
Discovery of Novel Antiangiogenic Marine Natural Product Scaffolds



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
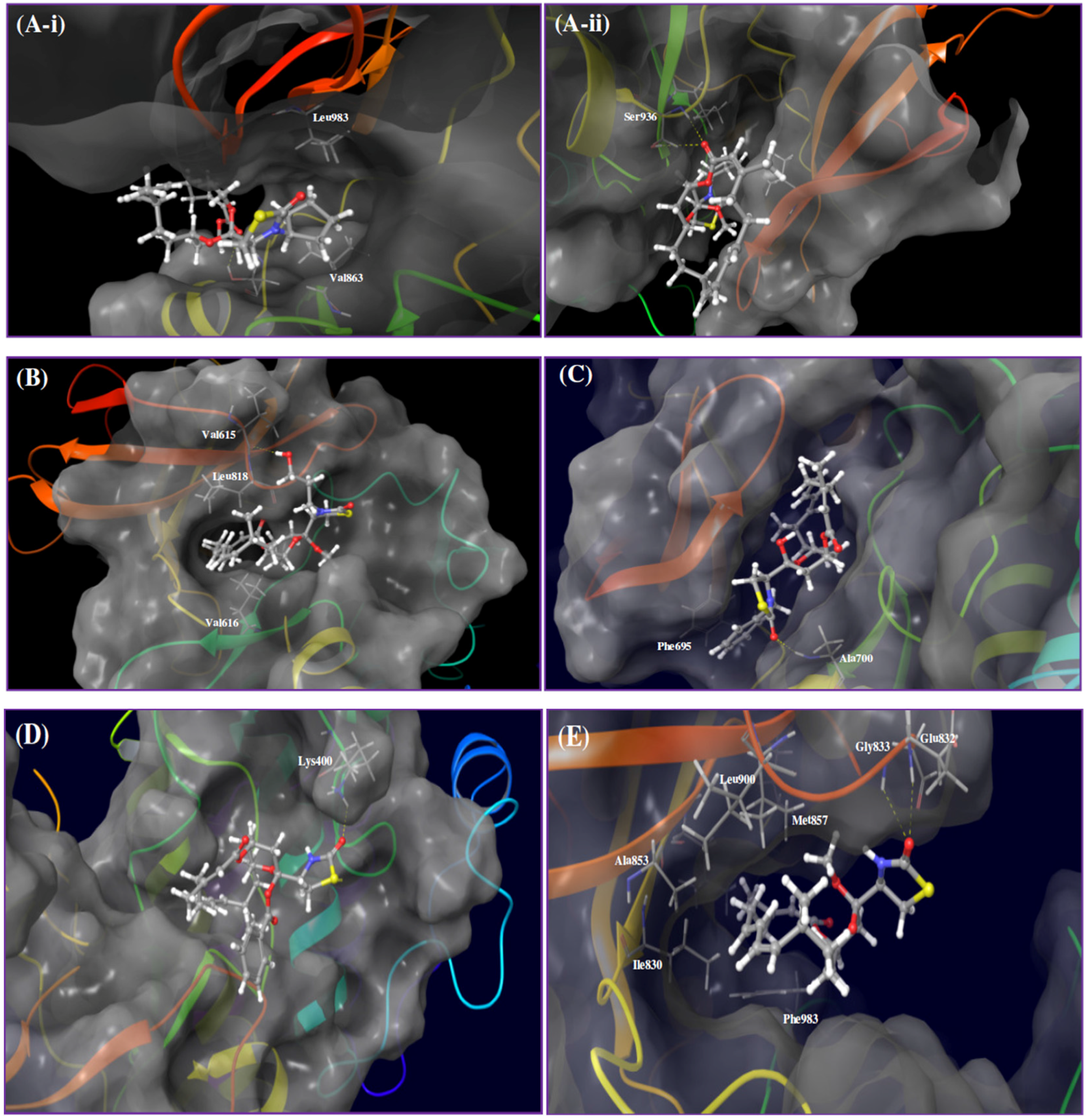
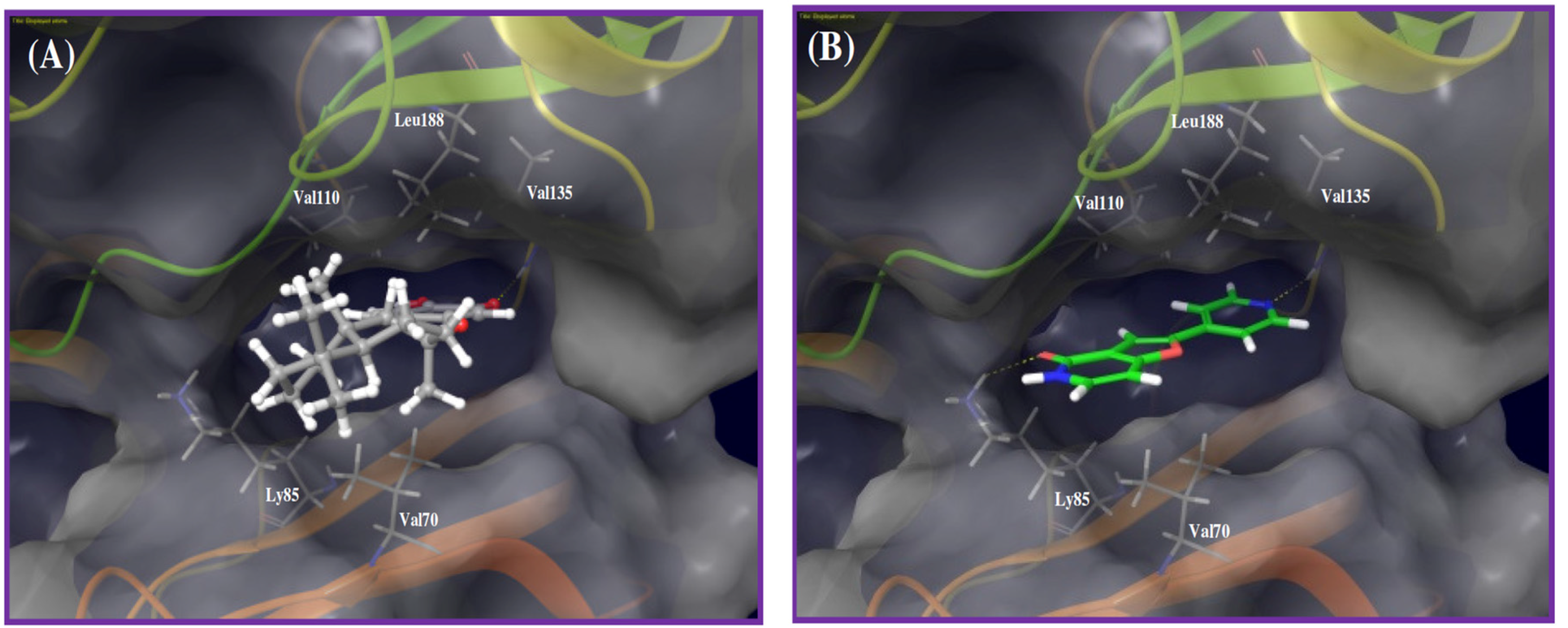
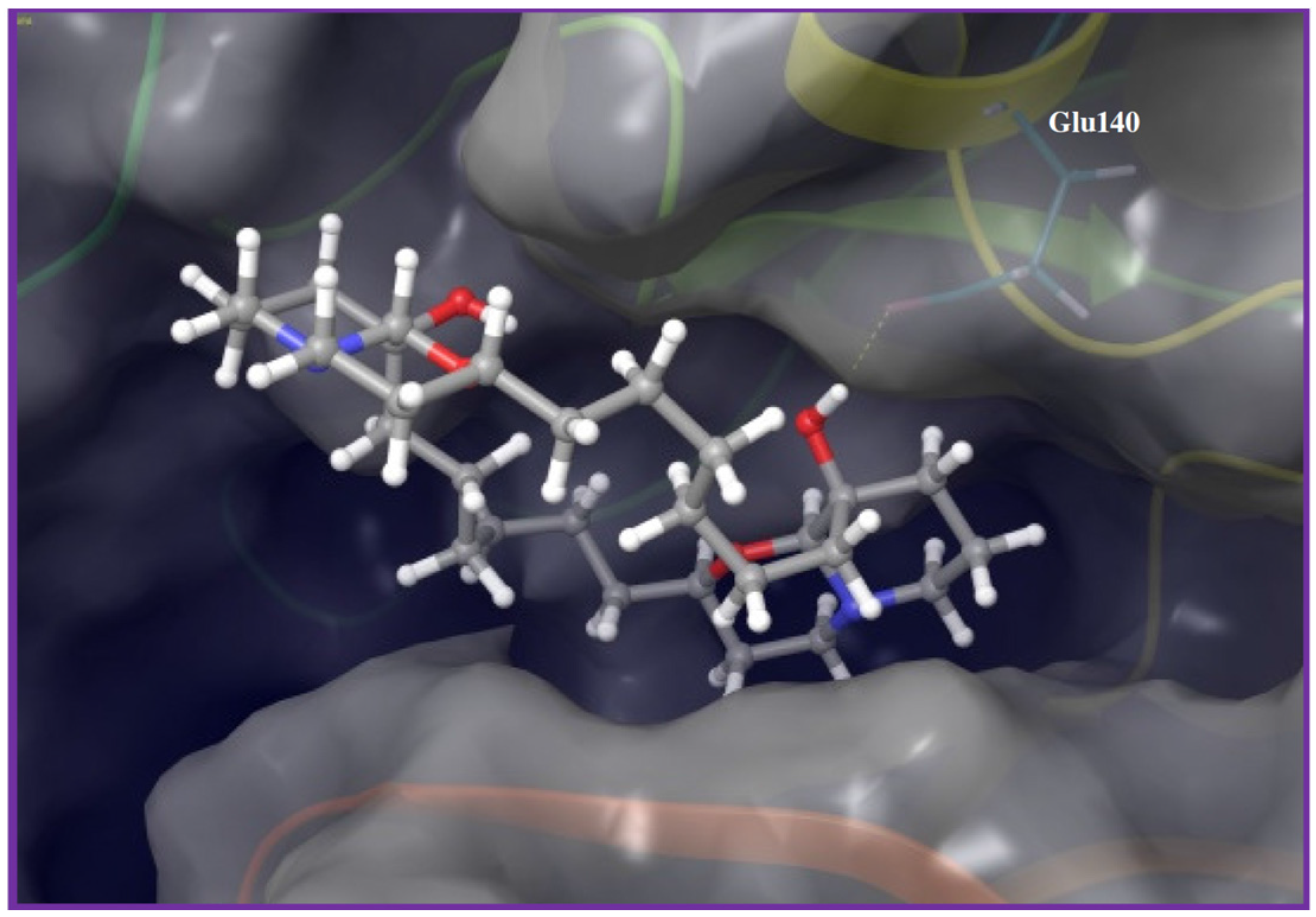
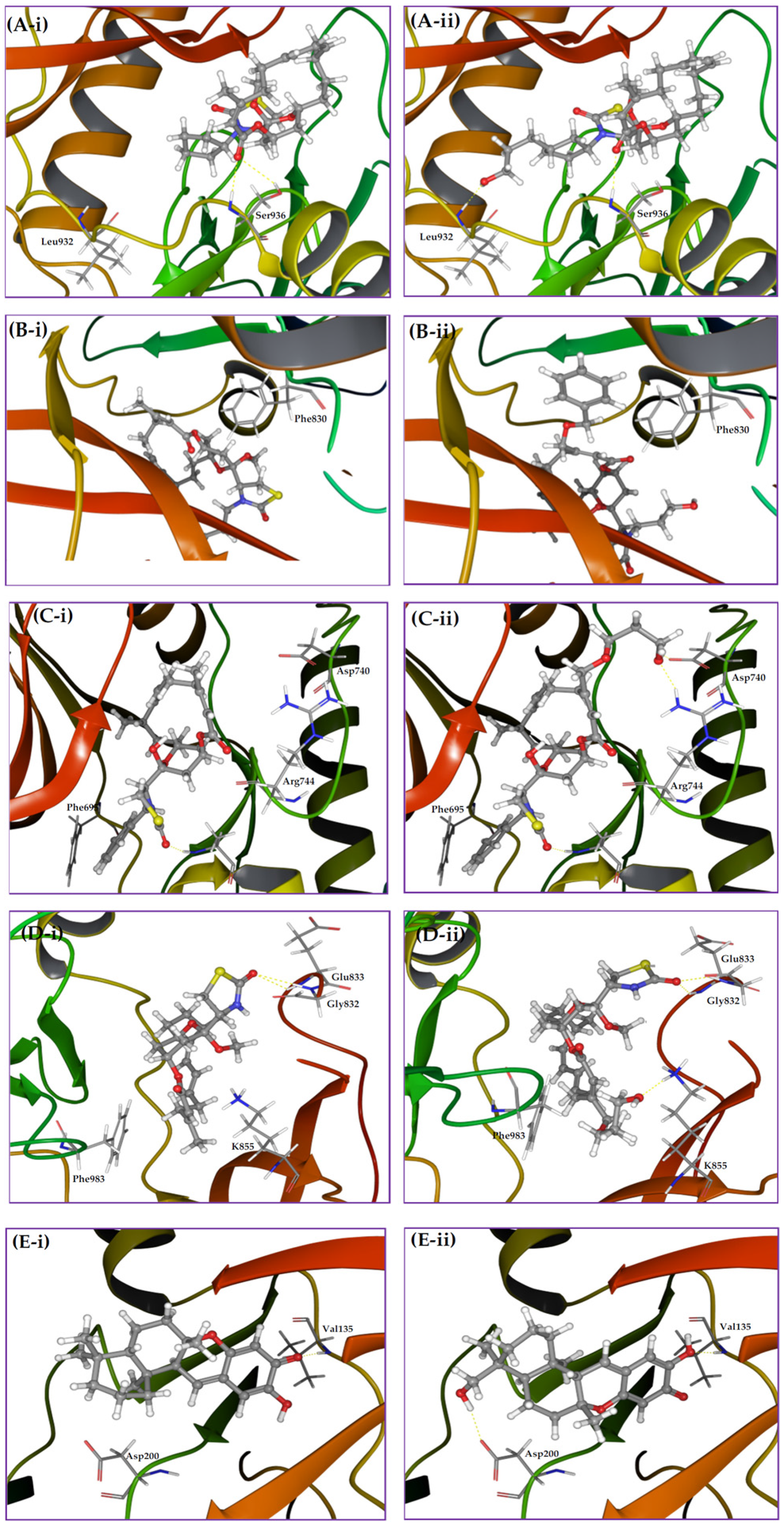
2. Results
3. Discussion
4. Materials and Methods
4.1. General Experimental Procedures
4.2. Test Compounds
4.3. Biological Experiments
4.3.1. Cell Lines
4.3.2. Endothelial Colony-Forming/Adipose-Derived Stem Cells (ECFC-ADSC) Coculture
4.3.3. Endothelial and Nuclear Staining
4.3.4. Cellular Imaging
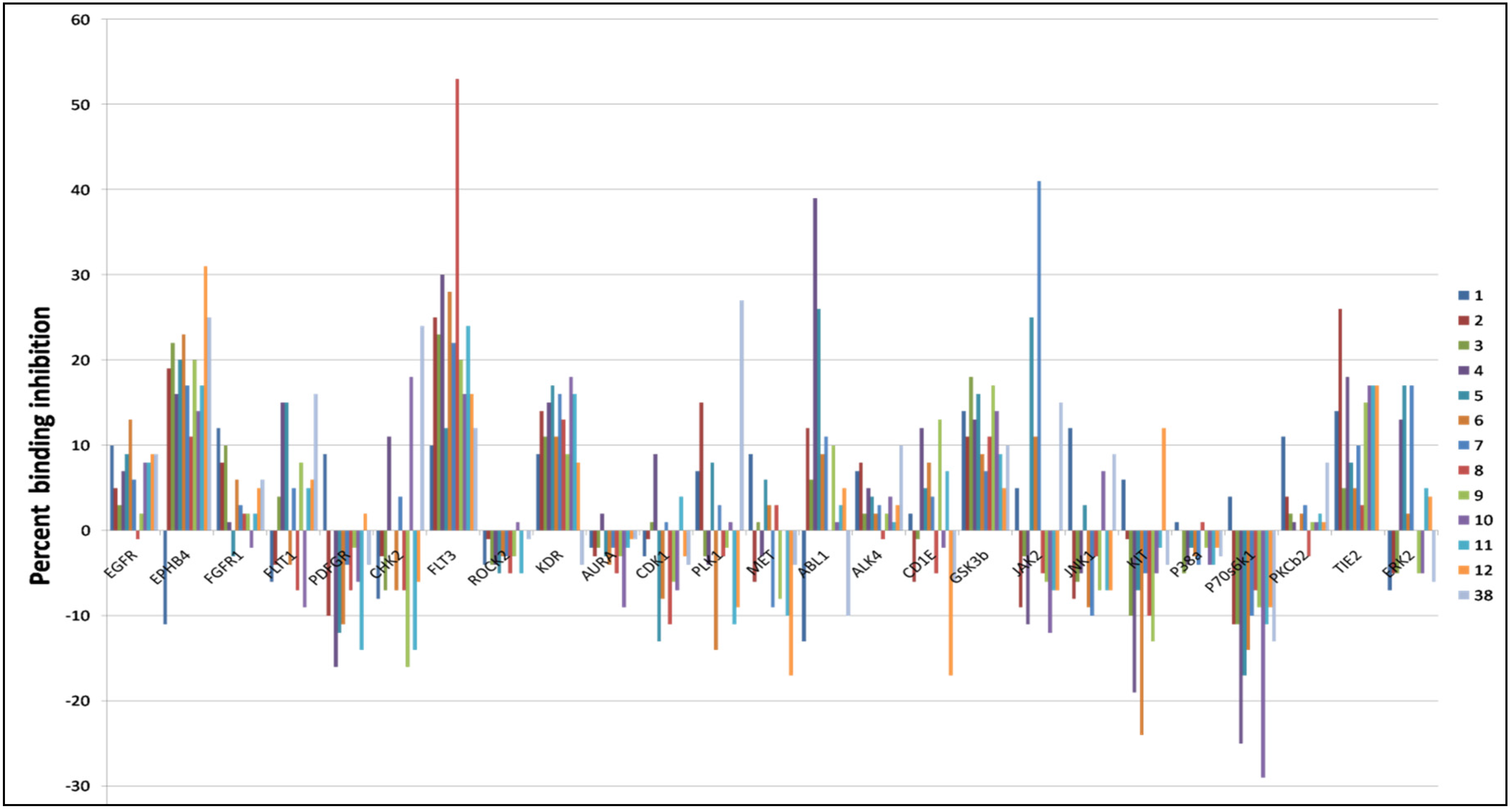
4.4. Biochemical Kinase Profiling
4.4.1. Molecular Modeling
4.4.2. Protein Structure Preparation
4.4.3. Ligand Structure Preparation
4.4.4. Molecular Docking
4.5.Statistical Validation of the Assays
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| MNPs | Marine natural products |
| FDA | United States Food and Drug Administration |
| EMA | European Medicines Agency |
| OIDD | The Lilly Open Innovation Drug Discovery Program |
| TZD | Thiazolidinon-2-yl |
| THP | Tetrahydropyran |
| JAK | Janus kinase |
| STATs | Signal transducers and activators of transcription |
| VEGF | Vascular endothelial growth factor |
| FLT3 | Fms-like tyrosine kinase 3 |
| RTK | Receptor tyrosine kinase |
| RAS | Rat sarcoma viral oncogene homolog |
| PI3K | Phosphatidylinositol-4,5-biphopshate 3-kinase |
| Ephrin | Eph receptor interacting protein |
| ABL1 | Abelson murine leukemia viral oncogene homolog 1 |
| CML | Chronic myeloid leukemia |
| bFGF | Basic fibroblast growth factor |
| TIE | Tyrosine kinases with immunoglobulin-like and EGF-like domains |
| VEGFRs | Vascular endothelial growth factor receptors |
| Grb2 | Growth factor receptor-bound protein 2 |
| EGF | Epidermal growth factor |
| Ang | Angiopoietin |
| GSK3-β | Glycogen synthase kinase-3-beta |
| PLK | Polo-like kinase |
| SAR | Structure-activity relationship |
| ECFCs | Endothelial colony-forming cells |
| ADSCs | Human adipose-derived stem cells |
| cLogP | Calculated logarithm of partition coefficient |
