5-Alkylresorcinol Derivatives from the Bryozoan Schizomavella mamillata: Isolation, Synthesis, and Antioxidant Activity


Abstract
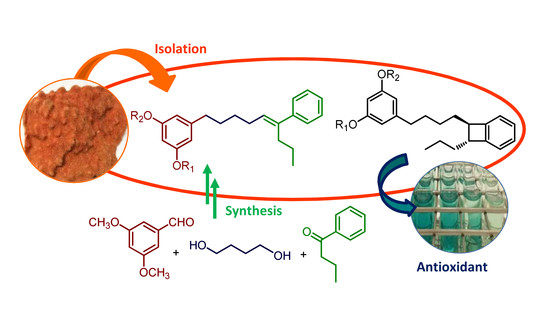
:
1. Introduction
2. Results and Discussion
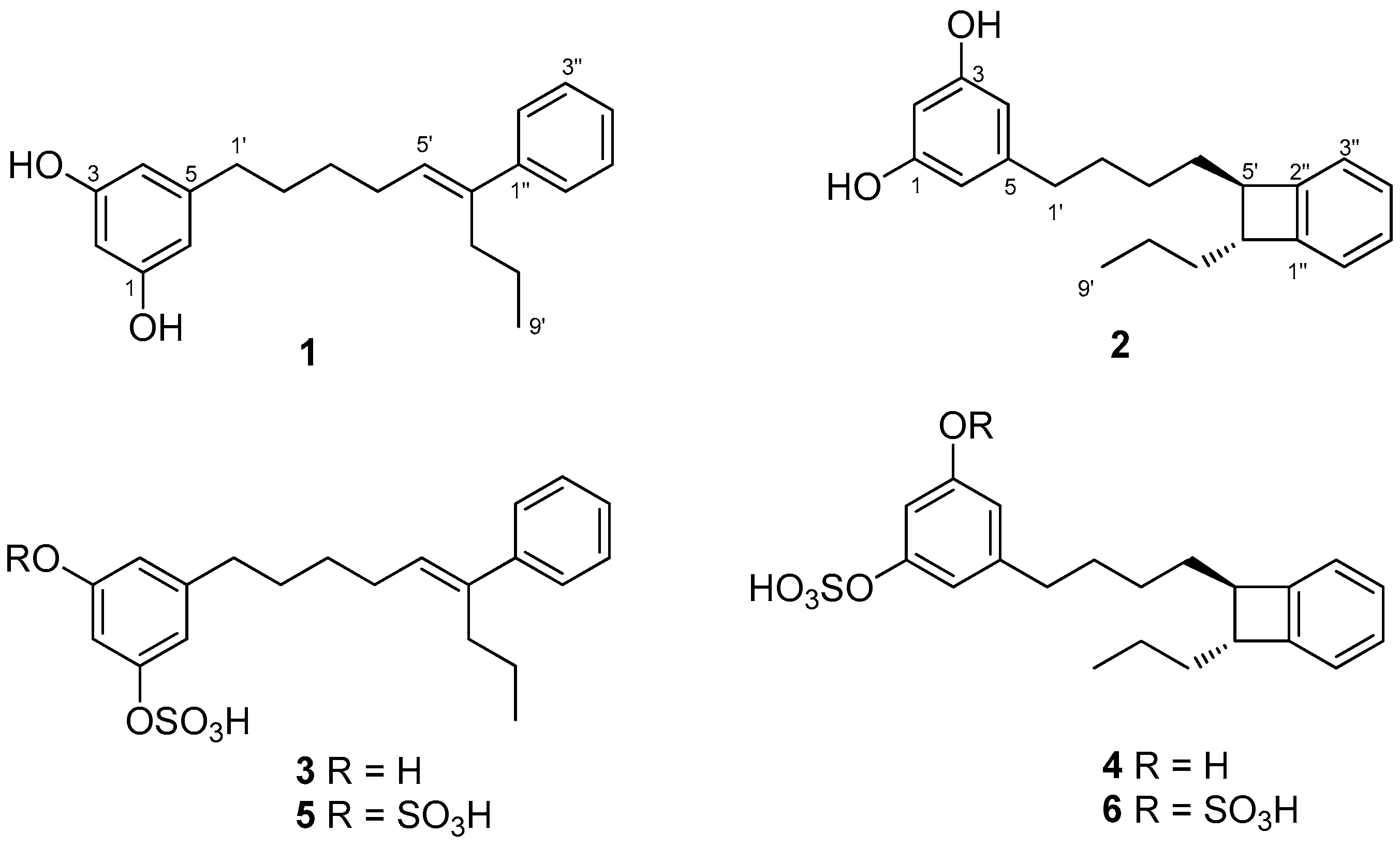
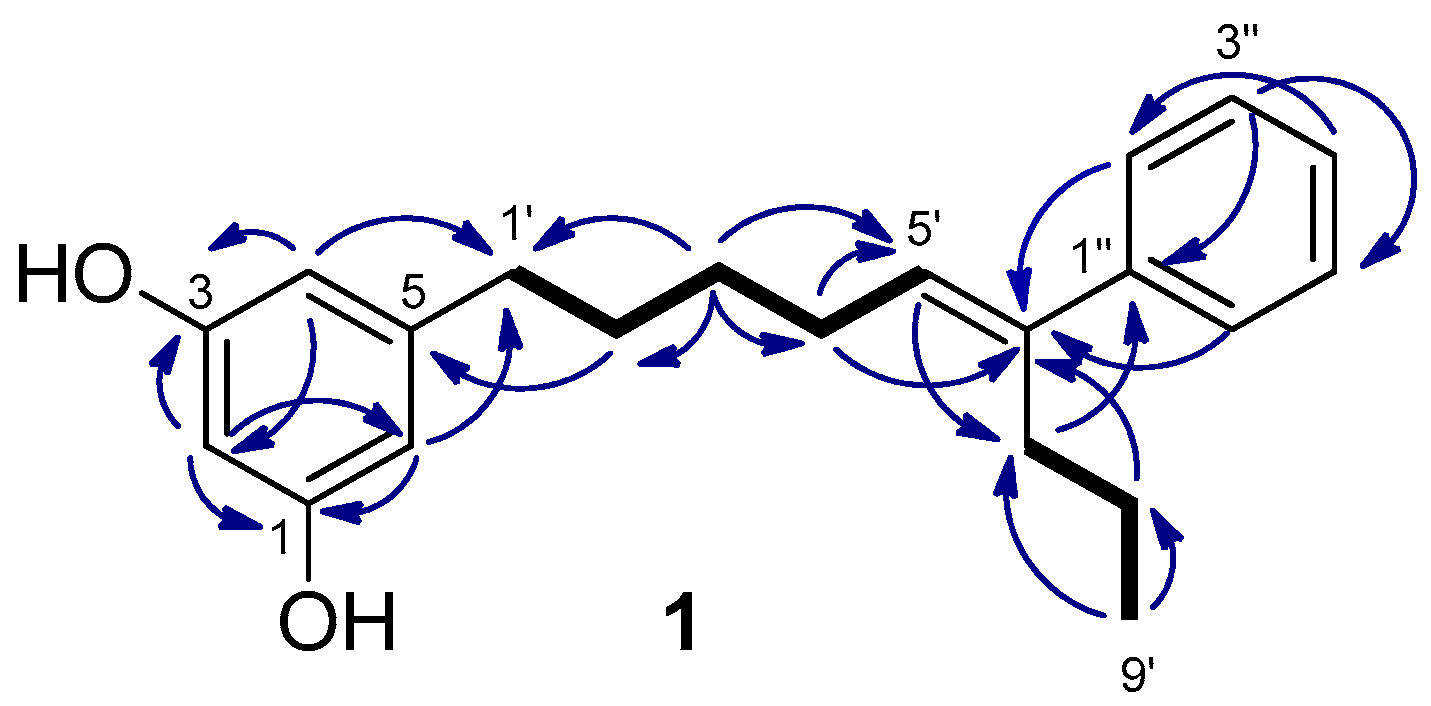
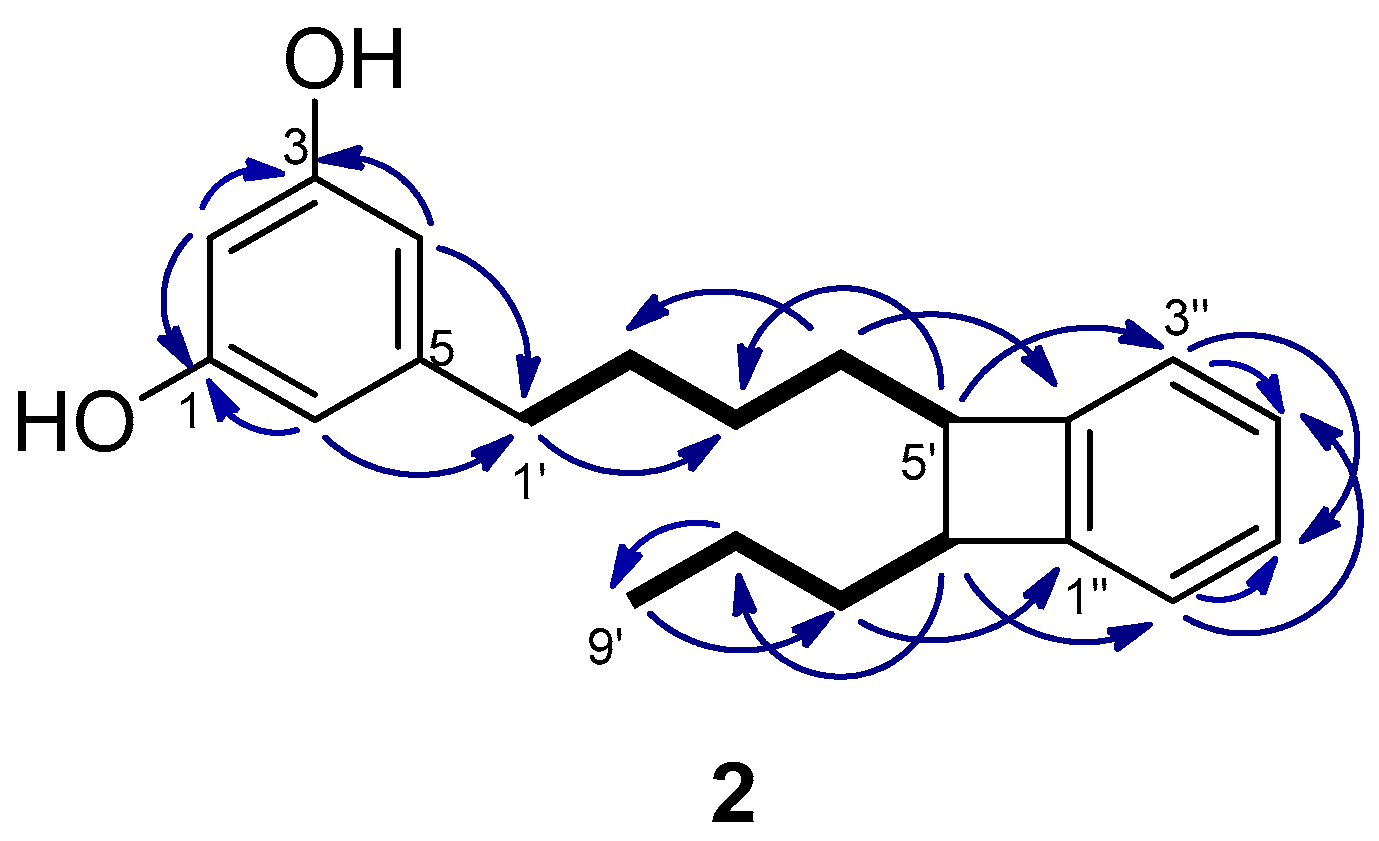
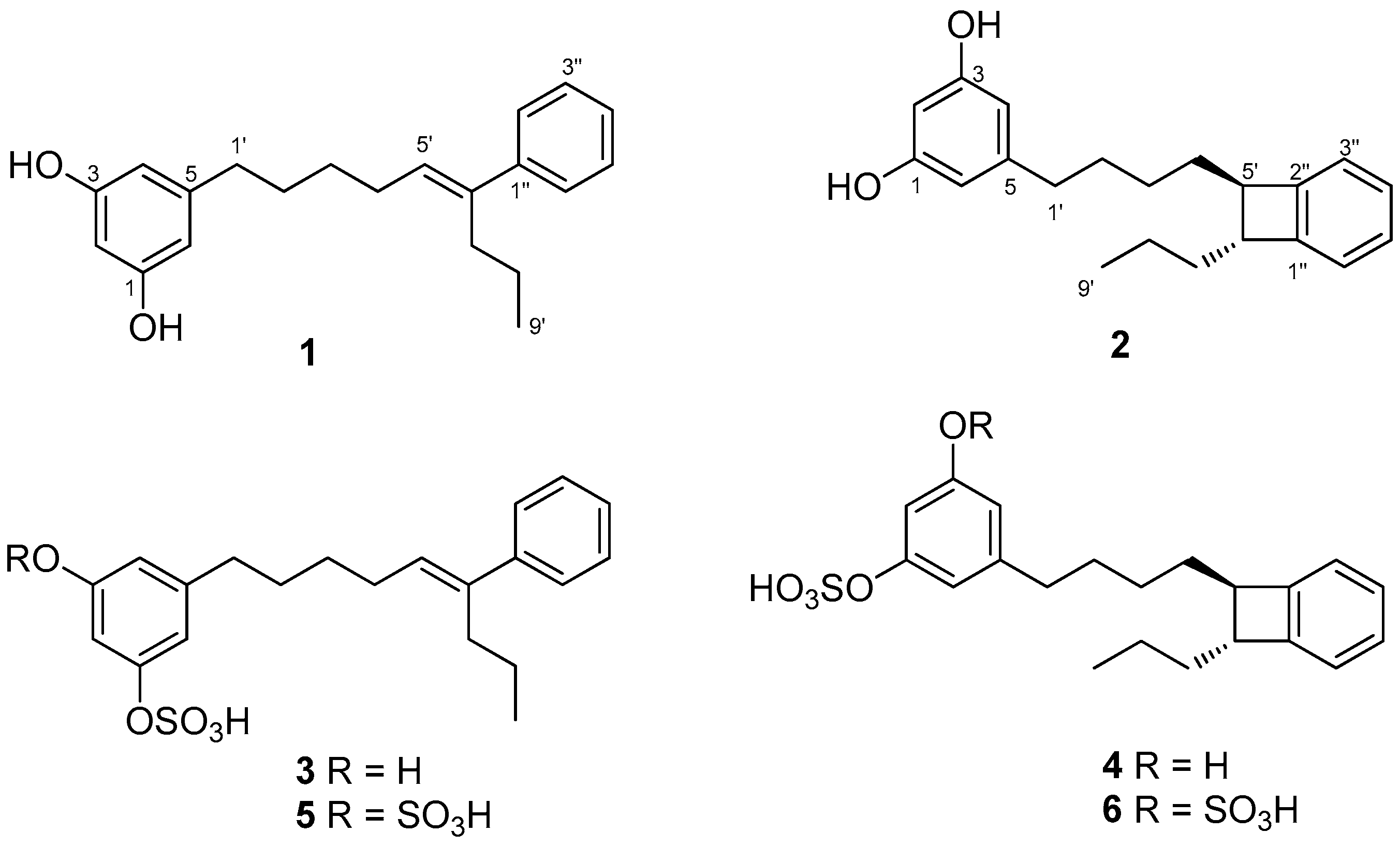
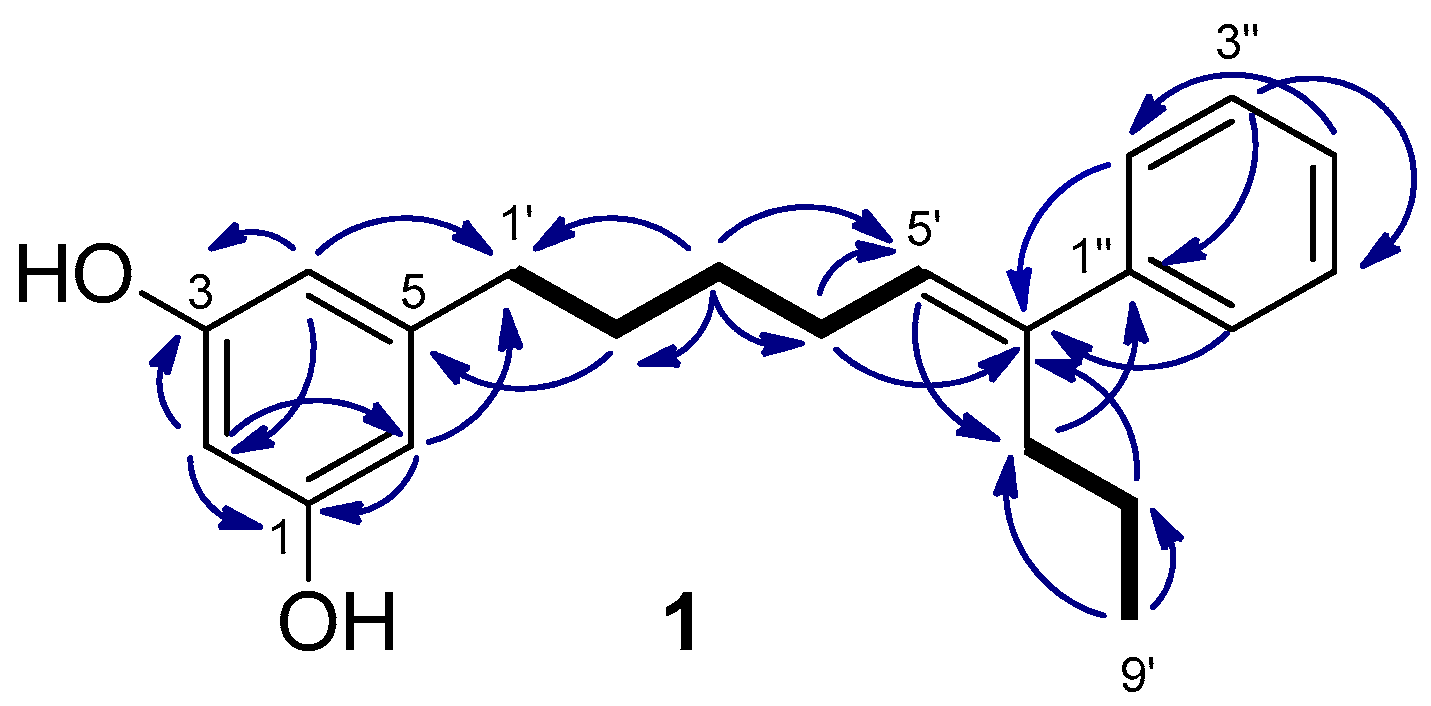
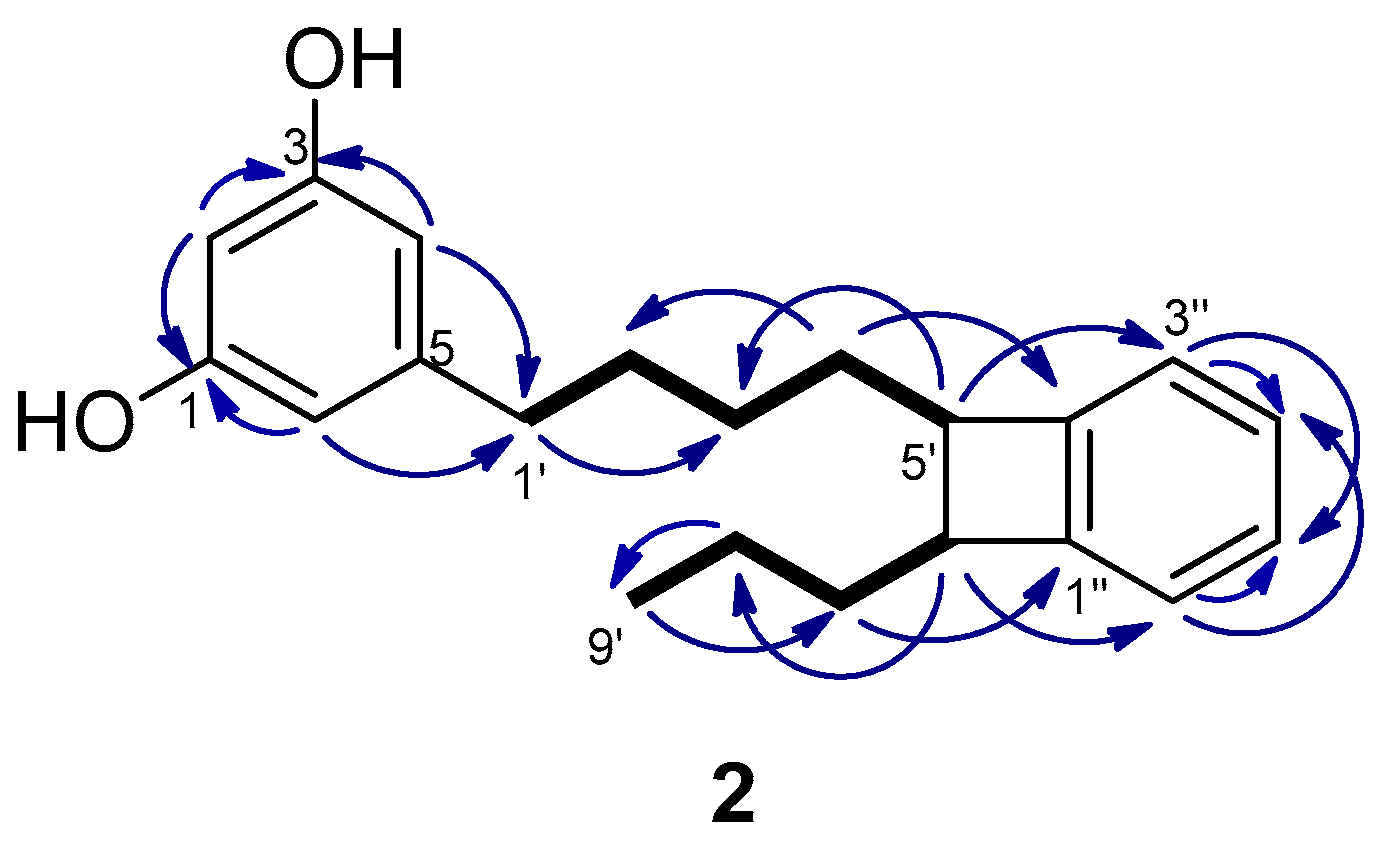
2.1. Isolation and Structure Determination
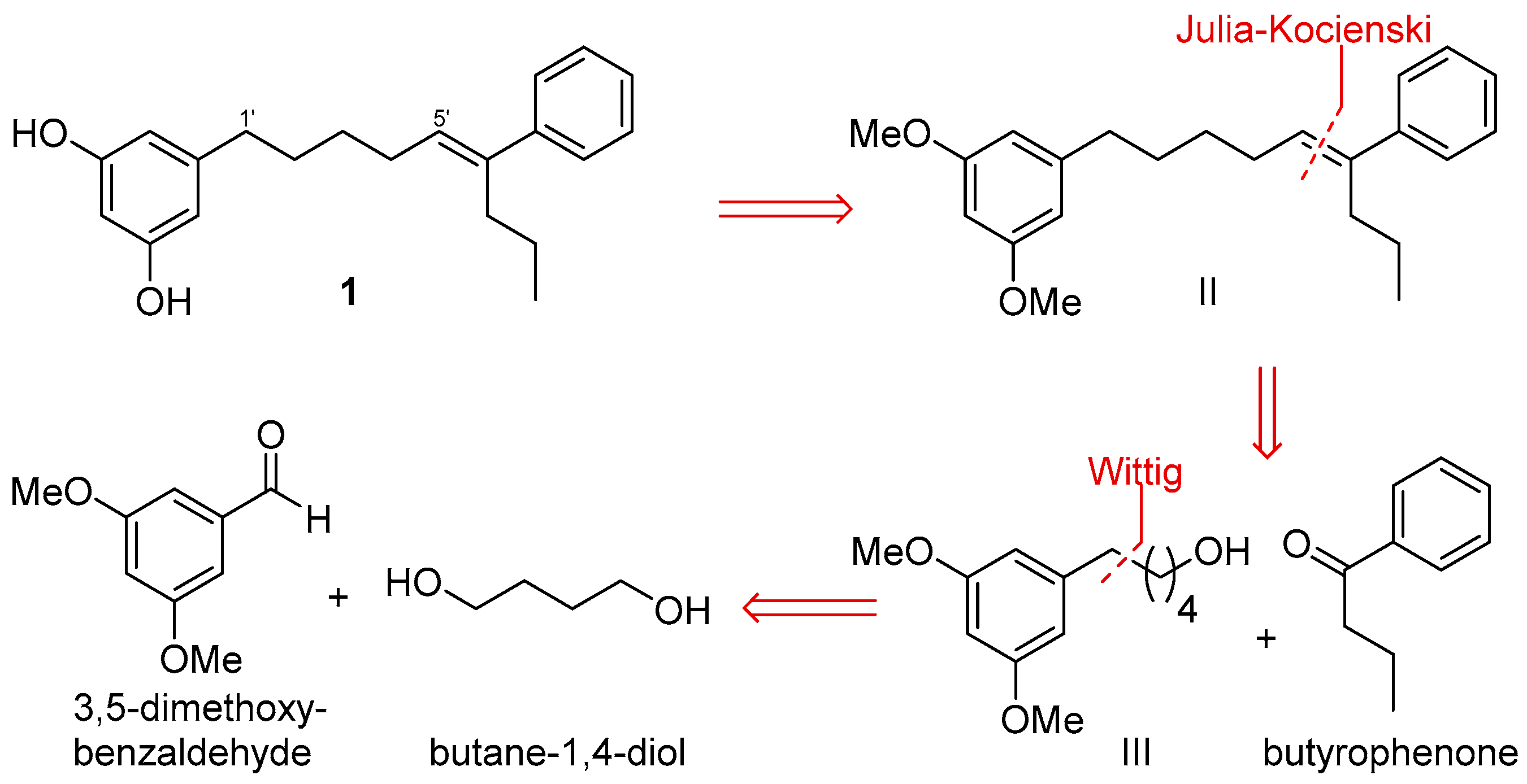
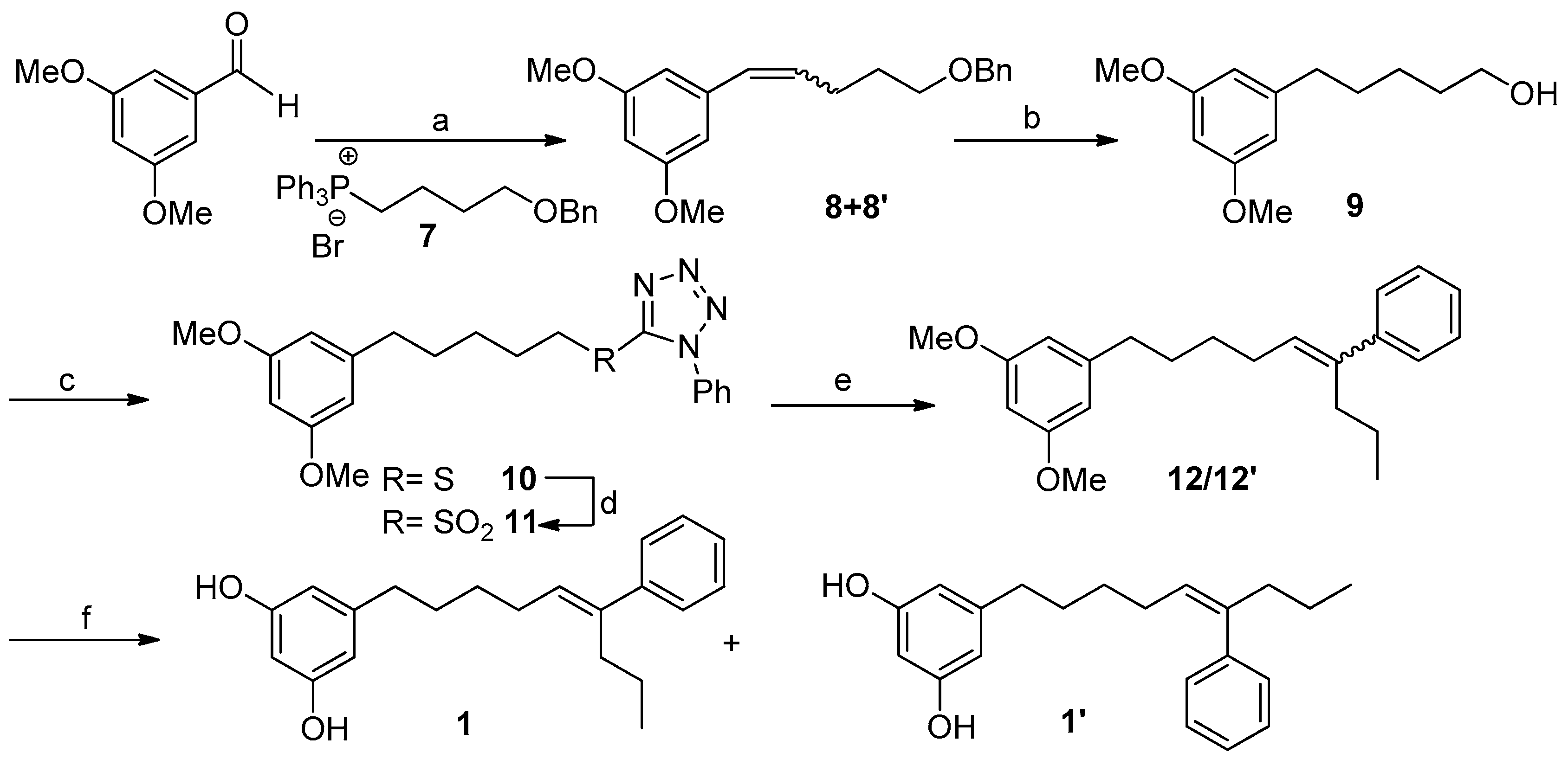
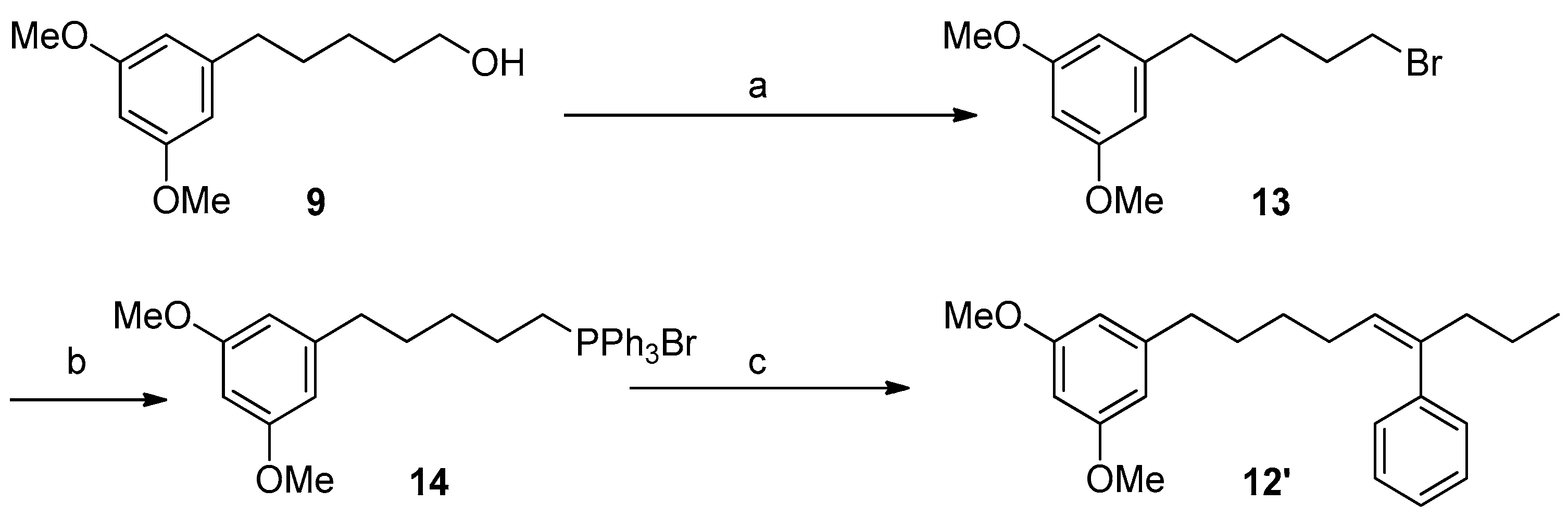
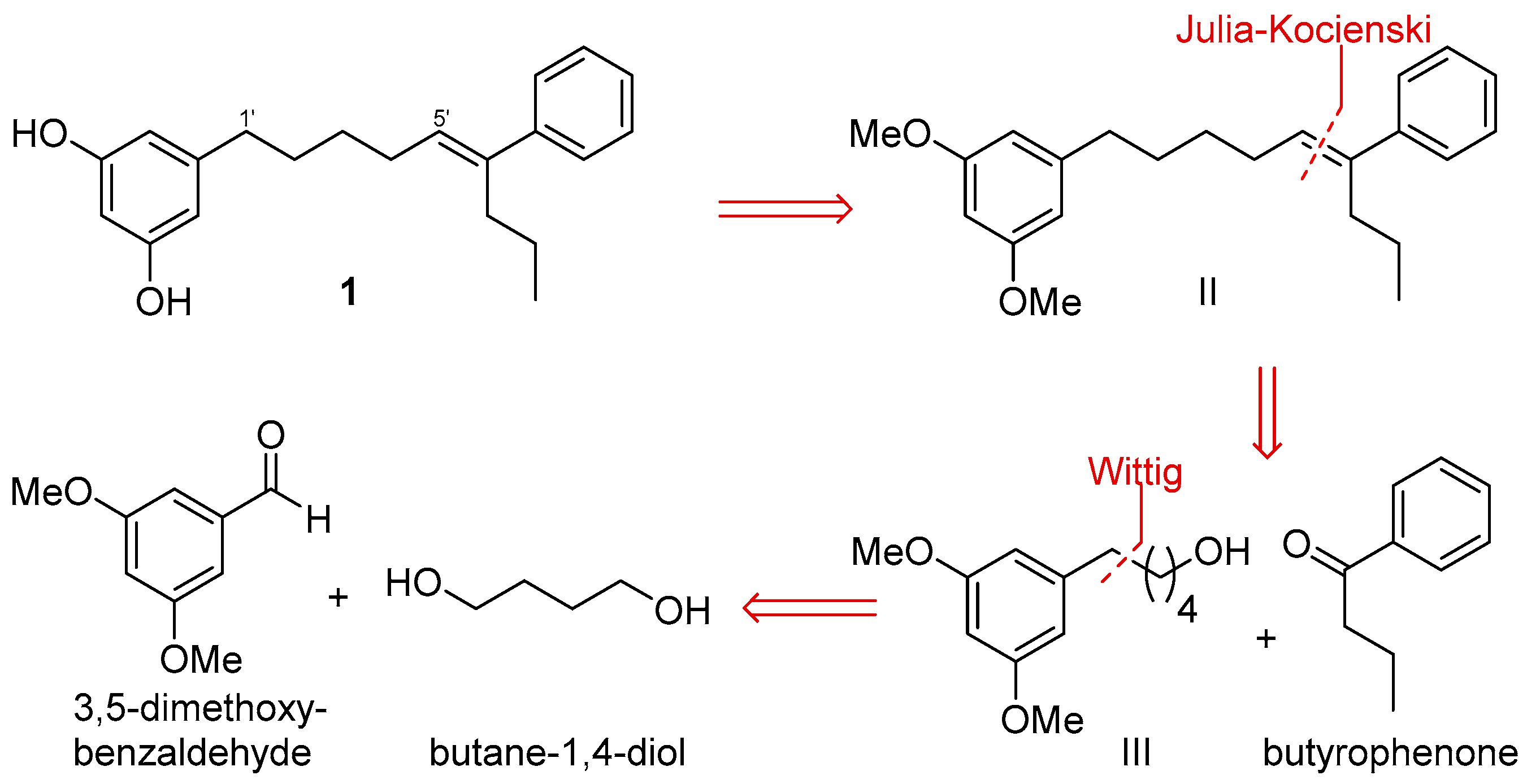
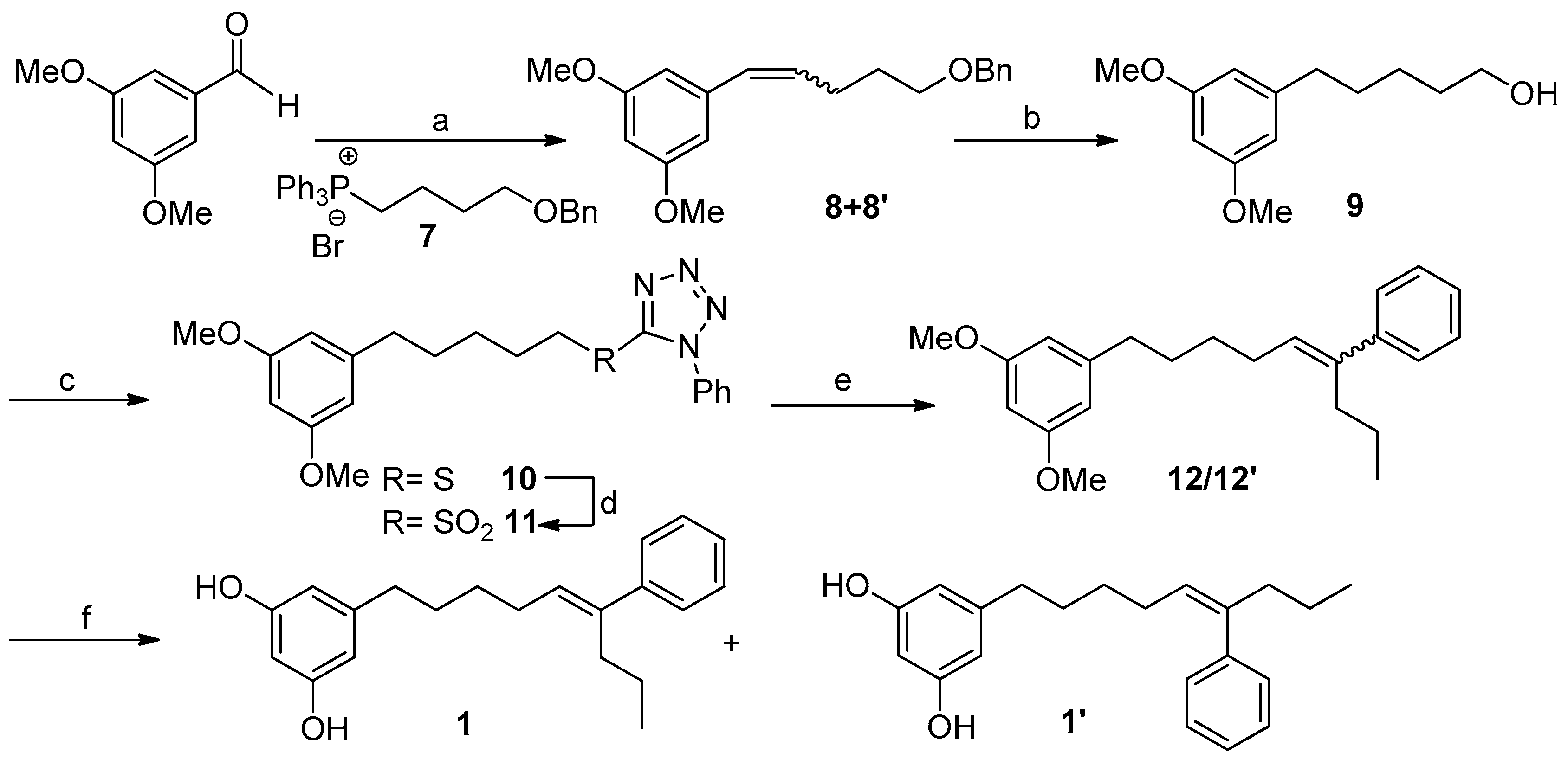
2.2. Synthesis of Schizol A (1)
2.3. Antioxidant Activity
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Biological Material
3.3. Extraction and Isolation
3.4. Hydrolysis of Schizol F (6)
3.5. Synthesis of Schizol A (1)
3.5.1. Synthesis of Compounds 8/8′
3.5.2. Synthesis of Compound 9
3.5.3. Synthesis of Compound 10
3.5.4. Synthesis of Compound 11
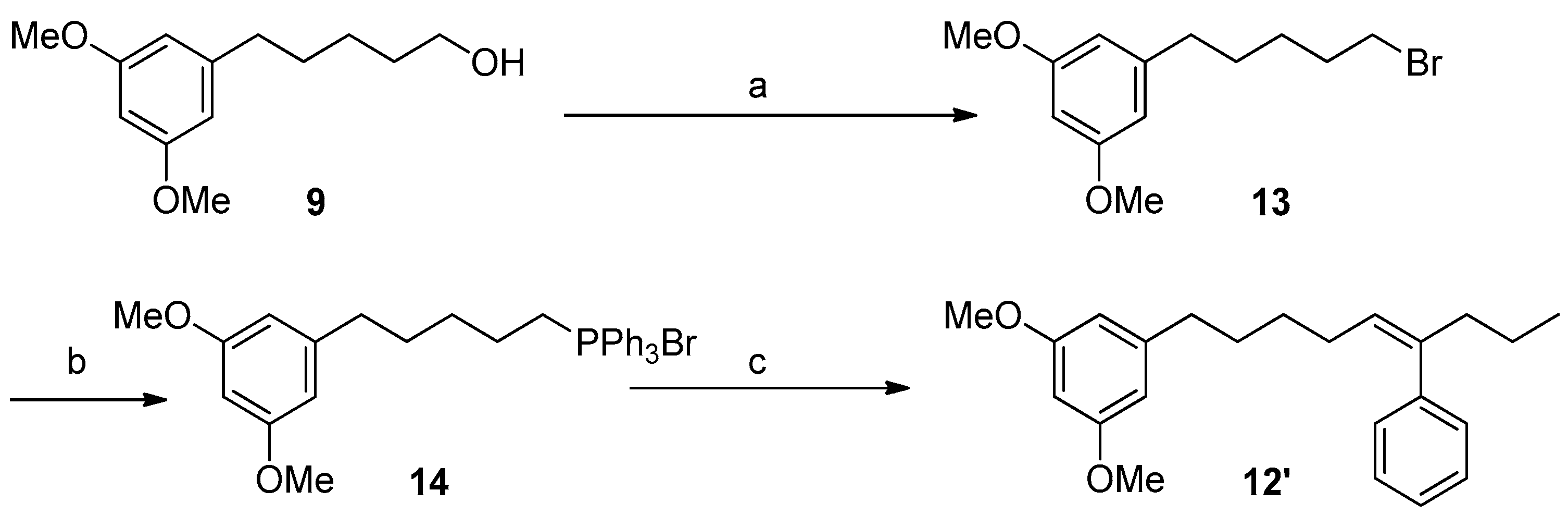
3.5.5. Synthesis of Compounds 12/12′ (Julia–Kocienski Reaction)
3.5.6. Synthesis of Compounds 1/1′
3.5.7. Synthesis of Compound 13
3.5.8. Synthesis of Compound 14
3.5.9. Synthesis of Compound 12′ (Wittig Reaction)
3.6. Antioxidant Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Christophersen, C. Secondary metabolites from marine bryozoans. A review. Acta Chem. Scand. B 1985, 39, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2017, 34, 235–294, and previous reviews of this series. [Google Scholar] [CrossRef] [PubMed]
- Sharp, J.H.; Winson, M.K.; Porter, J.S. Bryozoan metabolites: An ecological perspective. Nat. Prod. Rep. 2007, 24, 659–673. [Google Scholar] [CrossRef] [PubMed]
- Rochfort, S.J.; Moore, S.; Craft, C.; Martin, N.H.; Van Wagoner, R.M.; Wright, J.L.C. Further studies on the chemistry of the flustra alkaloids from the bryozoan Flustra foliacea. J. Nat. Prod. 2009, 72, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.A.; Sykes, M.; Avery, V.M.; Camp, D.; Quinn, R.J. Convolutamines I and J, antitrypanosomal alkaloids from the bryozoan Amathia tortusa. Bioorg. Med. Chem. 2011, 19, 6615–6619. [Google Scholar] [CrossRef] [PubMed]
