
Laucysteinamide A, a Hybrid PKS/NRPS Metabolite from a Saipan Cyanobacterium, cf. Caldora penicillata



Abstract
:1. Introduction
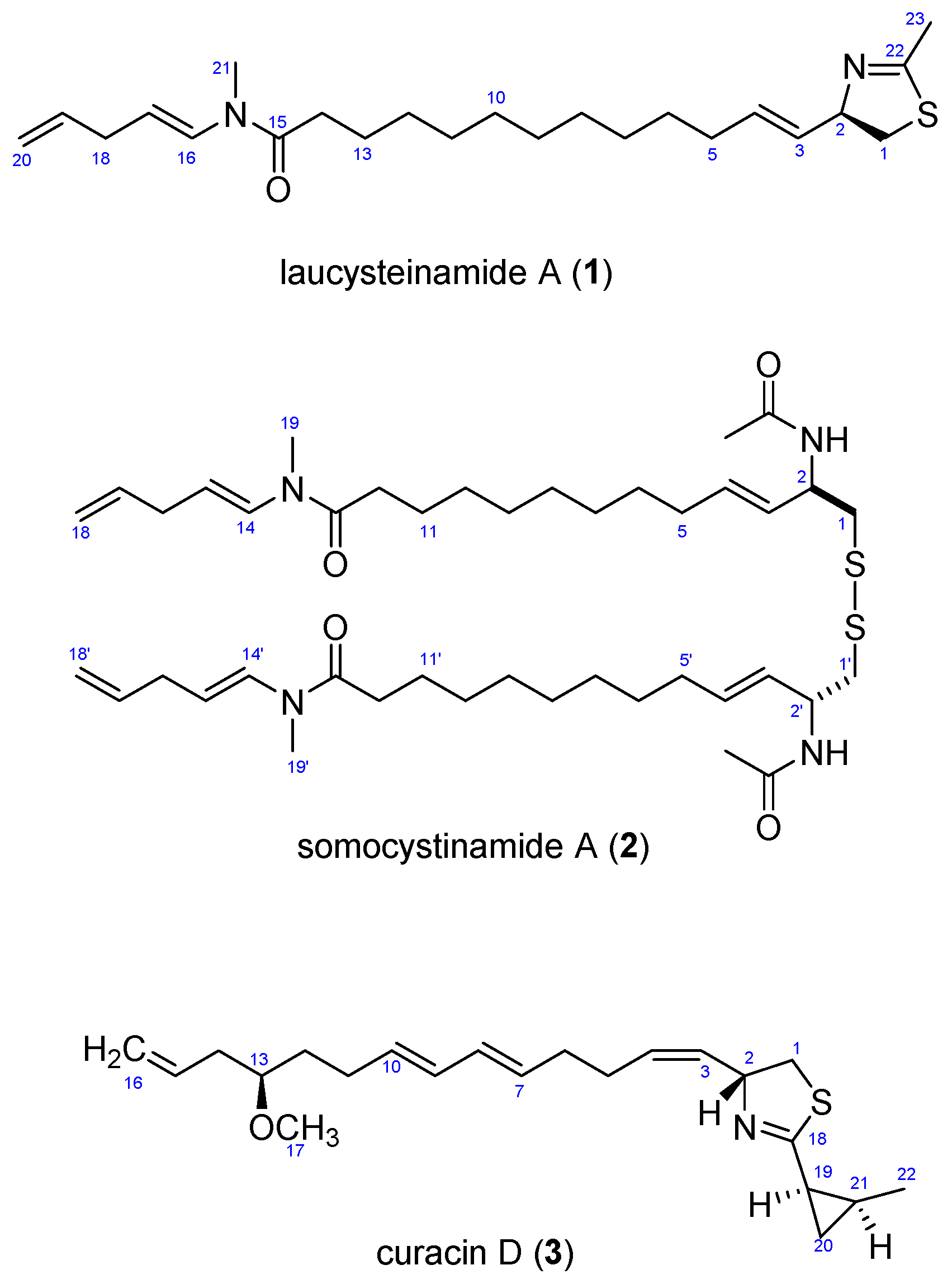
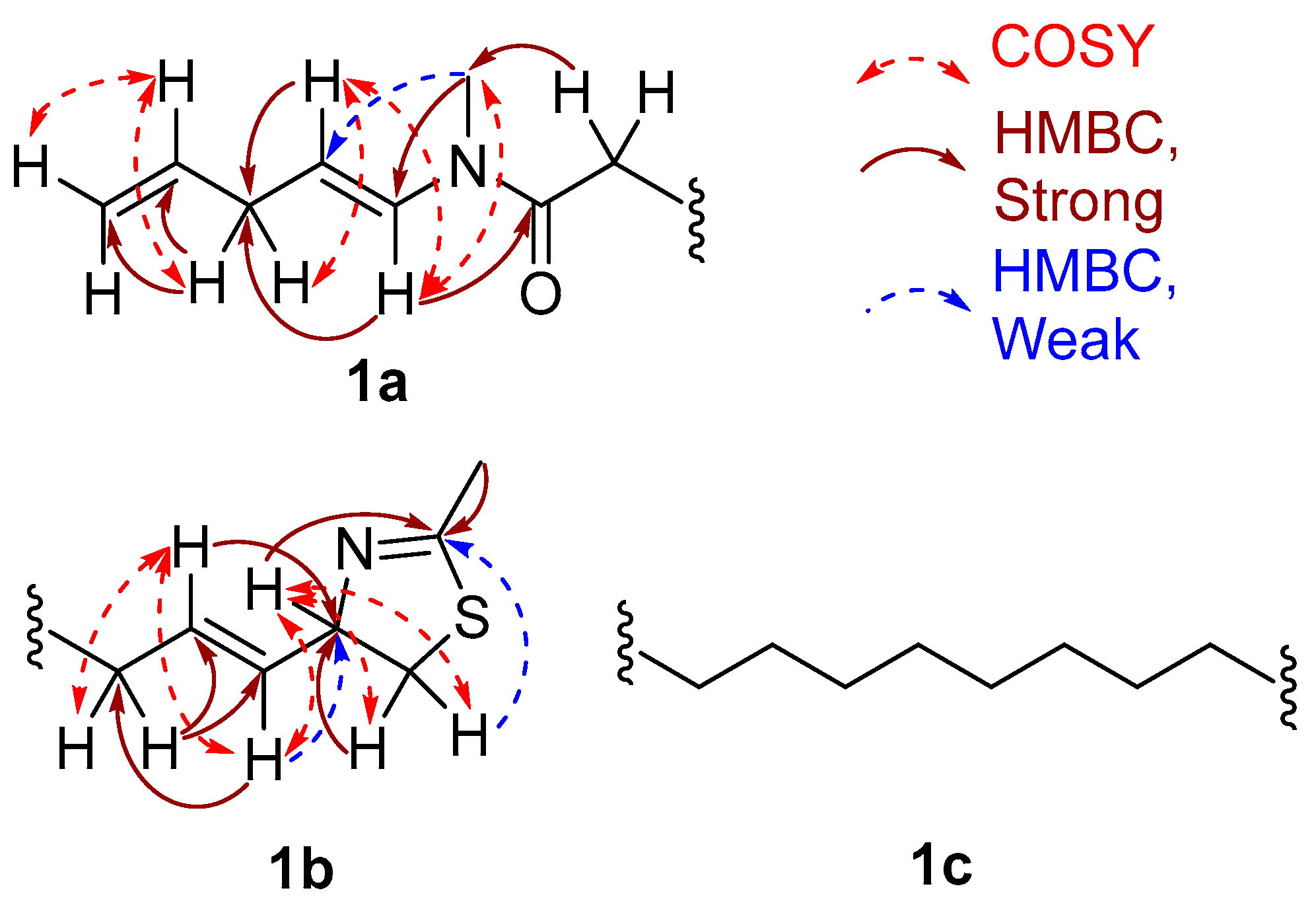
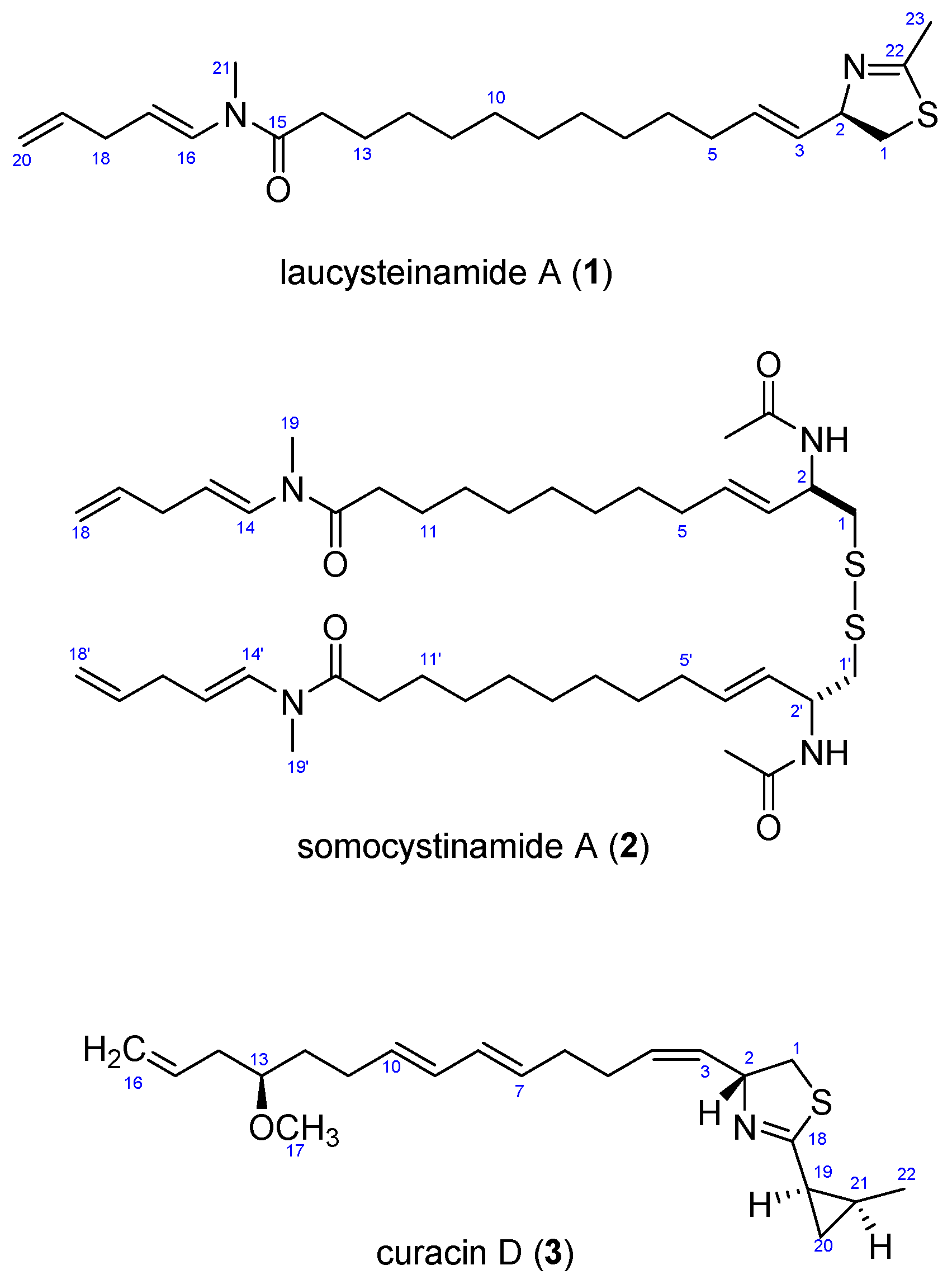
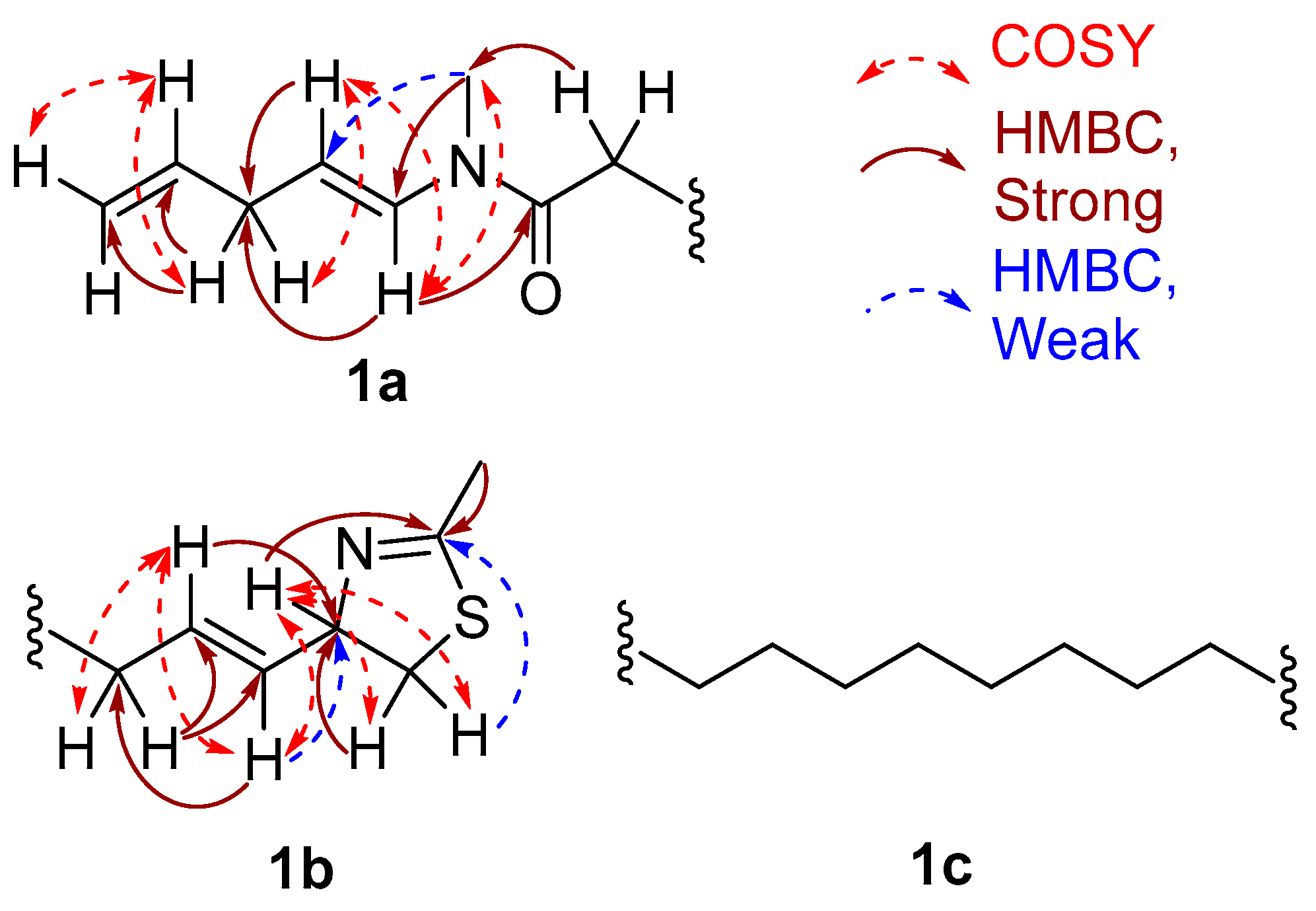
2. Results and Discussion
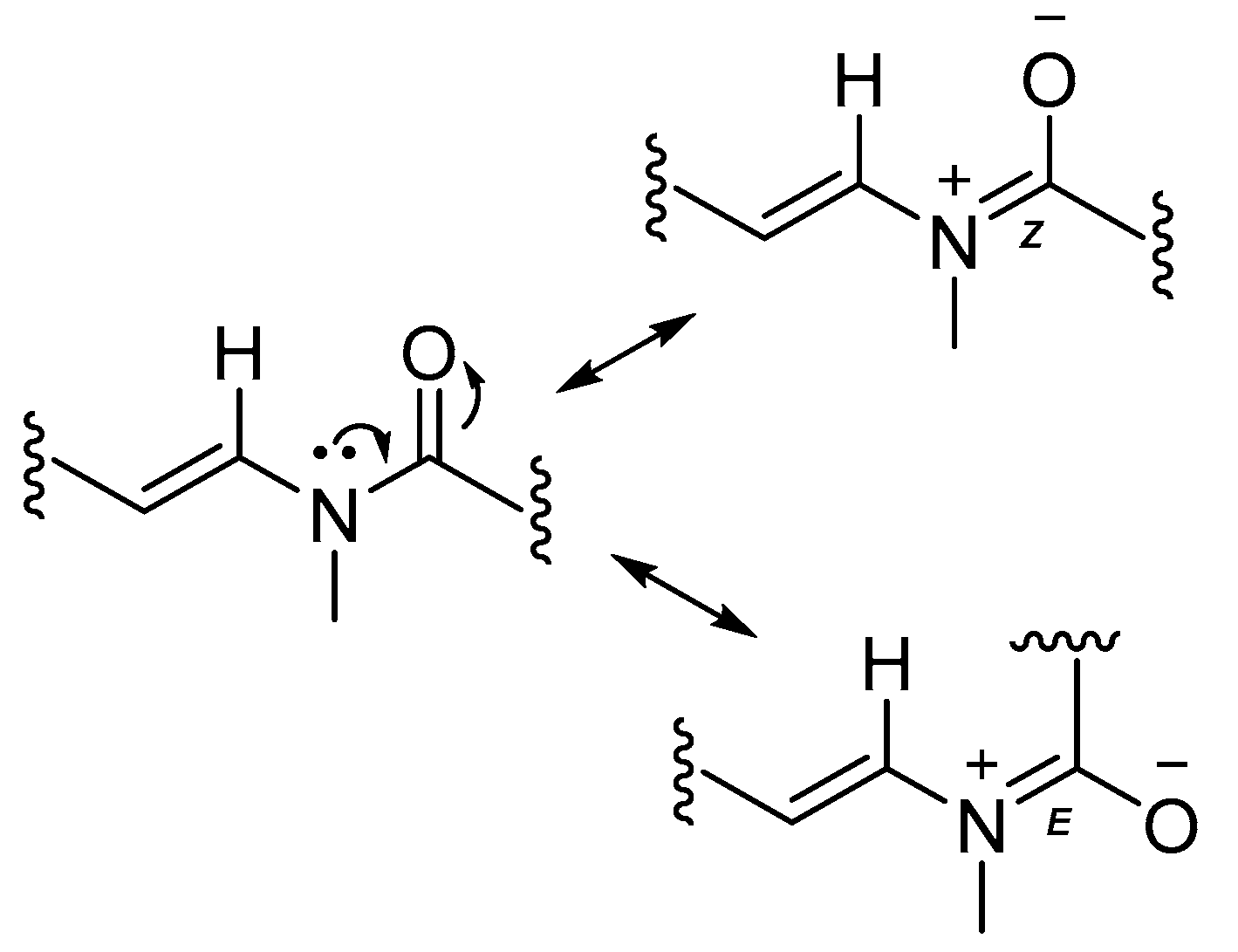
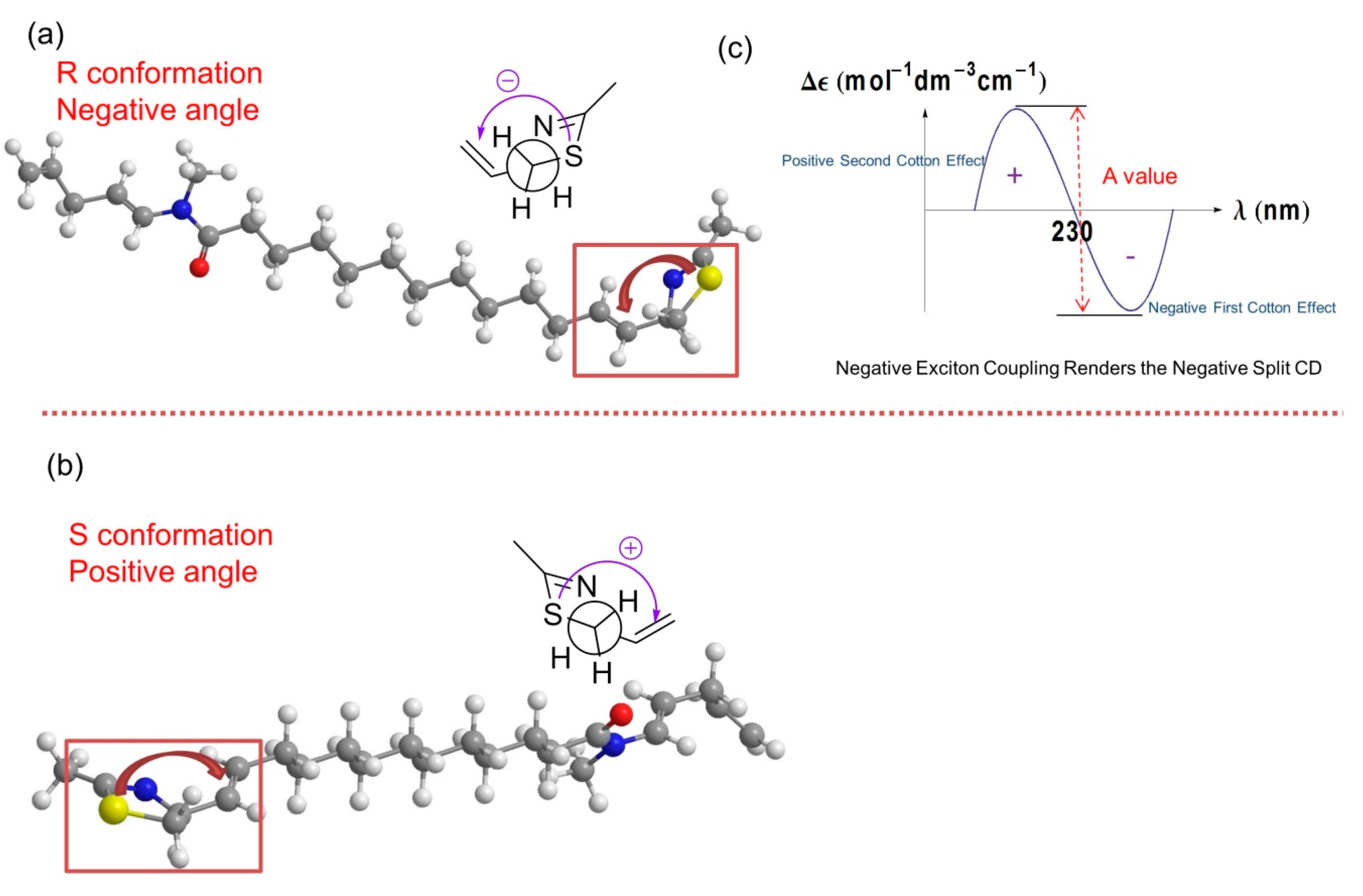
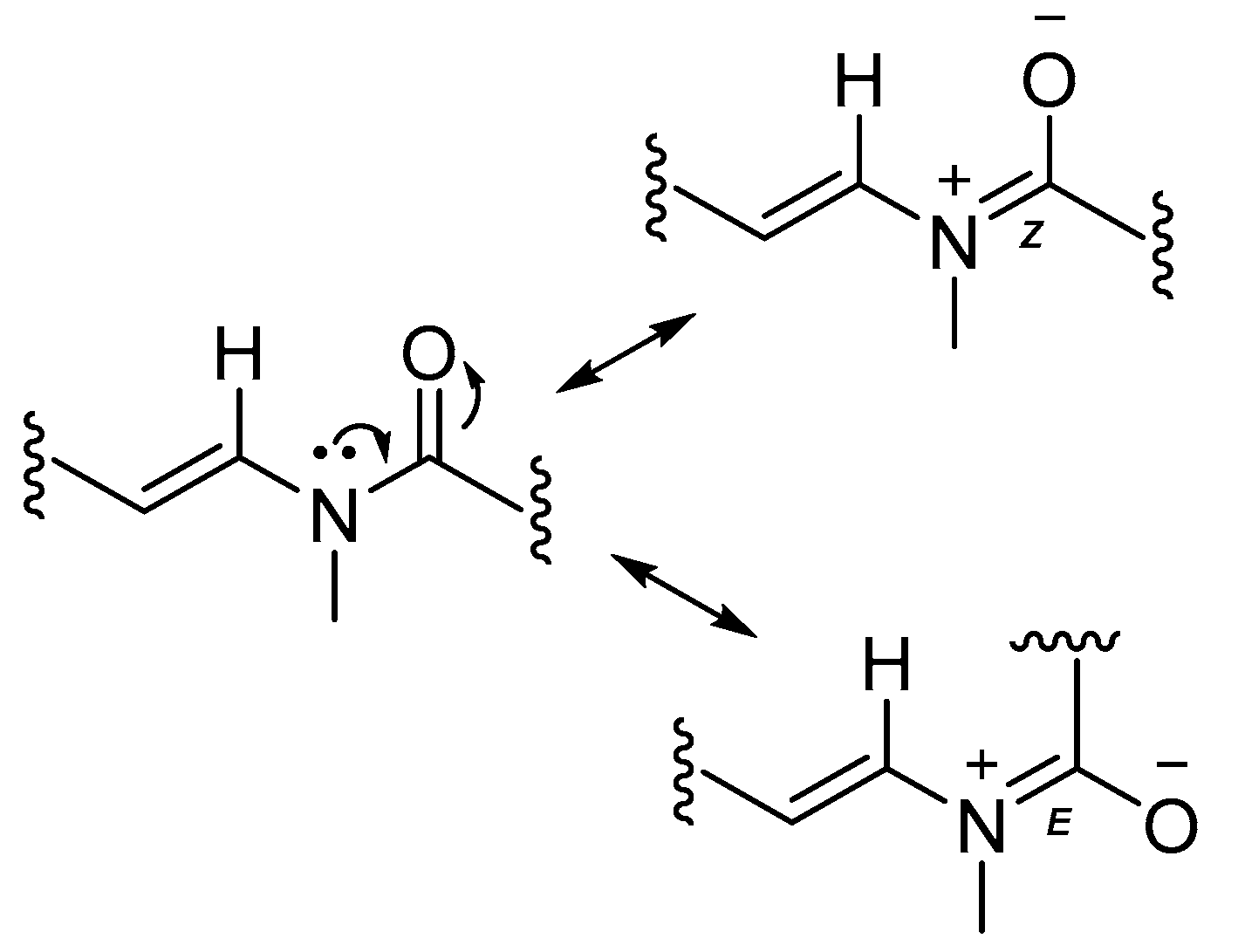
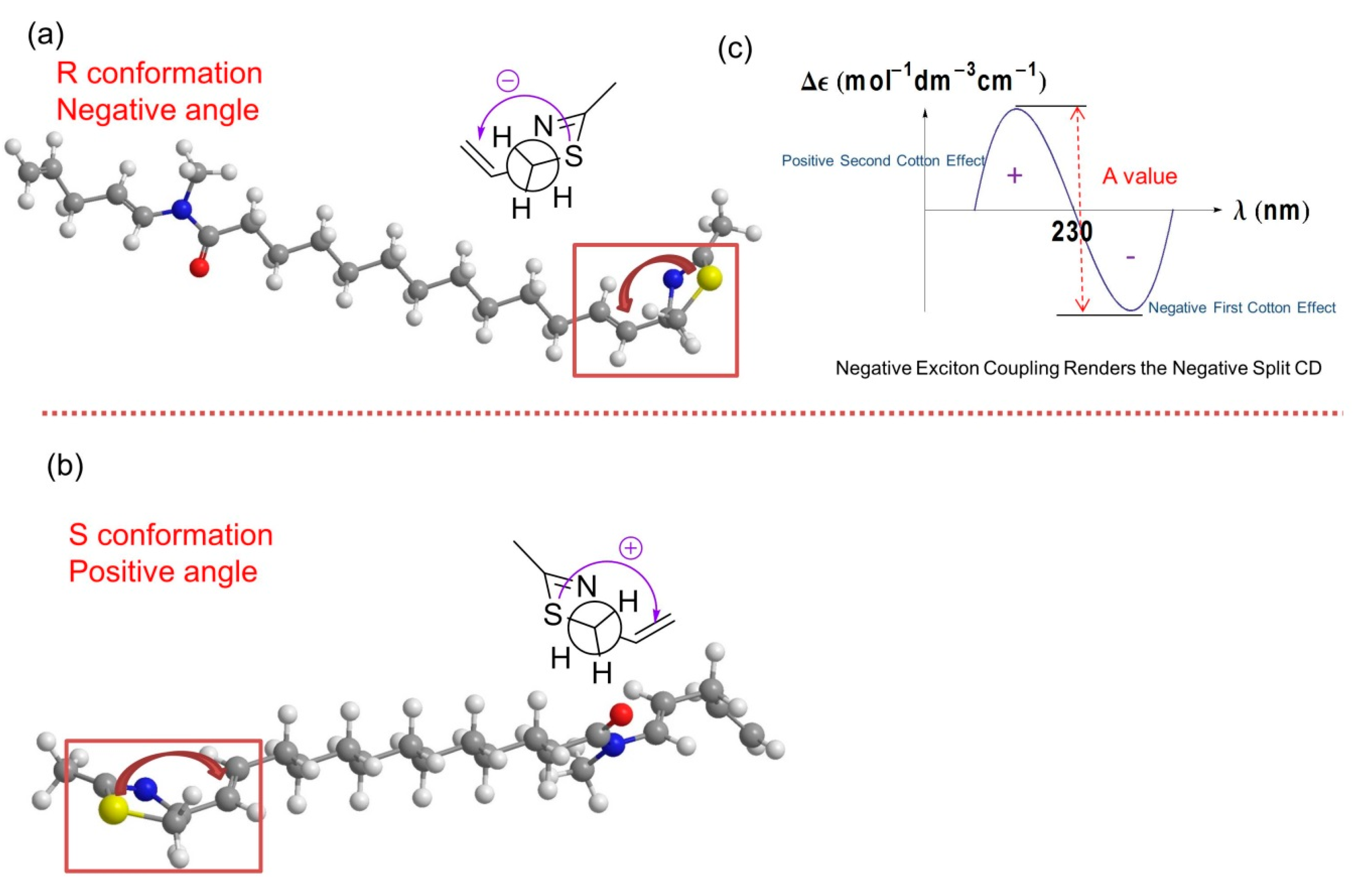
2.1. Stereochemistry
2.2. Bioactivity
2.3. Biosynthetic Considerations
3. Experimental Section
3.1. General Experimental Procedures
3.2. Sample Material
3.3. Extraction and Isolation
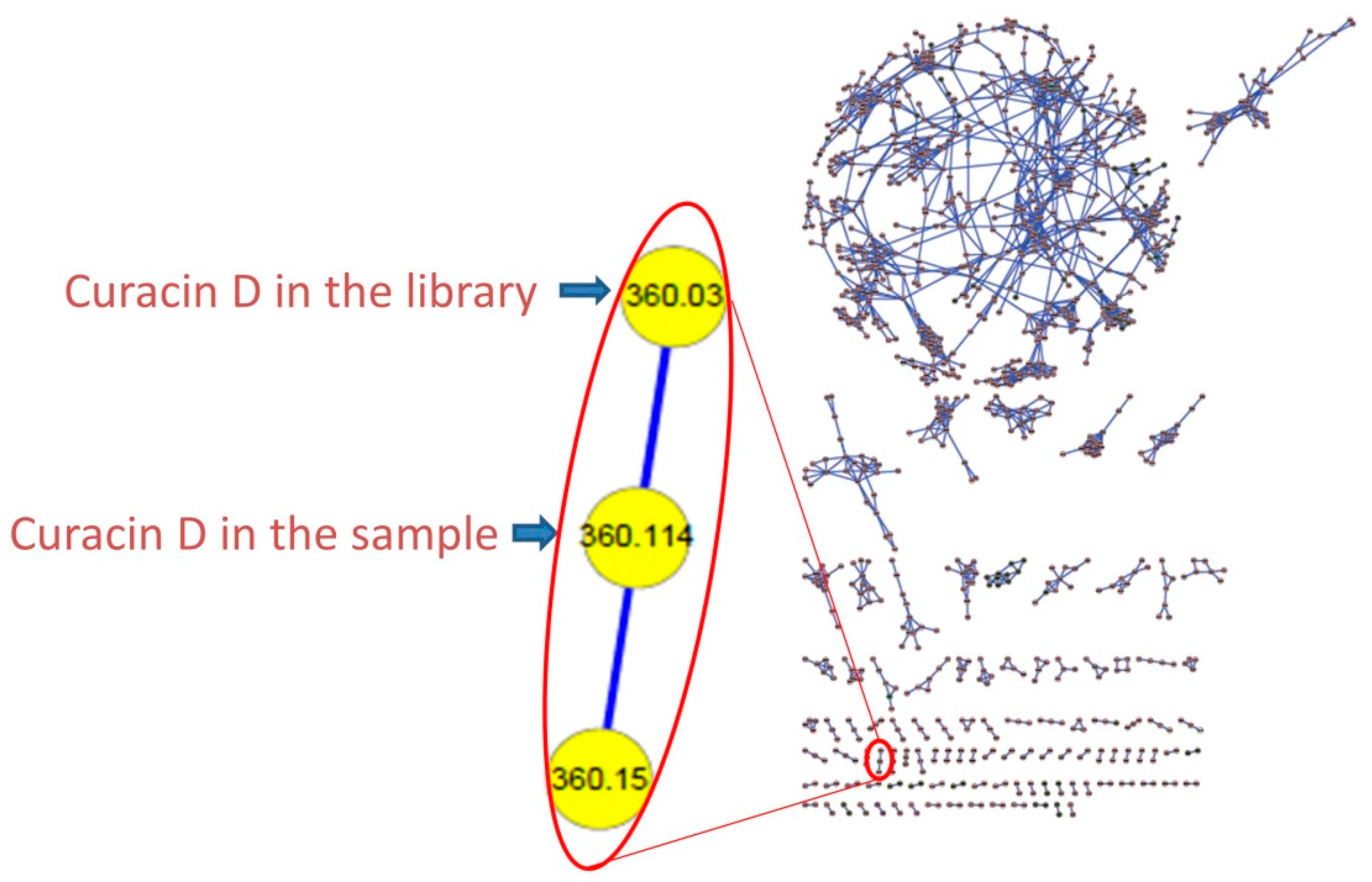
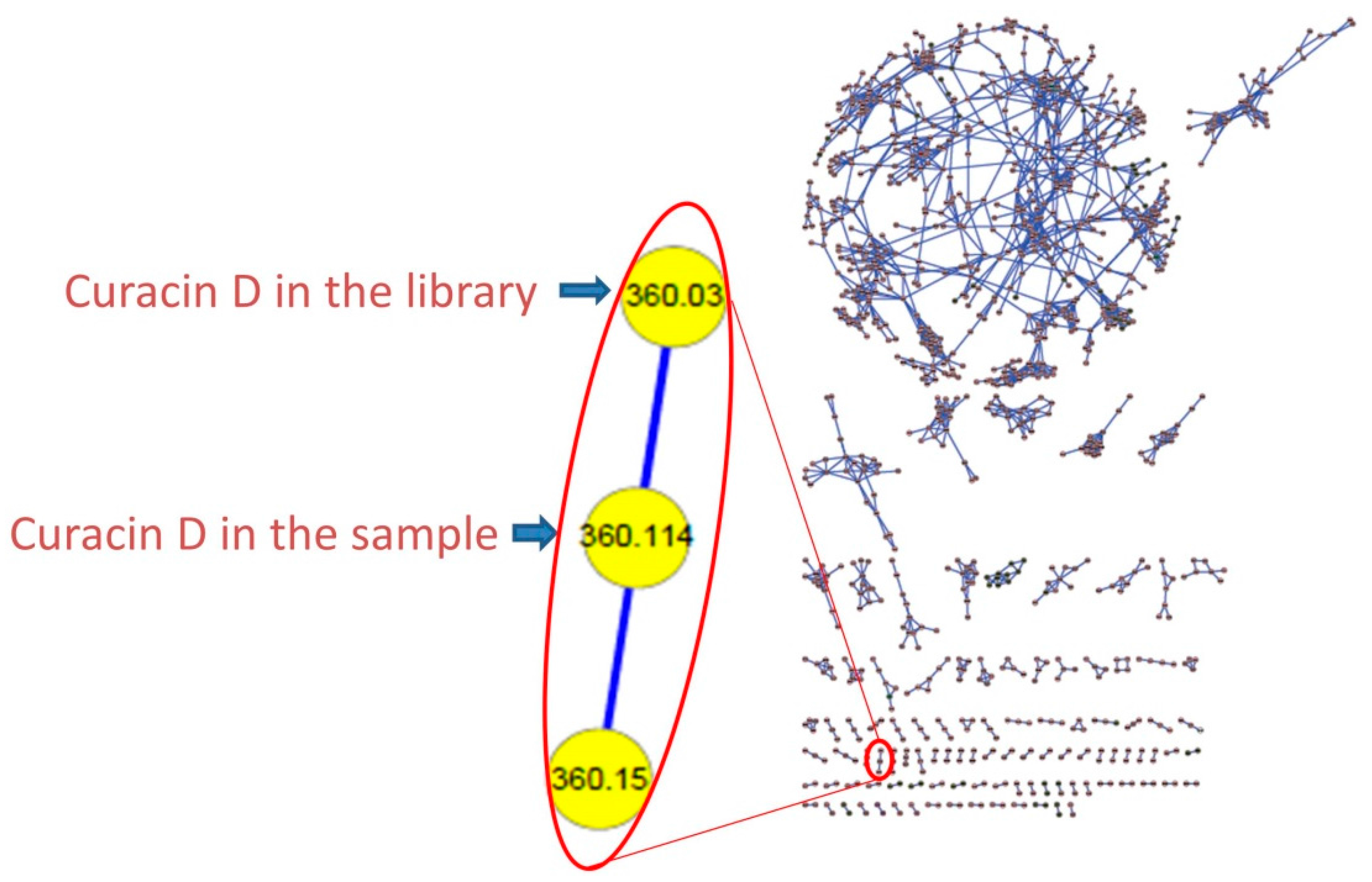
3.4. Molecular Networking
3.5. Biological Testing