| HBD | Hydrogen bond donor |
| HBA | Hydrogen bond acceptor |
References
- Montaser, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 12, 1475–1489. [Google Scholar] [CrossRef] [PubMed]
- Martins, A.; Vieira, H.; Gaspar, H.; Santos, S. Marketed marine natural products in the pharmaceutical and cosmeceutical industries: Tips for success. Mar. Drugs 2014, 12, 1066–1101. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, J.S.; Daranas, A.H.; Norte, M.; Fernandez, J.J. Marine macrolides, a promising source of antitumor compounds. Anticancer Agents Med. Chem. 2009, 9, 122–137. [Google Scholar] [CrossRef] [PubMed]
- Spector, I.; Shochet, N.R.; Kashman, Y.; Groweiss, A. Latrunculins: Novel marine toxins that disrupt microfilament organization in cultured cells. Science 1983, 219, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Groweiss, A.; Shmueli, U.; Kashman, Y. Marine toxins of Latrunculia magnifica. J. Org. Chem. 1983, 48, 3512–3516. [Google Scholar] [CrossRef]
- Kashman, Y.; Groweiss, A.; Lidor, R.; Blasberger, D.; Carmely, S. Latrunculins: NMR study, two new toxins and a synthetic approach. Tetrahedron 1985, 41, 1905–1914. [Google Scholar] [CrossRef]
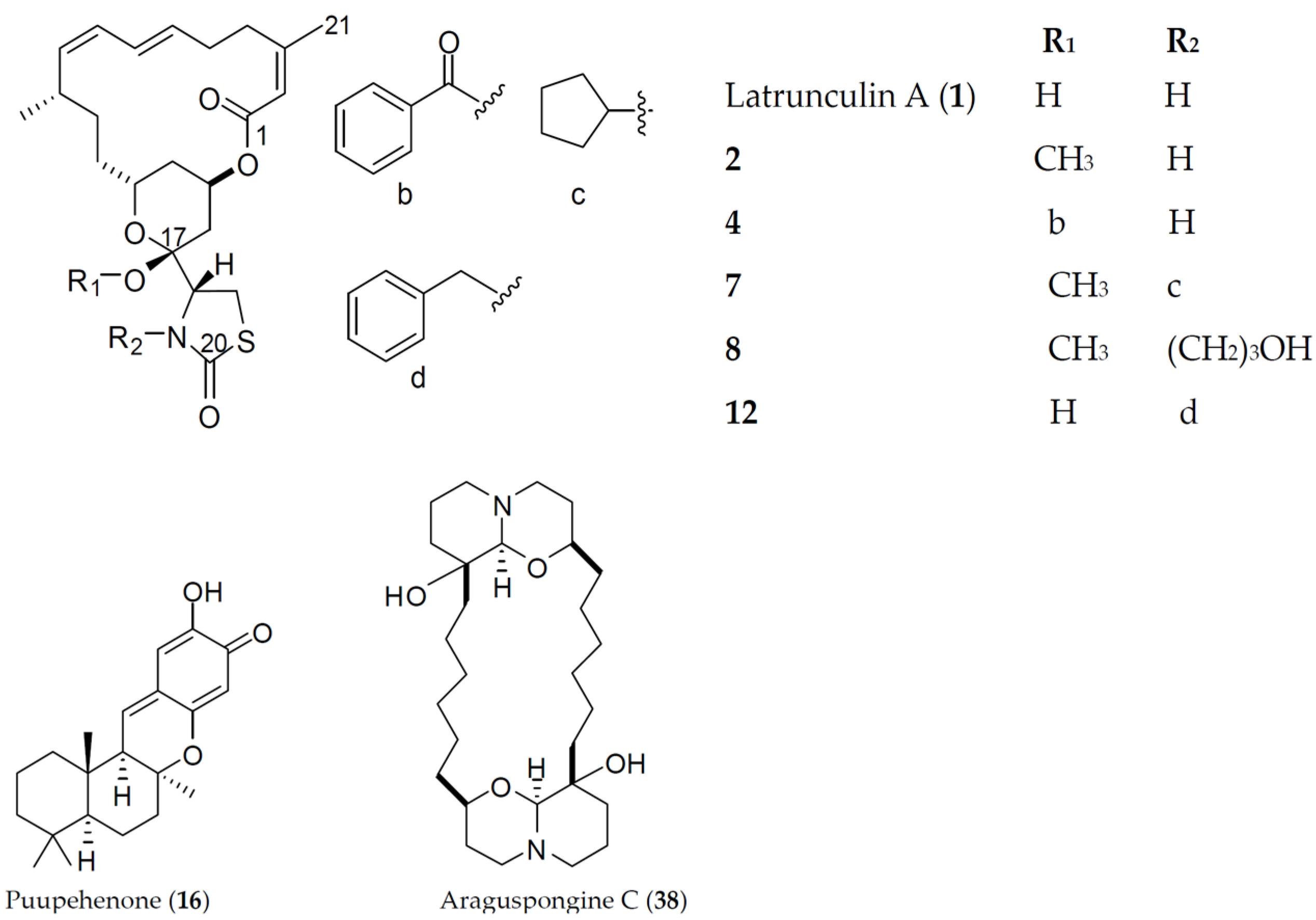
- El Sayed, K.A.; Youssef, D.T.A.; Marchetti, D. Bioactive natural and semisynthetic latrunculins. J. Nat. Prod. 2006, 69, 219–223. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, K.A.; Khanfar, M.A.; Shallal, H.M.; Muralidharan, A.; Awate, B.; Youssef, D.T.A.; Liu, Y.; Zhou, Y.D.; Nagle, D.G.; Shah, S. Latrunculin A and its C-17-O-carbamates inhibit prostate tumor cell invasion and HIF-1 activation in breast tumor cells. J. Nat. Prod. 2008, 71, 396–402. [Google Scholar]
- Khanfar, M.A.; Youssef, D.T.A.; El Sayed, K.A. Semisynthetic latrunculin derivatives as inhibitors of metastatic breast cancer: Biological evaluations, preliminary structure-activity relationship and molecular modeling studies. ChemMedChem 2010, 5, 274–285. [Google Scholar] [CrossRef] [PubMed]
- Gordaliza, M. Cytotoxic terpene quinones from marine sponges. Mar. Drugs 2010, 8, 2849–2870. [Google Scholar] [CrossRef] [PubMed]