- Carroll, A.R.; Duffy, S.; Sykes, M.; Avery, V.M. Wilsoniamines A and B: Novel alkaloids from the temperate Australian bryozoan, Amathia wilsoni. Org. Biomol. Chem. 2011, 9, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Carroll, A.R.; Wild, S.J.; Duffy, S.; Avery, V.M. Kororamide A, a new tribrominated indole alkaloid from the Australian bryozoan Amathia tortuosa. Tetrahedron Lett. 2012, 53, 2873–2875. [Google Scholar] [CrossRef]
- Dashti, Y.; Vial, M.L.; Wood, S.A.; Mellick, G.D.; Roullier, C.; Quinn, R.J. Kororamide B, a brominated alkaloid from Amathia tortuosa and its effects on Parkinson’s disease cells. Tetrahedron 2015, 71, 7879–7884. [Google Scholar] [CrossRef]
- Newman, D.J. The bryostatins. In Anticancer Agents from Natural Products; Cragg, G.M., Kingston, D.G.I., Newman, D.J., Eds.; Taylor & Francis: Boca Raton, FL, USA, 2005; pp. 137–150. [Google Scholar]
- Nelson, T.J.; Sun, M.-K.; Lim, C.; Sen, A.; Khan, T.; Chirila, F.V.; Alkon, D.L. Bryostatin effects on congnitive function and PKCε in Alzheimer’s disease phase IIa and expanded access trials. J. Alzheimers Dis. 2017, 58, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Herald, C.L.; Doubek, D.L.; Herald, D.L.; Arnold, E.; Clardy, J. Isolation and structure of bryostatin 1. J. Am. Chem. Soc. 1982, 104, 6846–6848. [Google Scholar] [CrossRef]
- Sudek, S.; Lopanik, N.B.; Waggoner, L.E.; Hildebrand, M.; Anderson, C.; Liu, H.; Patel, A.; Sherman, D.H.; Haygood, M.G. Identification of the putative bryostatin polyketide synthase gene cluster from “Candidatus Endobugula sertula”, the uncultivated microbial symbiont of the marine bryozoan Bugula neritina. J. Nat. Prod. 2007, 70, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Eisenbarth, S.; Gehling, M.; Harder, A.; Steffan, B. Pentaporins A, B and C: Disulfides from the marine bryozoan Pentapora fascialis. Tetrahedron 2002, 58, 8461–8464. [Google Scholar] [CrossRef]
