both sodium influx and membrane potential [5,25]. In this study, we examined the ability of five VGSC agonists which bound to topologically distinctive neurotoxin sites to stimulate the membrane depolarization in hNav1.7-HEK-293 cells. Consistent with the previous studies [23,25], veratridine (neurotoxin site 2) only produced minimal response on the membrane depolarization whereas PbTx-2 (neurotoxin site 5) was without effect in hNav1.7-HEK-293 cells. We further demonstrated that deltamethin, which bound to an undefined neurotoxin site delaying the inactivation of the VGSCs [32], produced minimal response on the membrane potential changes. In addition, a scorpion toxin, BmK NT1 which likely bound to neurotoxin site 3 and prolonged the inactivation of the VGSCs in neurons [22] only produced marginal response to stimulate membrane depolarization. However, ATX produced an efficacious response on the membrane depolarization. Although the sodium channel expression density may partially account for the response discrepancy between neuronal and heterologously expression system [33], the hNav1.7-HEK-293 cells had little β-subunits co-expression. Sodium channel β-subunits regulate α-subunit function at multiple levels including mRNA expression, channel stabilization/trafficking and direct channel modulation [34]. In addition, the resting membrane potential for HEK-293 cells is relatively depolarized (−35 ± 5 mV) [35]. At this depolarized resting membrane potential, most hNav1.7 channels are in inactivated state [35]. Veratridine, BmK NT1, PbTx-2 and deltamethrin were demonstrated to primarily delay the VGSCs inactivation kinetics, but not the activation kinetics [22,32,36], an alternative explanation for these four VGSC agonists only producing marginal response in hNav1.7-HEK-293 cells. Although the detailed electrophysiological characterization of ATX on the VGSC remained to be established, the efficacious response on the membrane depolarization in hNav1.7-HEK-293 cells highly suggested that ATX response was not dependent on the β-subunits. The unique efficacious response of ATX also suggested that ATX may interact preferentially with the inactivated state of VGSC α-subunits.
The ATX response in hNav1.7-HEK-293 cells was from activation of hNav1.7 inasmuch as TTX completely suppressed the ATX-induced membrane depolarization. Furthermore, in null HEK-293 cells, ATX produced marginal membrane depolarization. The marginal membrane depolarization possibly was derived from the endogenously expressed VGSCs in null HEK-293 cells [37]. A rapid throughput assay to identify Nav1.7 antagonists has been developed by co-application of veratridine and a scorpion toxin SVqq to achieve the robust fluorescence signals through activating VGSCs. However, this co-application of two agonists resulted in a low Z′ value (0.15–0.45) [25], which was not suitable for the HTS assay. We demonstrated here that ATX produced a robust as well as consistent fluorescence change in a whole 96-well plate with a Z′ value of 0.7589. The Z′ value greater than 0.5 was thought to be suitable for the HTS assay [25].
In this study, we further demonstrated that an array of VGSC antagonists, including SKA-19, riluzole, phenytoin, lamotrigine, carbamazepine, and lidocaine all concentration-dependently suppressed ATX-induced membrane depolarization. It has been demonstrated that the IC50 values generated from a fluorescence-based assay is typically five-fold less potent than that generated from patch-clamp [24,30]. However, we demonstrated that the IC50 values for these VGSC inhibitors generated in this study were consistent with that from patch-clamp experiments [24,28,29,30,31,38,39] (Table 1). Riluzole has been reported to suppress TTX-sensitive VGSC current in inactivated state with IC50 values of 2 μM in dorsal root ganglion neurons, in which Nav1.7 was the major TTX-sensitive VGSC subtype. At resting state, riluzole displayed a much lower affinity with an IC50 value of 90 μM, suggesting riluzole preferred to bind to the inactivated state of VGSCs [29]. We demonstrated that riluzole inhibited the ATX-induced membrane potential changes with an IC50 value of 3.58 μM which was comparable to its affinity in the inactivated state [29]. SKA-19, a thioanalog of riluzole, is a use- and state-dependent VGSC antagonist with IC50 value of 5.8 ± 2.6 μM. We demonstrated here that SKA-19 suppressed the ATX (10 nM)-induced membrane depolarization in hNav1.7-HEK-293 cells with an IC50 value of 2.02 μM, a value similar to that from voltage-patch clamp experiment [28]. The anticonvulsants phenytoin, lamotrigine, and carbamazepine were also preferred to bind to inactivated state of VGSCs [40,41,42,43,44,45,46,47]. The affinities for these anticonvulsants on the inactivated state are much higher than that on the closed and open states [48]. We demonstrated that the IC50 values for lamotrigine, carbamazepine and phenytoin suppression of ATX-induced membrane depolarization in hNav1.7-HEK-293 cells were 66.3, 77.7 and 18.7 μM, respectively, which were more consistent with their affinities on the inactivated state. The local anesthetics, lidocaine has been reported to affect the steady-state fast inactivation of Nav1.7 channels with an IC50 value of 110 ± 20 μM [31] which was also consistent with current finding (150.6 μM, 92.9–244.0 μM, 95% CI). Considered together, it appeared that the IC50 values generated from current fluorescence-based HTS assay were more consistent with their respective affinity in the inactivated state.