- Kohmoto, S.; McConnell, O.J.; Wright, A.; Thompson, W.; Lui, M.; Sander, K.M. Puupehenone, a cytotoxic metabolite from a deep water marine sponge, Stronglyophora hartmani. J. Nat. Prod. 1987, 50, 336. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.E.; González-Iriarte, M.; Barrero, A.F.; Salvador-Tormo, N.; Muñzo-Chápuli, R.; Medina, M.A.; Quesada, A.R. Study of puupehenone and related compounds as inhibitors of angiogenesis. Int. J. Cancer 2004, 110, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Vicencio, J.M.; Ortiz, C.; Criollo, A.; Jones, A.W.; Kepp, O.; Galluzzi, L.; Joza, N.; Vitale, I.; Morselli, E.; Tailler, M.; et al. The inositol 1,4,5-trisphosphate receptor regulates autophagy through its interaction with Beclin 1. Cell Death Differ. 2009, 16, 1006–1017. [Google Scholar] [CrossRef] [PubMed]
- Akl, M.R.; Ayoub, N.M.; Ebrahim, H.Y.; Mohyeldin, M.M.; Orabi, K.Y.; Foudah, A.I.; El Sayed, K.A. Araguspongine C induces autophagic death in breast cancer cells through suppression of c-Met and HER2 receptor tyrosine kinase signaling. Mar. Drugs 2015, 13, 288–311. [Google Scholar] [CrossRef] [PubMed]
- Otrock, Z.K.; Mahfouz, R.A.; Makarem, J.A.; Shamselddine, A.I. Understanding the biology of angiogenesis: Review of the most important molecular mechanisms. Blood Cells Mol. Dis. 2007, 39, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Hanahan, D. Modes of resistance to antiangiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Verheul, H.M.W.; Pinedo, H.M. Possible molecular mechanisms involved in the toxicity of angiogenesis inhibition. Nat. Rev. Cancer 2007, 7, 475–485. [Google Scholar]
- Pawson, T.; Nash, P. Protein-protein interactions define specificity in signal transduction. Genes Dev. 2000, 14, 1027–1047. [Google Scholar] [PubMed]
- Cook, K.M.; Figg, W.D. Angiogenesis inhibitors—Current strategies and future prospects. CA Cancer J. Clin. 2010, 60, 222–243. [Google Scholar] [CrossRef] [PubMed]