- Hashmi, A.S.K.; Wieteck, M.; Braun, I.; Rudolph, M.; Rominger, F. Gold vinylidene complexes: Intermolecular C(sp3)–H insertions and cyclopropanations pathways. Angew. Chem. Int. Ed. 2012, 51, 10633–10637. [Google Scholar] [CrossRef] [PubMed]
- Barron, D.; Ibrahim, R.K. Synthesis of flavonoid sulfates: I. Stepwise sulfation of positions 3, 7, and 4′ using N,N′-dicyclohexylcarbodiimide and tetrabutilammonium hydrogen sulfate. Tetrahedron 1987, 43, 5197–5202. [Google Scholar] [CrossRef]
- Kozubeck, A.; Tyman, J.H.P. Bioactive phenolic lipids. In Studies in Natural Products Chemistry; ur Rahman, A., Ed.; Elsevier B.V.: Amsterdam, The Netherlands, 2005; Volume 30, pp. 111–190. [Google Scholar]
- González, M.J.T.G.; DeOliveira, C.J.C.; Fernandes, J.O.; Kijoa, A.; Herz, W. Further alkyl and alkenylphenols of Knema laurina and Knema austrosiamensis: Location of the double bond in the alkenyl chains. Phytochemistry 1996, 43, 1333–1337. [Google Scholar] [CrossRef]
- Du, Y.; Oshima, R.; Yamauchi, Y.; Kumanotani, J.; Miyakoshi, T. Long chain phenols from the burmese lac tree, Melanorrhoea usitate. Phytochemistry 1986, 25, 2211–2218. [Google Scholar] [CrossRef]
- Kikuchi, H.; Ito, I.; Takahashi, K.; Ishigaki, H.; Iizumi, K.; Kubohara, Y.; Oshima, Y. Isolation, synthesis, and biological activity of chlorinated alkylresorcinols from Dictyostelium cellular slime molds. J. Nat. Prod. 2017, 80, 2716–2722. [Google Scholar] [CrossRef] [PubMed]
- Kouno, I.; Komori, T.; Kawasaki, T. Zur struktur der neuen typen homo-isoflavanone aus bulben von Scilla scilloides Druce. Tetrahedron Lett. 1973, 14, 4569–4572. [Google Scholar] [CrossRef]
- Waller, C.P.; Thumser, A.E.; Langat, M.K.; Crouch, N.R.; Mulholland, D.A. COX-2 inhibitory activity of homoisoflavanones and xanthones from the bulbs of the Southern African Ledebouria socialis and Ledebouria ovatifolia (Hyacinthaceae: Hyacinthoideae). Phytochemistry 2013, 95, 284–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutanyatta, J.; Matapa, B.G.; Shushu, D.D.; Abegaz, B.M. Homoisoflavonoids and xanthones from the tubers of wild and in vitro regenerated Ledebouria graminifolia and cytotoxic activities of some of the homoisoflavonoids. Phytochemistry 2003, 62, 797–804. [Google Scholar] [CrossRef]