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2015, 32, 116–211. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.G.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Isolation and structure determination of obyanamide, a novel cytotoxic cyclic depsipeptide from the marine cyanobacterium Lyngbya confervoides. J. Nat. Prod. 2002, 65, 29–31. [Google Scholar] [CrossRef] [PubMed]
- Kingston, D.G.I. Tubulin-interactive natural products as anticancer agents. J. Nat. Prod. 2009, 72, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Marquez, B.; Verdier-Pinard, P.; Hamel, E.; Gerwick, W.H. Curacin D, an antimitotic agent from the marine cyanobacterium Lyngbya majuscula. Phytochemistry 1998, 49, 2387–2389. [Google Scholar] [CrossRef]
- Nogle, L.M.; Gerwick, W.H. Somocystinamide A, a novel cytotoxic disulfide dimer from a Fijian marine cyanobacterial mixed assemblage. Org. Lett. 2002, 4, 1095–1098. [Google Scholar] [CrossRef] [PubMed]
- Engene, N.; Tronholm, A.; Salvador-Reyes, L.A.; Luesch, H.; Paul, V.J. Caldora penicillata Gen. Nov., Comb. Nov (Cyanobacteria), a Pantropical Marine Species with Biomedical Relevance. J. Phycol. 2015, 51, 670–681. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Sanchez, L.M.; Rath, C.M.; Liu, X.T.; Boudreau, P.D.; Bruns, N.; Glukhov, E.; Wodtke, A.; de Felicio, R.; Fenner, A.; et al. Molecular networking as a dereplication strategy. J. Nat. Prod. 2013, 76, 1686–1699. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R.; Hall, C.D.; El-Dien, B.; El-Gendy, M.; Draghici, B. Tautomerism in drug discovery. J. Comput. Aid Mol. Des. 2010, 24, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Gerwick, W.H.; Proteau, P.J.; Nagle, D.G.; Hamel, E.; Blokhin, A.; Slate, D.L. Structure