- Ihle, J.N.; Gilliland, D.G. Jak2: Normal function and role in hematopoietic disorders. Curr. Opin. Genet. Dev. 2007, 17, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, E.M.; Wallace, T.A.; Godeny, M.D.; von Der Linden, D.; Sayeski, P.P. Jak2 tyrosine kinase: A true JAK of all trades? Cell Biochem. Biophys. 2004, 41, 207–232. [Google Scholar] [CrossRef]
- Chen, Z.; Han, Z.C. STAT3: A critical transcription activator in angiogenesis. Med. Res. Rev. 2008, 28, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Bar-Nata, M.; Nelson, E.; Xiang, M.; Frank, D. STAT signaling in the pathogenesis and treatment of myeloid malignancies. JAKSTAT 2012, 1, 55–64. [Google Scholar]
- Roux, P.; Blenis, J. ERK and P38 MAPK-activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.; Cantley, L. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Solanilla, A.; Grosset, C.; Lemercier, C.; Dupouy, M.; Mahon, F.X.; Schweitzer, K.; Reiffers, J.; Weksler, B.; Ripoche, J. Expression of Flt3-ligand by the endothelial cell. Leukemia 2000, 14, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Martiny-Baron, G.; Holzer, P.; Billy, E.; Schnell, C.; Brueggen, J.; Ferretti, M.; Schmiedeberg, N.; Wood, J.M.; Furet, P.; Imbach, P. The small molecule specific EphB4 kinase inhibitor NVP-BHG712 inhibits VEGF driven angiogenesis. Angiogenesis 2010, 13, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Park, I.; Lee, H. EphB/ephrinB signaling in cell adhesion and migration. Mol. Cells 2015, 38, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Noren, N.K.; Lu, M.; Freeman, A.L.; Koolpe, M.; Pasquale, E.B. Interplay between EphB4 on tumor cells and vascular ephrin-B2 regulates tumor growth. Proc. Natl. Acad. Sci. USA 2004, 101, 5583–5588. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, R.A. Cycling, stressed-out and nervous: Cellular functions of c-Abl. Trends Cell Biol. 1999, 9, 179–186. [Google Scholar] [CrossRef]
- Pendergast, A.M. The Abl family kinases: Mechanisms of regulation and signaling. Adv. Cancer Res. 2002, 85, 51–100. [Google Scholar] [PubMed]
- Yan, W.; Bentley, B.; Shao, R. Distinct angiogenic mediators are required for basic fibroblast growth factor- and vascular endothelial growth factor-induced angiogenesis: The role of cytoplasmic tyrosine kinas c-Abl in tumor angiogenesis. Mol. Biol. Cell 2008, 19, 2278–2288. [Google Scholar] [CrossRef] [PubMed]