It has been reported that riluzole, lidocaine, phenytoin bound to distinct sites of the sodium channels [49,50,51,52]. For example, riluzole interacts with amino acids residues TYR 1787, LEU 1843 and GLN 1799 located in the transmembrane segment S6 of domain IV of the α-subunit [53]. Lidocaine binds to the local anesthetics site located in the channel pore [39,54]. Phenytoin binds to the S6 segments of domains III and IV of the Na+ channel α-subunit [40,47,55,56]. In addition, TTX binds to another neurotoxin site distinct to those use- and steady-state blockers. The comparable IC50 values between current study and reported previously suggesting that the fluorescence-based assay developed here was capable of identifying the hNav1.7 inhibitors bound to distinct neurotoxin sites on the α-subunits of VGSCs.
It has been reported that fluorescence-based assays are often subject to high false positive hits [57,58]. Further study was required to screen a chemical library to determine the liability of the assay. Nevertheless, the HTS assay developed here may represent a useful alternative for the primary screen to identify hNav1.7 antagonists with novel pharmacophores.
4. Materials and Methods
4.1. Materials
FMP blue dye was obtained from Molecular Devices (Sunnyvale, CA, USA). ATX was synthesized as described previously and was characterized to be above 95% purity [59]. G-418, penicillin, streptomycin, heat inactivated fetal bovine serum, poly-d-lysine (molecular weight >300,000), riluzole, veratridine and carbamazepine, deltamethrin, PbTx-2, TTX, and lamotrigine were obtained from Sigma-Aldrich (St. louis, MO, USA). SKA-19 was provided by Prof. Wulff at the University of California, Davis as described previously [28] and was characterized to be greater than 95% purity. Lidocaine was purchased from Abcam (Cambridge, MA, USA). The HEK-293 stably expressed Nav1.7 was a generous gift from Dr. Lossin (University of California, Davis) and was the same line used as described previously [28].
4.2. Cell Culture
Human Embryonic Kidney 293 (HEK-293) cells were cultured in DMEM with glutamine supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin and 0.1 mg/mL streptomycin. HEK-293 cells stably expressed hNav1.7 were cultured in DMEM supplemented with 10% FBS, 100 units/mL penicillin, 0.1 mg/mL streptomycin, and 500 μg/mL G-418. All cells were grown routinely as monolayers on poly-d-lysine coated T-75 flask in an atmosphere of 5% CO2 and 95% humidity at 37 °C.
