Genome Sequences of Marine Shrimp Exopalaemon carinicauda Holthuis Provide Insights into Genome Size Evolution of Caridea

Abstract
:1. Introduction
2. Results
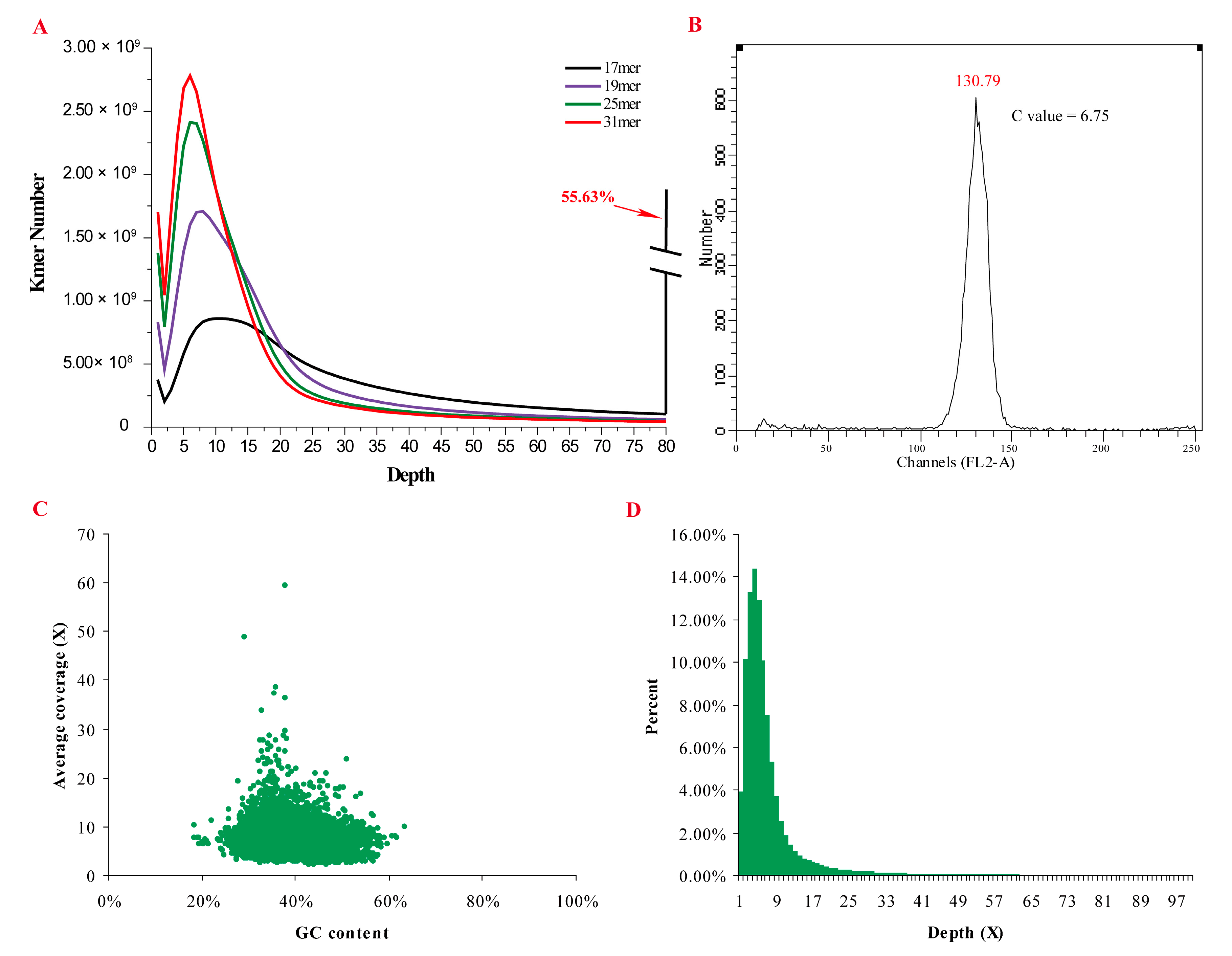
2.1. Genomic Characteristics
2.2. Genome Assembly and Validation