- Corsaro, M.M.; Lanzetta, R.; Manzino, A.; Parrilli, M. Homoisoflavanones from Chionodoxa luciliae. Phytochemistry 1992, 31, 1395–1397. [Google Scholar] [CrossRef]
- Barrow, R.A.; Capon, R.J. Alkyl and alkenyl resorcinols from an Australian marine sponge, Haliclona sp. (Haplosclerida: Haliclonidae). Aust. J. Chem. 1991, 44, 1393–1405. [Google Scholar] [CrossRef]
- Anthoni, U.; Nielsen, P.H.; Pereira, M.; Christophersen, C. Bryozoan secondary metabolites: A chemotaxonomical challenge. Comp. Biochem. Physiol. 1990, 96B, 431–437. [Google Scholar] [CrossRef]
- Franks, A.; Haywood, P.; Holmström, C.; Egan, S.; Kjelleberg, S.; Kumar, N. Isolation and structure elucidation of a novel yellow pigment from the marine bacterium Pseudoalteromonas tunicata. Molecules 2005, 10, 1286–1291. [Google Scholar] [CrossRef] [PubMed]
- Afonso, T.B.; Costa, M.S.; Rezende de Castro, R.; Freitas, S.; Silva, A.; Schneider, M.P.C.; Martins, R.; Leao, P.N. Bartolosides E-K from a marine coccoid cyanobacterium. J. Nat. Prod. 2016, 79, 2504–2513. [Google Scholar] [CrossRef] [PubMed]
- Leao, P.N.; Nakamura, H.; Costa, M.; Pereira, A.R.; Martins, R.; Vasconcelos, V.; Gerwick, W.H.; Balskus, E.P. Biosynthesis-assisted structural elucidation of the bartolosides, chlorinated aromatic glycolipids from cyaanobacteria. Angew. Chem. Int. Ed. 2015, 54, 11063–11067, Corrigendum in Angew. Chem. Int. Ed. 2016, 55, 14895. [Google Scholar] [CrossRef] [PubMed]
- Fürstner, A.; Seidel, G. Shortcut synthesis of naturally occurring 5-alkylresorcinols with DNA-cleaving properties. J. Org. Chem. 1997, 62, 2332–2336. [Google Scholar] [CrossRef] [PubMed]
- Vyvyan, J.R.; Holst, C.L.; Johnson, A.L.; Schwenk, C.M. Total synthesis of gibbilimbols A-D. J. Org. Chem. 2002, 67, 2263–2265. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Soroka, D.N.; Sang, S. Synthesis and inhibitory activities against colon cancer cell growth and proteasome of alkylresorcinols. J. Agric. Food Chem. 2012, 60, 8624–8631. [Google Scholar] [CrossRef] [PubMed]