4.3. Membrane Potential Change Detection
Membrane potential changes in HEK-293 or HEK-293 stably expressed hNav1.7 were determined using the FMP blue dye (Molecular Devices, Sunnyvale, CA, USA). The cells were plated onto poly-d-lysine (10 μg/mL) coated, 96-well, black-walled, clear-bottom plates at an initial density of 20,000 cells/well and cultured for 6 h. Cells were removed of their medium and 150 μL of 1× dye solution (1 bottle dissolved in 50 mL Locke’s buffer, in mM: 8.6 HEPES, 5.6 KCl, 154 NaCl, 5.6 Glucose, 1.0 MgCl2, 2.3 CaCl2, 0.0001 glycine, pH 7.4) was added to each well. Cells were then incubated at room temperature for 30 min. The plate was then transferred to a FLIPR®TETRA (Molecular Devices, Sunnyvale, CA, USA) chamber. Cells were excited at 510–545 nm and emission at 565–625 nm was recorded at 1 s intervals. After recording the basal fluorescence for 120 s, 50 μL of sodium channel agonists at different concentrations (prepared in 1× dye at 4× final drug concentrations) or vehicle (0.4% DMSO, 4×) were added to different wells by an automated, programmable pipetting system. The fluorescence was recorded for additional 5–6 min at a sampling rate of 1 s. To examine the VGSC antagonist response on ATX-stimulated membrane depolarization, after recording the basal fluorescence for 120 s, different concentrations of VGSC antagonists were added to corresponding wells and the fluorescence was recorded for additional 5 min followed by an addition of 40 nM (4×, final concentration, 10 nM) ATX. The fluorescence signals were presented as F/F0, where F was defined as the fluorescence at different time points; F0 was the basal fluorescence averaged from initial 5 data points.
4.4. Data Analysis
Time-response and concentration-response relationships curves were generated using GraphPad Prism 5 software (GraphPad Software, Inc., San Diego, CA, USA). The EC50 value for VGSC agonists-induced membrane depolarization was determined by non-linear regression analysis using a logistic equation. The IC50 values of VGSC antagonists against ATX-induced membrane depolarization was determined by non-linear regression analysis using a logistic equation. Z′ factor was calculated using the following equation as described previously [60]: Z′ = 1 − [3 SD of sample + 3 SD of control]/[mean of sample − mean of control]. Each experiment was repeated at least twice in independent cultures performed at least in triplicate.
5. Conclusions
The current study investigated an array of VGSC agonists to stimulate the membrane depolarization in hNav1.7-HEK-293 cells. We demonstrated that ATX but not other VGSC agonists tested produced efficacious response on the membrane depolarization. We further demonstrated that ATX can serve as a probe to develop an HTS assay for identifying hNav1.7 antagonist with distinct binding sites.
Acknowledgments
This work was supported by National Natural Science Foundation of China (No. 81473539, 81570696, and 31270985); National High Technology Research and Development Program of China (863 Program, 2015AA020314); Excellent Youth Foundation of Jiangsu Scientific Committee (BK20140029); Jiangsu Provincial Natural Science Foundation (BK20141357); A program project from State Key Laboratory of Natural Medicines, China Pharmaceutical University (SKLNMZZJQ201402); A program project from State Key Laboratory of Environmental Chemistry and Ecotoxicology, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences (KF-2015-13) and the Fundamental Research Funds for the Central Universities (Z114037).
Author Contributions
Fang Zhao and Xichun Li performed the experiments, analyzed the data and wrote the manuscript; Fan Zhang, Liang Jin and Boyang Yu analyzed the data and wrote the manuscript; Masayuki Inoue synthesized the antillatoxin; Zhengyu Cao designed the experiments, performed the data analysis and wrote the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| ATX | antillatoxin |