of curacin a, a novel antimitotic, antiproliferative, and brine shrimp toxic natural product from the marine cyanobacterium Lyngbya majuscula. J. Org. Chem. 1994, 59, 1243–1245. [Google Scholar] [CrossRef]
- Rocha, G.B.; Freire, R.O.; Simas, A.M.; Stewart, J.J.P. RM1: A reparameterization of AM1 for H, C, N, O, P, S, F, Cl, Br, and I. J. Comput. Chem. 2006, 27, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Chen, S.L.; Nakanishi, K. Quantitative definition of exciton chirality and distant effect in exciton chirality method. J. Am. Chem. Soc. 1975, 97, 5345–5352. [Google Scholar] [CrossRef]
- Gehret, J.J.; Gu, L.C.; Gerwick, W.H.; Wipf, P.; Sherman, D.H.; Smith, J.L. Terminal alkene formation by the thioesterase of curacin A biosynthesis: structure of a decarboxylating thioesterase. J. Biol. Chem. 2011, 286, 14445–14454. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.X.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Alley, M.C.; Scudiero, D.A.; Monks, A.; Hursey, M.L.; Czerwinski, M.J.; Fine, D.L.; Abbott, B.J.; Mayo, J.G.; Shoemaker, R.H.; Boyd, M.R. Feasibility of drug screening with panels of human-tumor cell-lines using a microculture tetrazolium assay. Cancer Res. 1988, 48, 589–601. [Google Scholar] [PubMed]
- Meyer, B.N.; Ferrigni, N.R.; Putnam, J.E.; Jacobsen, L.B.; Nichols, D.E.; Mclaughlin, J.L. Brine shrimp—A convenient general bioassay for active-plant constituents. Planta Med. 1982, 45, 31–34. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δH Mult. (J in Hz) | δC, Mult. (J in Hz) | COSY | H2BC | HMBC |
|---|---|---|---|---|---|