- Ramsauer, M.; D’Amore, P.A. Getting Tie(2)d up in angiogenesis. J. Clin. Investig. 2002, 110, 1615–1617. [Google Scholar] [CrossRef] [PubMed]
- Seegar, T.C.; Eller, B.; Tzvetkova-Robev, D.; Kolev, M.V.; Henderson, S.C.; Nikolov, D.B.; Barton, W.A. Tie1-Tie2 interactions mediate functional differences between angiopoietin ligands. Mol. Cell 2010, 37, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Papapetropoulos, A.; Fulton, D.; Mahboubi, K.; Kalb, R.G.; O’Connor, D.S.; Li, F.; Altieri, D.C.; Sessa, W.C. Angiopoietin-1 inhibits endothelial cell apoptosis via the Akt/survivin pathway. J. Biol. Chem. 2000, 275, 9102–9105. [Google Scholar] [CrossRef] [PubMed]
- Kontos, C.D.; Cha, E.H.; York, J.D.; Peters, K.G. The endothelial receptor tyrosine kinase Tie1 activates phosphatidylinositol 3-kinase and Akt to inhibit apoptosis. Mol. Cell Biol. 2002, 22, 1704–1713. [Google Scholar] [CrossRef] [PubMed]
- Thurston, G. Role of angiopoietins and Tie receptor tyrosine kinases in angiogenesis and lymphangiogenesis. Cell Tissue Res. 2003, 314, 61–80. [Google Scholar] [CrossRef] [PubMed]
- Phukan, S.; Babu, V.S.; Kannoji, A.; Hariharan, R.; Balaji, V.N. GSK3β: Role in therapeutic landscape and development of modulators. Br. J. Pharmacol. 2010, 160, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Skurk, C.; Maatz, H.; Rocnik, E.; Bialik, A.; Force, T.; Walsh, K. Angiogenesis through activation of vascular endothelial growth factor signaling in endothelial cells. Circ. Res. 2005, 96, 308–310. [Google Scholar] [CrossRef] [PubMed]
- Llamazares, S.; Moreira, A.; Tavares, A.; Girdham, C.; Spruce, B.A.; Gonzalez, C.; Karess, R.E.; Glover, D.M.; Sunkel, C.E. polo encodes a protein kinase homolog required for mitosis in Drosophila. Genes Dev. 1991, 5, 2153–2165. [Google Scholar] [CrossRef] [PubMed]