| FBS | fetal bovine serum |
| FLIPR | fluorescence imaging plate reader |
| FMP | FLIPR membrane potential |
| HEK | human embryonic kidney |
| hNav1.7 | human voltage-gated sodium channel subtype 1.7 |
| HTS | high throughput screen |
| PbTxs | brevetoxins |
| SBFI/AM | sodium-binding benzofuran isophthalate/acetoxymethyl ester |
| SKA-19 | 2-Amino-6-trifluoromethylthiobenzothiazole |
| Tl+ | thallium |
| TTX | tetrodotoxin |
| VGSCs | voltage-gated sodium channels |
References
- Frank, H.Y.; Yarov-Yarovoy, V.; Gutman, G.A.; Catterall, W.A. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol. Rev. 2005, 57, 387–395. [Google Scholar]
- Murray, J.K.; Ligutti, J.; Liu, D.; Zou, A.; Poppe, L.; Li, H.; Andrews, K.L.; Moyer, B.D.; McDonough, S.I.; Favreau, P.; et al. Engineering Potent and Selective Analogues of GpTx-1, a Tarantula Venom Peptide Antagonist of the NaV1. 7 Sodium Channel. J. Med. Chem. 2015, 58, 2299–2314. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Cestèle, S.; Yarov-Yarovoy, V.; Frank, H.Y.; Konoki, K.; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.L.; Barchi, R.L.; Caldwell, J.H.; Hofmann, F.; Howe, J.R.; Hunter, J.C.; Kallen, R.G.; Mandel, G.; Meisler, M.H.; Netter, Y.B.; et al. Nomenclature of voltage-gated sodium channels. Neuron 2000, 28, 365–368. [Google Scholar] [CrossRef]
- Cao, Z.; George, J.; Gerwick, W.H.; Baden, D.G.; Rainier, J.D.; Murray, T.F. Influence of lipid-soluble gating modifier toxins on sodium influx in neocortical neurons. J. Pharmacol. Exp. Ther. 2008, 326, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Ruigt, G.F.; Neyt, H.C.; Van der Zalm, J.M.; Van den Bercken, J. Increase of sodium current after pyrethroid insecticides in mouse neuroblastoma cells. Brain Res. 1987, 437, 309–322. [Google Scholar] [CrossRef]
- Estacion, M.; Han, C.; Choi, J.-S.; Hoeijmakers, J.; Lauria, G.; Drenth, J.; Gerrits, M.M.; Dib-Hajj, S.D.; Faber, C.G.; Merkies, I.; et al. Intra-and interfamily phenotypic diversity in pain syndromes associated with a gain-of-function variant of NaV1. 7. Mol. Pain 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Cummins, T.R.; Howe, J.R.; Waxman, S.G. Slow closed-state inactivation: A novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J. Neurosci. 1998, 18, 9607–9619. [Google Scholar] [PubMed]
- Toledo-Aral, J.J.; Moss, B.L.; He, Z.-J.; Koszowski, A.G.; Whisenand, T.; Levinson, S.R.; Wolf, J.J.; Silos-Santiago, I.; Halegoua, S.; Mandel, G. Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc. Natl. Acad. Sci. USA 1997, 94, 1527–1532. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, Y.; Li, S.; Xu, Z.; Li, H.; Ma, L.; Fan, J.; Bu, D.; Liu, B.; Fan, Z.; et al. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J. Med. Genet. 2004, 41, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Dib-Hajj, S.D.; Rush, A.M.; Cummins, T.R.; Hisama, F.M.; Novella, S.; Tyrrell, L.; Marshall, L.; Waxman, S.G. Gain-of-function mutation in Nav1. 7 in familial erythromelalgia induces bursting of sensory neurons. Brain 2005, 128, 1847–1854. [Google Scholar] [CrossRef] [PubMed]
- Nassar, M.A.; Stirling, L.C.; Forlani, G.; Baker, M.D.; Matthews, E.A.; Dickenson, A.H.; Wood, J.N. Nociceptor-specific gene deletion reveals a major role for Nav1. 7 (PN1) in acute and inflammatory pain. Proc. Natl. Acad. Sci. USA 2004, 101, 12706–12711. [Google Scholar] [CrossRef] [PubMed]