| 1 | 3.01, dd (10.8, 8.4) | 40.3 | 2.79, 4.80 | 79.2 | 32.7, 79.2, 164.7 |
| 2.79, dd (10.8, 8.4) | 3.01, 4.80 | 79.2 | 164.7 | ||
| 2 | 4.8, ddd (6.6) | 79.2 | 2.79, 3.01, 1.97, 5.53 | 40.3 | 164.7, 130.1, 132.4 |
| 3 | 5.53, dd (15.6, 6.6) | 130.1 | 1.97, 4.80 | 79.2, 132.4 | 32.7, 132.4 |
| 4 | 5.69, dt (15.6, 7.8) | 132.4 | 1.97 | 32.7, 130.1 | 29.9, 32.7, 130.1 |
| 5 | 1.97, m | 32.7 | 4.80, 5.53/5.69 | 132.4 | 40.3 |
| 6 | 1.25, m | 29.9 | 30.0 | ||
| 7 to 12 | 1.25–1.4, m | 30.0 | |||
| 13 | 1.69, m | 25.3 | 1.28, 1.93, 2.09 | 33.6 | 30.0 |
| 14 | 2.09, t (7.2) | 33.6 | 1.69 | 25.3 | 30.0 |
| 1.93, t (7.8) | 34.7 | 1.69 | 25.3 | 170.2 | |
| 15 | 170.5 | ||||
| 170.2 | |||||
| 16 | 6.42, d (13.8) | 130.1 | 4.61 | 107.2 | 34.7, 29.6, 170.5 |
| 7.80, d (14.4) | 129.0 | 4.66 | 106.9 | 31.4, 34.7 | |