- Barr, F.A.; Silljé, H.H.; Nigg, E.A. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 2004, 5, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Kushner, E.J.; Ferro, L.S.; Liu, J.Y.; Durrant, J.R.; Rogers, S.L.; Dudley, A.C.; Bautch, V.L. Excess centrosomes disrupt endothelial cell migration via centrosome scattering. J. Cell Biol. 2014, 206, 257–272. [Google Scholar] [CrossRef] [PubMed]
- De Cárcer, G.; Manning, G.; Malumbres, M. From Plk1 to Plk5: Functional evolution of polo-like kinases. Cell Cycle 2011, 10, 2255–2262. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W.; Schmidt, M.; Gekeler, V.; Denkert, C.; Stephan, C.; Jung, K.; Loening, S.; Dietel, M.; Kristiansen, G. Polo-like kinase 1 is overexpressed in prostate cancer and linked to higher tumor grades. Prostate 2004, 60, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Weichert, W.; Kristiansen, G.; Winzer, K.J.; Schmidt, M.; Gekeler, V.; Noske, A.; M€uller, B.M.; Niesporek, S.; Dietel, M.; Denkert, C. Polo-like kinase isoforms in breast cancer: Expression patterns and prognostic implications. Virchows Arch. 2005, 446, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Astrinidis, A.; Senapedis, W.; Henske, E.P. Hamartin, the tuberous sclerosis complex 1 gene product, interacts with polo-like kinase 1 in a phosphorylation-dependent manner. Hum. Mol. Genet. 2006, 15, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Valianou, M.; Cox, A.M.; Pichette, B.; Hartley, S.; Paladhi, U.R.; Astrinidis, A. Pharmacological inhibition of Polo-like kinase 1 (PLK1) by BI-2536 decreases the viability and survival of hamartin and tuberin deficient cells via induction of apoptosis and attenuation of autophagy. Cell Cycle 2015, 14, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Orabi, K.Y.; El Sayed, K.A.; Hamann, M.T.; Dunbar, D.C.; Al-Said, M.S.; Higa, T.; Kelly, M. Araguspongines K and L, new bioactive bis-1-oxaquinolizidine N-oxide alkaloids from Red Sea specimens of Xestospongia exigua. J. Nat. Prod. 2002, 12, 1782–1785. [Google Scholar] [CrossRef] [PubMed]
- Nasu, S.S.; Yeung, B.K.S.; Hamann, M.T.; Scheuer, P.J.; Kelly-Borges, M.; Goins, K. Puupehenone-related metabolites from two Hawaiian sponges, Hyrtios spp. J. Org. Chem. 1995, 60, 7290–7292. [Google Scholar] [CrossRef]