- Minett, M.S.; Nassar, M.A.; Clark, A.K.; Passmore, G.; Dickenson, A.H.; Wang, F.; Malcangio, M.; Wood, J.N. Distinct Nav1. 7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, J.; Bowlby, M.; Peri, R.; Vasilyev, D.; Arias, R. High-throughput electrophysiology: An emerging paradigm for ion-channel screening and physiology. Nat. Rev. Drug Discov. 2008, 7, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Farre, C.; Stoelzle, S.; Haarmann, C.; George, M.; Brüggemann, A.; Fertig, N. Automated ion channel screening: Patch clamping made easy. Expert Opin. Ther. Targets 2007, 11, 557–565. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-P.; Mangano, T.; Hufeisen, S.; Setola, V.; Roth, B.L. Identification of human Ether-a-go-go related gene modulators by three screening platforms in an academic drug-discovery setting. Assay Drug Dev. Technol. 2010, 8, 727–742. [Google Scholar] [CrossRef] [PubMed]
- Niswender, C.M.; Johnson, K.A.; Luo, Q.; Ayala, J.E.; Kim, C.; Conn, P.J.; Weaver, C.D. A novel assay of Gi/o-linked G protein-coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol. Pharmacol. 2008, 73, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Lewis, L.M.; Bhave, G.; Chauder, B.A.; Banerjee, S.; Lornsen, K.A.; Redha, R.; Fallen, K.; Lindsley, C.W.; Weaver, C.D.; Denton, J.S. High-throughput screening reveals a small-molecule inhibitor of the renal outer medullary potassium channel and Kir7. 1. Mol. Pharmacol. 2009, 76, 1094–1103. [Google Scholar] [CrossRef] [PubMed]
- Delpire, E.; Days, E.; Lewis, L.M.; Mi, D.; Kim, K.; Lindsley, C.W.; Weaver, C.D. Small-molecule screen identifies inhibitors of the neuronal K-Cl cotransporter KCC2. Proc. Natl. Acad. Sci. USA 2009, 106, 5383–5388. [Google Scholar] [CrossRef] [PubMed]
- Carmosino, M.; Rizzo, F.; Torretta, S.; Procino, G.; Svelto, M. High-throughput fluorescent-based NKCC functional assay in adherent epithelial cells. BMC Cell Biol. 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Shafer, T.J.; Crofton, K.M.; Gennings, C.; Murray, T.F. Additivity of pyrethroid actions on sodium influx in cerebrocortical neurons in primary culture. Environ. Health Perspect. 2011, 119, 1236–1249. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; He, Y.; Qiao, J.; Zhang, C.; Cao, Z. The natural scorpion peptide, BmK NT1 activates voltage-gated sodium channels and produces neurotoxicity in primary cultured cerebellar granule cells. Toxicon 2016, 109, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Gerwick, W.H.; Murray, T.F. Antillatoxin is a sodium channel activator that displays unique efficacy in heterologously expressed rNav1. 2, rNav1. 4 and rNav1. 5 alpha subunits. BMC Neurosci. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, S.; Dekermendjian, K.; Julien, R.; Huang, J.; Lund, P.-E.; Krupp, J.; Kronqvist, R.; Larsson, O.; Bostwick, R. Cellular HTS assays for pharmacological characterization of NaV1. 7 modulators. Assay Drug Dev. Technol. 2008, 6, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Felix, J.P.; Williams, B.S.; Priest, B.T.; Brochu, R.M.; Dick, I.E.; Warren, V.A.; Yan, L.; Slaughter, R.S.; Kaczorowski, G.J.; Smith, M.M.; et al. Functional assay of voltage-gated sodium channels using membrane potential-sensitive dyes. Assay Drug Dev. Technol. 2004, 2, 260–268. [Google Scholar] [CrossRef] [PubMed]
- Berman, F.; Gerwick, W.; Murray, T. Antillatoxin and kalkitoxin, ichthyotoxins from the tropical cyanobacterium Lyngbya majuscula, induce distinct temporal patterns of NMDA receptor-mediated neurotoxicity. Toxicon 1999, 37, 1645–1648. [Google Scholar] [CrossRef]
- Li, W.; Berman, F.; Okino, T.; Yokokawa, F.; Shioiri, T.; Gerwick, W.; Murray, T. Antillatoxin is a marine cyanobacterial toxin that potently activates voltage-gated sodium channels. Proc. Natl. Acad. Sci. USA 2001, 98, 7599–7604. [Google Scholar] [CrossRef] [PubMed]