- Parikka, K.; Wahala, K. An expedient synthesis of 5-n-alkylresorcinols and novel 5-n-alkylresorcinols haptens. Beilstein J. Org. Chem. 2009, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Byrne, P.A.; Gilheany, D.G. The modern interpretation of the Wittig reaction mechanism. Chem. Soc. Rev. 2013, 42, 6670–6696. [Google Scholar] [CrossRef] [PubMed]
- Blakemore, P.R.; Cole, W.J.; Kocienski, P.J.; Morley, A. A stereoselective synthesis of trans-1,2-disubstituted alkenes based on the condensation of aldehydes with metallated 1-phenyl-1H-tetrazol-5-yl sulfones. Synlett 1998, 26–28. [Google Scholar] [CrossRef]
- Zhou, J.; Gao, B.; Xu, Z.; Ye, T. Total synthesis and stereochemical assignment of callyspongiolide. J. Am. Chem. Soc. 2016, 138, 6948–6951. [Google Scholar] [CrossRef] [PubMed]
- Mitsunobu, O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis 1981. [Google Scholar] [CrossRef]
- Paquette, L.A.; Chang, S.-K. The polyol domain of amphidinol 3. A stereoselective synthesis of the entire C(1)-C(30) sector. Org. Lett. 2005, 7, 3111–3114. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Zajc, B. Stereoselective synthesis of conjugated fluoro enynes. J. Org. Chem. 2012, 77, 8417–8427. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Singh, G.; Todaro, L.J.; Yang, L.; Zajc, B. E- or Z-selective synthesis of 4-fluorovinyl-1,2,3-triazoles with fluorinated second-generation Julia-Kocienski reagents. Org. Biomol. Chem. 2015, 13, 1536–1549. [Google Scholar] [CrossRef] [PubMed]
- Salih, M.Q.; Beaudry, C.M. Enantioselective Ullmann Ether couplings: syntheses of (-)-myricatomentogenin, (-)-jugcathanin, (+)-galeon, and (+)-pterocarine. Org. Lett. 2013, 15, 4540–4543. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.P.; Aithagani, S.K.; Yadav, M.; Singh, V.P.; Vishwakarma, R.A. Iron-catalyzed cross-coupling of electron-deficient heterocycles and quinone with organoboron species via innate C-H functionalization: Application in total synthesis of pyrazine alkaloid botryllazine A. J. Org. Chem. 2013, 78, 2639–2648. [Google Scholar] [CrossRef] [PubMed]