- Lee, J.A.; Chu, S.; Willard, F.S.; Cox, K.L.; Sells Galvin, R.J.; Peery, R.B.; Oliver, S.E.; Oler, J.; Meredith, T.D.; Heidler, S.A.; et al. Open Innovation for Phenotypic Drug Discovery: The PD2 assay panel. J. Biomol. Screen. 2011, 16, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Clayton, J.R.; Walgren, R.A.; Zhao, B.; Evans, R.J.; Smith, M.C.; Heinz-Taheny, K.M.; Kreklau, E.L.; Bloem, L.; Pitou, C.; et al. Discovery and characterization of LY2784544, a small-molecule tyrosine kinase inhibitor of JAK2V617F. Blood Cancer J. 2013, 3, e109. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, K.; Sang, X.; Mastalerz, H.A.; Johnson, W.L.; Zhang, G.; Liu, Q.; Batt, D.; Lombardo, L.J.; Vyas, D.; Trianor, G.L.; et al. 9H-Carbazole-1-carboxamides as potent and selective JAK2 inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 2809–2812. [Google Scholar] [CrossRef] [PubMed]
- Zorn, J.A.; Wang, Q.; Fujimura, E.; Barros, T.; Kuriyan, J. Crystal structure of the FLT3 kinase domain bound to the inhibitor quizartinib (AC220). PLoS ONE 2015, 10, e0121177. [Google Scholar] [CrossRef] [PubMed]
- Overman, R.C.; Debreczeni, J.E.; Truman, C.M.; Mcalister, M.S.; Attwood, T.K. Completing the structural family portrait of the human EphB tyrosine kinase domains. Protein Sci. 2014, 23, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Pemovska, T.; Johnson, E.; Kontro, M.; Repasky, G.A.; Chen, J.; Wells, P.; Cronin, C.N.; McTigue, M.; Kallioniemi, O.; Porkka, K.; Murray, B.W.; Wennerberg, K. Axitinib effectively inhibits BCR-ABL1 (T315I) with a distinct binding conformation. Nature 2015, 519, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Luke, R.W.; Ballard, P.; Buttar, D.; Campbell, L.; Curwen, J.; Emery, S.C.; Griffen, A.M.; Hassall, L.; Hayter, B.R.; Jones, C.D.; et al. Novel thienopyrimidine and thiazolopyrimidine with activity against Tie-2 in vitro and in vivo. Bioorg. Med. Chem. Lett. 2009, 19, 6670–6674. [Google Scholar] [CrossRef] [PubMed]