- Coleman, N.; Nguyen, H.M.; Cao, Z.; Brown, B.M.; Jenkins, D.P.; Zolkowska, D.; Chen, Y.-J.; Tanaka, B.S.; Goldin, A.L.; Rogawski, M.A.; et al. The riluzole derivative 2-amino-6-trifluoromethylthio-benzothiazole (SKA-19), a mixed KCa2 activator and NaV blocker, is a potent novel anticonvulsant. Neurotherapeutics 2015, 12, 234–249. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-H.; Huang, C.-S.; Nagata, K.; Yeh, J.Z.; Narahashi, T. Differential action of riluzole on tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels. J. Pharmacol. Exp. Ther. 1997, 282, 707–714. [Google Scholar] [PubMed]
- Castle, N.; Printzenhoff, D.; Zellmer, S.; Antonio, B.; Wickenden, A.; Silvia, C. Sodium channel inhibitor drug discovery using automated high throughput electrophysiology platforms. Comb. Chem. High Throughput Screen. 2009, 12, 107–122. [Google Scholar] [CrossRef] [PubMed]
- Sheets, P.L.; Jarecki, B.W.; Cummins, T.R. Lidocaine reduces the transition to slow inactivation in Nav1. 7 voltage-gated sodium channels. Br. J. Pharmacol. 2011, 164, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Shafer, T.J.; Meyer, D.A.; Crofton, K.M. Developmental neurotoxicity of pyrethroid insecticides: Critical review and future research needs. Environ. Health Perspect. 2005, 113, 123–136. [Google Scholar] [CrossRef] [PubMed]
- LePage, K.T.; Dickey, R.W.; Gerwick, W.H.; Jester, E.L.; Murray, T.F. On the use of neuro-2a neuroblastoma cells versus intact neurons in primary culture for neurotoxicity studies. Crit. Rev. Neurobiol. 2005, 17, 27–50. [Google Scholar] [CrossRef]
- Moran, O.; Nizzari, M.; Conti, F. Endogenous expression of the β1A sodium channel subunit in HEK-293 cells. FEBS Lett. 2000, 473, 132–134. [Google Scholar] [CrossRef]
- Chemin, J.; Monteil, A.; Briquaire, C.; Richard, S.; Perez-Reyes, E.; Nargeot, J.; Lory, P. Overexpression of T-type calcium channels in HEK-293 cells increases intracellular calcium without affecting cellular proliferation. FEBS Lett. 2000, 478, 166–172. [Google Scholar] [CrossRef]
- Ulbricht, W. Effects of veratridine on sodium currents and fluxes. In Reviews of Physiology Biochemistry and Pharmacology; Springer: Berlin, Germany; Heidelberg, Germany, 1998; Volume 133, pp. 1–54. [Google Scholar]
- He, B.; Soderlund, D.M. Human embryonic kidney (HEK293) cells express endogenous voltage-gated sodium currents and Na v 1.7 sodium channels. Neurosci. Lett. 2010, 469, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.Z.; Gilmore, E.S.; Estacion, M.; Eastman, E.; Taylor, S.; Melanson, M.; Dib-Hajj, S.D.; Waxman, S.G. A novel Nav1. 7 mutation producing carbamazepine-responsive erythromelalgia. Ann. Neurol. 2009, 65, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Chevrier, P.; Vijayaragavan, K.; Chahine, M. Differential modulation of Nav1. 7 and Nav1. 8 peripheral nerve sodium channels by the local anesthetic lidocaine. Br. J. Pharmacol. 2004, 142, 576–584. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-C. A common anticonvulsant binding site for phenytoin, carbamazepine, and lamotrigine in neuronal Na+ channels. Mol. Pharmacol. 1998, 54, 712–721. [Google Scholar] [PubMed]
- Clare, J.J.; Tate, S.N.; Nobbs, M.; Romanos, M.A. Voltage-gated sodium channels as therapeutic targets. Drug Discov. Today 2000, 5, 506–520. [Google Scholar] [CrossRef]
- Errington, A.C.; Stöhr, T.; Heers, C.; Lees, G. The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage-gated sodium channels. Mol. Pharmacol. 2008, 73, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-C.; Bean, B.P. Slow binding of phenytoin to inactivated sodium channels in rat hippocampal neurons. Mol. Pharmacol. 1994, 46, 716–725. [Google Scholar] [PubMed]
- Kuo, C.-C.; Chen, R.-S.; Lu, L.; Chen, R.-C. Carbamazepine inhibition of neuronal Na+ currents: Quantitative distinction from phenytoin and possible therapeutic implications. Mol. Pharmacol. 1997, 51, 1077–1083. [Google Scholar] [PubMed]