- Maurent, K.; Vanucci-Bacqué, C.; Saffon-Merceron, N.; Baltas, M. Bedos-Belval, F. Total synthesis of tedarene A. J. Nat. Prod. 2017, 80, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Struski, D.G.J.; Kozubek, A. Cereal grain alk(en)ylresorcinols protect lipids against ferrous ions-induced peroxidation. Z. Naturforsch. 1992, 47C, 47–50. [Google Scholar]
- Kozubek, A.; Nienartowicz, B. Cereal grain resorcinolic lipids inhibit H2O2-induced peroxidation of biological membranes. Acta Biochim. Polon. 1995, 42, 309–316. [Google Scholar] [PubMed]
- Parikka, K.; Rowland, I.R.; Welch, R.W.; Wähälä, K. In vitro antioxidant activity and antigenotoxicity of 5-n-alkylresorcinols. J. Agric. Food Chem. 2006, 54, 1646–1650. [Google Scholar] [CrossRef] [PubMed]
- Korycinska, M.; Czelna, K.; Jaromin, A.; Kozubek, A. Antioxidant activity of rye bran alkylresorcinols and extracts from whole-grain cereal products. Food Chem. 2009, 116, 1013–1018. [Google Scholar] [CrossRef]
- López, P.; Ferraro, G.; Anesini, C. Comparative antioxidant activity of an extract of Lithraea molleoides and an isolated 5-alkylresorcinol derivative. Effects on the proliferation of normal and tumoral lymphocytes. Phytother. Res. 2011, 25, 271–276. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δC, Type | δH, m (J in Hz) | δC, Type | δH, m (J in Hz) | |
| 1 | 159.3, C | 159.3, C | ||
| 2 | 101.0, CH | 6.07, t (2.2) | 101.0, CH | 6.08, t (2.2) |
| 3 | 159.3, C | 159.3, C | ||
| 4 | 107.9, CH | 6.13, d (2.1) | 108.0, CH | 6.14, d (2.2) |
| 5 | 146.2, C | 146.2, C | ||
| 6 | 107.9, CH | 6.13, d (2.1) | 108.0, CH | 6.14, d (2.2) |
| 1′ | 36.9, CH2 | 2.48, t (7.6) | 36.9, CH2 | 2.45, t (7.5) |
| 2′ | 32.1, CH2 | 1.64, m | 32.5, CH2 | 1.64, m |
| 3′ | 30.6, CH2 | 1.49, tt (7.7, 7.4) | 29.1, CH2 | 1.52, m |
| 4′ | 29.4, CH2 | 2.21, dt (7.4, 7.4) | 35.1, CH2 | 1.69, m |
| 5′ | 129.9, CH | 5.61, t (7.3) | 51.43, CH | 2.94, td (7.4, 2.1) |
| 6′ | 141.6, C | 51.37, CH | 2.95, td (7.4, 2.1) | |
| 7′ | 32.6, CH2 | 2.48, t (7.5) | 37.6, CH2 | 1.64, m |
| 8′ | 22.8, CH2 | 1.32, tq (7.5, 7.5) | 22.8, CH2 | 1.48, m |