| 17 | 4.61, dt (13.8, 6.6) | 107.2 | 2.60, 6.42 | 129.0 | 34.7/34.8, 34.7, 129.0, 137.5 |
| 4.66, dt (14.4, 7.2) | 106.9 | 2.68, 7.80 | 137.5 | 34.7/34.8 | |
| 18 | 2.60, t (6.1) | 34.7 | 4.61, 5.78 | 107.2/106.9 | 114.9/115.2, 107.2, 130.1, 137.5 |
| 2.68, t (6.1) | 34.8 | 4.66, 5.80 | 114.9/115.2, 106.9, 138.0 | ||
| 19 | 5.78, m | 137.5 | 2.60, 5.03 | 114.9 | 130.1 |
| 5.80, m | 138.0 | 2.68, 5.03 | 115.2 | 129.0 | |
| 20 | 5.03, m | 114.9 | 2.60/2.68 | 137.5/138.0 | 34.7/34.8 |
| 5.03, m | 115.2 | 2.60/2.68 | 137.5/138.0 | 34.7/34.8 | |
| 21 | 2.90, s | 29.6 | 6.48 | 130.1, 107.2/106.9, 170.5 | |
| 2.35, s | 31.4 | 129.0, 170.5 | |||
| 22 | 164.7 | 79.2 | |||
| 23 | 1.98, s | 20.3 | 164.7 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Naman, C.B.; Engene, N.; Gerwick, W.H. Laucysteinamide A, a Hybrid PKS/NRPS Metabolite from a Saipan Cyanobacterium, cf. Caldora penicillata. Mar. Drugs 2017, 15, 121. https://doi.org/10.3390/md15040121
Zhang C, Naman CB, Engene N, Gerwick WH. Laucysteinamide A, a Hybrid PKS/NRPS Metabolite from a Saipan Cyanobacterium, cf. Caldora penicillata. Marine Drugs. 2017; 15(4):121. https://doi.org/10.3390/md15040121
Chicago/Turabian StyleZhang, Chen, C. Benjamin Naman, Niclas Engene, and William H. Gerwick. 2017. "Laucysteinamide A, a Hybrid PKS/NRPS Metabolite from a Saipan Cyanobacterium, cf. Caldora penicillata" Marine Drugs 15, no. 4: 121. https://doi.org/10.3390/md15040121
APA StyleZhang, C., Naman, C. B., Engene, N., & Gerwick, W. H. (2017). Laucysteinamide A, a Hybrid PKS/NRPS Metabolite from a Saipan Cyanobacterium, cf. Caldora penicillata. Marine Drugs, 15(4), 121. https://doi.org/10.3390/md15040121