- Cardenas, C.A.; Cardenas, C.G.; de Armendi, A.J.; Scroggs, R.S. Carbamazepine interacts with a slow inactivation state of Na V 1.8-like sodium channels. Neurosci. Lett. 2006, 408, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Lenkowski, P.W.; Batts, T.W.; Smith, M.D.; Ko, S.-H.; Jones, P.J.; Taylor, C.H.; McCusker, A.K.; Davis, G.C.; Hartmann, H.A.; White, H.S.; et al. A pharmacophore derived phenytoin analogue with increased affinity for slow inactivated sodium channels exhibits a desired anticonvulsant profile. Neuropharmacology 2007, 52, 1044–1054. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Franceschetti, S.; Avanzini, G.; Mantegazza, M. Phenytoin inhibits the persistent sodium current in neocortical neurons by modifying its inactivation properties. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.; Sun, G.; Clare, J.J.; Werkman, T.R.; Wadman, W.J. Properties of human brain sodium channel α-subunits expressed in HEK293 cells and their modulation by carbamazepine, phenytoin and lamotrigine. Br. J. Pharmacol. 2014, 171, 1054–1067. [Google Scholar] [CrossRef] [PubMed]
- Diao, L.; Hellier, J.L.; Uskert-Newsom, J.; Williams, P.A.; Staley, K.J.; Yee, A.S. Diphenytoin, riluzole and lidocaine: Three sodium channel blockers, with different mechanisms of action, decrease hippocampal epileptiform activity. Neuropharmacology 2013, 73, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Bean, B.P.; Cohen, C.J.; Tsien, R.W. Lidocaine block of cardiac sodium channels. J. Gen. Physiol. 1983, 81, 613–642. [Google Scholar] [CrossRef] [PubMed]
- Starmer, C.; Grant, A.; Strauss, H. Mechanisms of use-dependent block of sodium channels in excitable membranes by local anesthetics. Biophys. J. 1984, 46, 15–27. [Google Scholar] [CrossRef]
- Tunnicliff, G. Basis of the antiseizure action of phenytoin. Gen. Pharmacol. Vasc. Syst. 1996, 27, 1091–1097. [Google Scholar] [CrossRef]
- Bello, O.S.; Gonzalez, J.; Capani, F.; Barreto, G.E. In silico docking reveals possible Riluzole binding sites on Nav1. 6 sodium channel: Implications for amyotrophic lateral sclerosis therapy. J. Theor. Biol. 2012, 315, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Hille, B. Local anesthetics: Hydrophilic and hydrophobic pathways for the drug-receptor reaction. J. Gen. Physiol. 1977, 69, 497–515. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, D.S.; McPhee, J.C.; Scheuer, T.; Catterall, W.A. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc. Natl. Acad. Sci. USA 1996, 93, 9270–9275. [Google Scholar] [CrossRef] [PubMed]
- McPhee, J.C.; Ragsdale, D.S.; Scheuer, T.; Catterall, W.A. A critical role for transmembrane segment IVS6 of the sodium channel α subunit in fast inactivation. J. Biol. Chem. 1995, 270, 12025–12034. [Google Scholar] [CrossRef] [PubMed]
- Bajorath, J. Integration of virtual and high-throughput screening. Nat. Rev. Drug Discov. 2002, 1, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Whiteaker, K.L.; Gopalakrishnan, S.M.; Groebe, D.; Shieh, C.-C.; Warrior, U.; Burns, D.J.; Coghlan, M.J.; Scott, V.E.; Gopalakrishnani, M. Validation of FLIPR membrane potential dye for high throughput screening of potassium channel modulators. J. Biomol. Screen. 2001, 6, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Okura, K.; Matsuoka, S.; Goto, R.; Inoue, M. The twisted side chain of antillatoxin is important for potent toxicity: Total synthesis and biological evaluation of antillatoxin and analogues. Angew. Chem., Int. Ed. 2010, 49, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Wu, Z. Alternative statistical parameter for HTS assay quality assessment. J. Biomol. Screen. 2007, 12. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).