- Gentile, G.; Bernasconi, G.; Pozzan, A.; Merlo, G.; Marzorati, P.; Bamborough, P.; Bax, B.; Bridges, A.; Brough, C.; Carter, P.; et al. Identification of 2-(4-pyridyl) thienopyridinones as GSK-3β inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 4823–4827. [Google Scholar] [CrossRef] [PubMed]
- Duffey, M.O.; Vos, T.J.; Adams, R.; Alley, J.; Anthony, J.; Barrett, C.; Bharathan, I.; Bowman, D.; Bump, N.J.; Chau, R.; et al. Discovery of a potent and orally bioavailable benzolactam-derived inhibitor of Polo-like kinase 1 (MLN0905). J. Med. Chem. 2012, 55, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Protein Data Bank. Available online: http://www.rcsb.org/pdb/home/home.do (accessed on 5 August 2015).
- Olsson, M.H.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory. Comp. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed]
- Iversen, P.W.; Eastwood, B.J.; Sittampalam, G.S.; Cox, K.L. A Comparison of assay performance measures in screening assays: Signal window, Z’ factor, and assay variability ratio. J. Biomol. Screen. 2006, 11, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Eastwood, B.J.; Farmen, M.W.; Iversen, P.W.; Craft, T.J.; Smallwood, J.K.; Garbison, K.E.; Delapp, N.W.; Smith, G.F. The minimum significant ratio: A statistical parameter to characterize the reproducibility of potency estimates from concentration-response assays and estimation by replicate-experiment studies. J. Biomol. Screen. 2006, 11, 253–261. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebrahim, H.Y.; El Sayed, K.A. Discovery of Novel Antiangiogenic Marine Natural Product Scaffolds. Mar. Drugs 2016, 14, 57. https://doi.org/10.3390/md14030057
Ebrahim HY, El Sayed KA. Discovery of Novel Antiangiogenic Marine Natural Product Scaffolds. Marine Drugs. 2016; 14(3):57. https://doi.org/10.3390/md14030057
Chicago/Turabian StyleEbrahim, Hassan Y., and Khalid A. El Sayed. 2016. "Discovery of Novel Antiangiogenic Marine Natural Product Scaffolds" Marine Drugs 14, no. 3: 57. https://doi.org/10.3390/md14030057
APA StyleEbrahim, H. Y., & El Sayed, K. A. (2016). Discovery of Novel Antiangiogenic Marine Natural Product Scaffolds. Marine Drugs, 14(3), 57. https://doi.org/10.3390/md14030057