| 9′ | 14.2, CH3 | 0.86, t (7.4) | 14.6, CH3 | 0.97, t (7.4) |
| 1′′ | 144.8, C | 148.99 c, C | ||
| 2′′ | 127.4, CH | 7.29, dd (8.2, 1.4) | 149.05 c, C | |
| 3′′ | 129.2, CH | 7.25, dd (8.0, 7.4) | 123.13 d, CH | 6.99, m |
| 4′′ | 127.5, CH | 7.17, tt (7.2, 1.5) | 127.9, CH | 7.11, m |
| 5′′ | 129.2, CH | 7.25, dd (8.0, 7.4) | 127.9, CH | 7.11, m |
| 6′′ | 127.4, CH | 7.29, dd (8.2, 1.4) | 123.07 d, CH | 7.02, m |
| Position | 3 | 4 | ||
|---|---|---|---|---|
| δC, Type | δH, m (J in Hz) | δC, Type | δH, m (J in Hz) | |
| 1 | 154.7, C | 154.6, C | ||
| 2 | 107.1, CH | 6.60, dd (2.2, 2.2) | 107.1, CH | 6.60, dd (2.4, 2.0) |
| 3 | 159.0, C | 159.0, C | ||
| 4 | 112.9, CH2 | 6.45, br dd (1.8, 1.5) | 113.0, CH2 | 6.45, br dd (1.6, 1.6) |
| 5 | 145.8, C | 145.9, C | ||
| 6 | 113.6, CH | 6.64, br dd (1.8, 1.5) | 113.6, CH | 6.63, br dd (1.6, 1.6) |
| 1′ | 36.8, CH2 | 2.55, t (7.7) | 36.9, CH2 | 2.54, (t, 7.5) |
| 2′ | 32.1, CH2 | 1.66, m | 32.5, CH2 | 1.66, m |
| 3′ | 30.6, CH2 | 1.48, tt (7.7, 7.3) | 29.2, CH2 | 1.52, m |
| 4′ | 29.4, CH2 | 2.22, dt (7.3, 7.3) | 35.1, CH2 | 1.69, m |
| 5′ | 129.9, CH | 5.61, t (7.3) | 51.44, CH | 2.94, td (7.7, 2.0) |
| 6′ | 141.6, C | 51.37, CH | 2.95, td (7.7, 2.0) | |
| 7′ | 32.6, CH2 | 2.49, t (7.5) | 37.6, CH2 | 1.64, m |
| 8′ | 22.8, CH2 | 1.33, tq (7.7, 7.3) | 22.8, CH2 | 1.49, m |
| 9′ | 14.2, CH3 | 0.86, t (7.3) | 14.6, CH3 | 0.97, t (7.5) |
| 1′′ | 144.7, C | 149.01 c, C | ||
| 2′′ | 127.4, CH | 7.29, dd (8.1, 1.3) | 149.06 c, C | |
| 3′′ | 129.2, CH | 7.26, dd (8.1, 7.3) | 123.18 d, CH | 7.01, m |
| 4′′ | 127.4, CH | 7.16, tt (7.3, 1.3) | 127.95 e, CH | 7.10, m |
| 5′′ | 129.2, CH | 7.26, dd (8.1, 7.3) | 127.92 e, CH | 7.10, m |
| 6′′ | 127.4, CH | 7.29, dd (8.1, 1.3) | 123.07 d, CH | 7.01, m |
| Position | 5 | 6 | ||
|---|---|---|---|---|
| δC, Type | δH, m (J in Hz) | δC, Type | δH, m (J in Hz) | |
| 1 | 154.2, C | 154.0, C | ||
| 2 | 113.3, CH | 7.04, m | 113.5, CH | 7.11, m |
| 3 | 154.2, C | 154.0, C | ||
| 4 | 118.8, CH | 7.04, m | 119.1, CH2 | 7.01, br s |
| 5 | 145.6, C | 145.8, C | ||
| 6 | 118.8, CH | 7.04, m | 119.1, CH | 7.01, br s |
| 1′ | 36.8, CH2 | 2.64, t (7.7) | 36.8, CH2 | 2.64, t (7.7) |
| 2′ | 32.1, CH2 | 1.70, m | 32.5, CH2 | 1.69, m |
| 3′ | 30.7, CH2 | 1.51, m | 29.3, CH2 | 1.54, m |
| 4′ | 29.4, CH2 | 2.24, dt (7.5, 7.3) | 35.1, CH2 | 1.71, m |
| 5′ | 129.9, CH | 5.62, t (7.3) | 51.44, CH | 2.95, br t (7.3) |
| 6′ | 141.6, C | 51.37, CH | 2.97, br t (7.3) | |
| 7′ | 32.6, CH2 | 2.50, t (7.7) | 37.6, CH2 | 1.65, m |
| 8′ | 22.8, CH2 | 1.32, m | 22.8, CH2 | 1.50, m |
| 9′ | 14.2, CH3 | 0.86, t (7.4) | 14.6, CH3 | 0.98, t (7.4) |
| 1′′ | 144.8, C | 149.0 c, C | ||
| 2′′ | 127.5, CH | 7.30, dd (8.1, 1.2) | 149.1 c, C | |
| 3′′ | 129.1, CH | 7.25, dd (8.1, 7.5) | 123.2 d, CH | 7.02, m |
| 4′′ | 127.4, CH | 7.16, m | 128.0 e, CH | 7.11, m |
| 5′′ | 129.1, CH | 7.25, dd (8.1, 7.5) | 127.9 e, CH | 7.11, m |
| 6′′ | 127.5, CH | 7.30, dd (8.1, 1.2) | 123.1 d, CH | 7.02, m |
| Compound | Trolox | 1 | 2 | 3 | 4 |
|---|---|---|---|---|---|
| EC50 (μM ± SD, n = 3) | 10.43 ± 0.06 | 6.24 ± 0.03 | 7.66 ± 0.24 | 30.25 ± 0.10 | 22.59 ± 0.53 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortega, M.J.; Pantoja, J.J.; De los Reyes, C.; Zubía, E. 5-Alkylresorcinol Derivatives from the Bryozoan Schizomavella mamillata: Isolation, Synthesis, and Antioxidant Activity. Mar. Drugs 2017, 15, 344. https://doi.org/10.3390/md15110344
Ortega MJ, Pantoja JJ, De los Reyes C, Zubía E. 5-Alkylresorcinol Derivatives from the Bryozoan Schizomavella mamillata: Isolation, Synthesis, and Antioxidant Activity. Marine Drugs. 2017; 15(11):344. https://doi.org/10.3390/md15110344
Chicago/Turabian StyleOrtega, María J., Juan J. Pantoja, Carolina De los Reyes, and Eva Zubía. 2017. "5-Alkylresorcinol Derivatives from the Bryozoan Schizomavella mamillata: Isolation, Synthesis, and Antioxidant Activity" Marine Drugs 15, no. 11: 344. https://doi.org/10.3390/md15110344
APA StyleOrtega, M. J., Pantoja, J. J., De los Reyes, C., & Zubía, E. (2017). 5-Alkylresorcinol Derivatives from the Bryozoan Schizomavella mamillata: Isolation, Synthesis, and Antioxidant Activity. Marine Drugs, 15(11), 344. https://doi.org/10.3390/md15110344