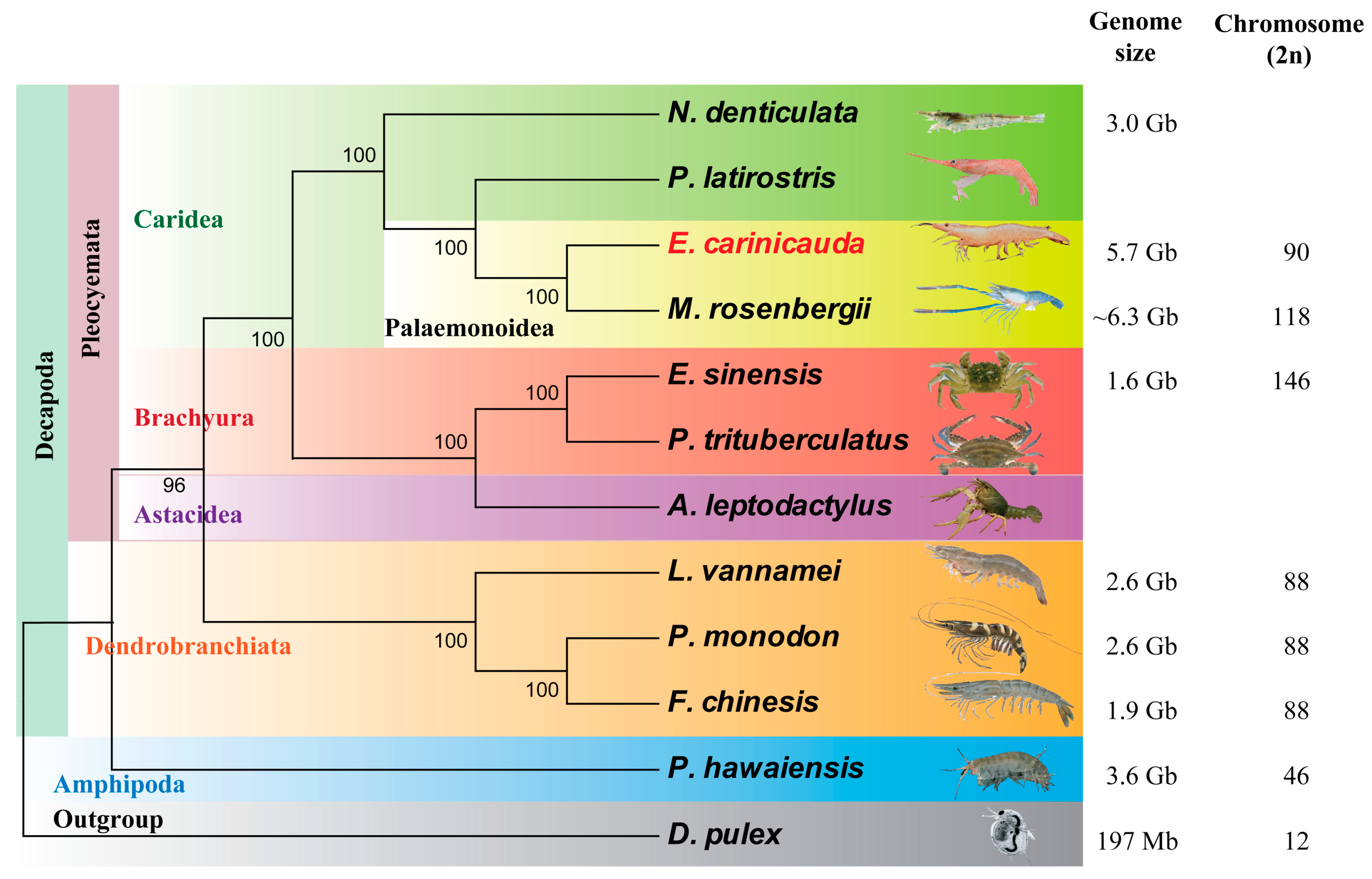
2.3. Phylogenetic Location
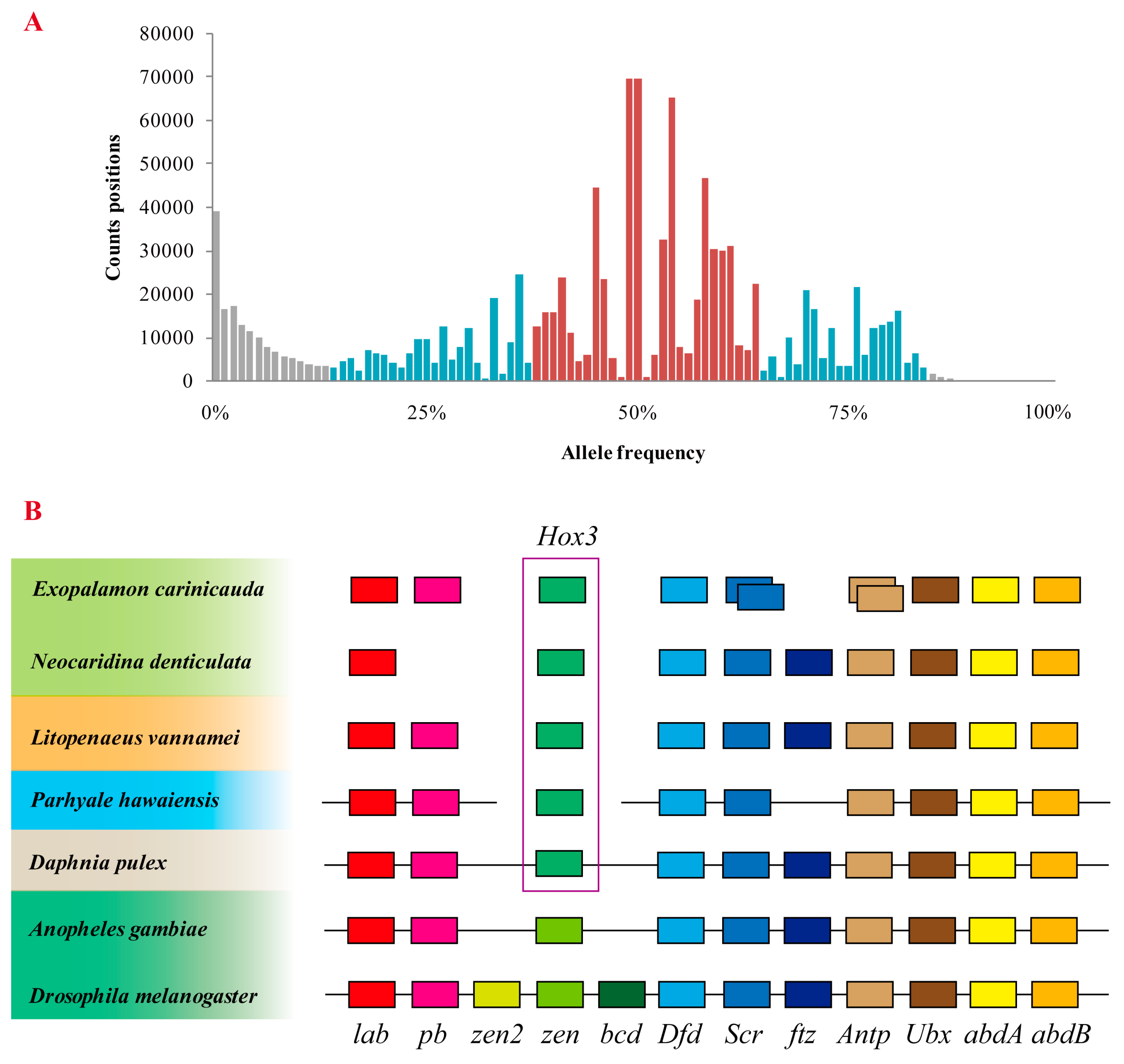
2.4. Assessment of Genome Duplication
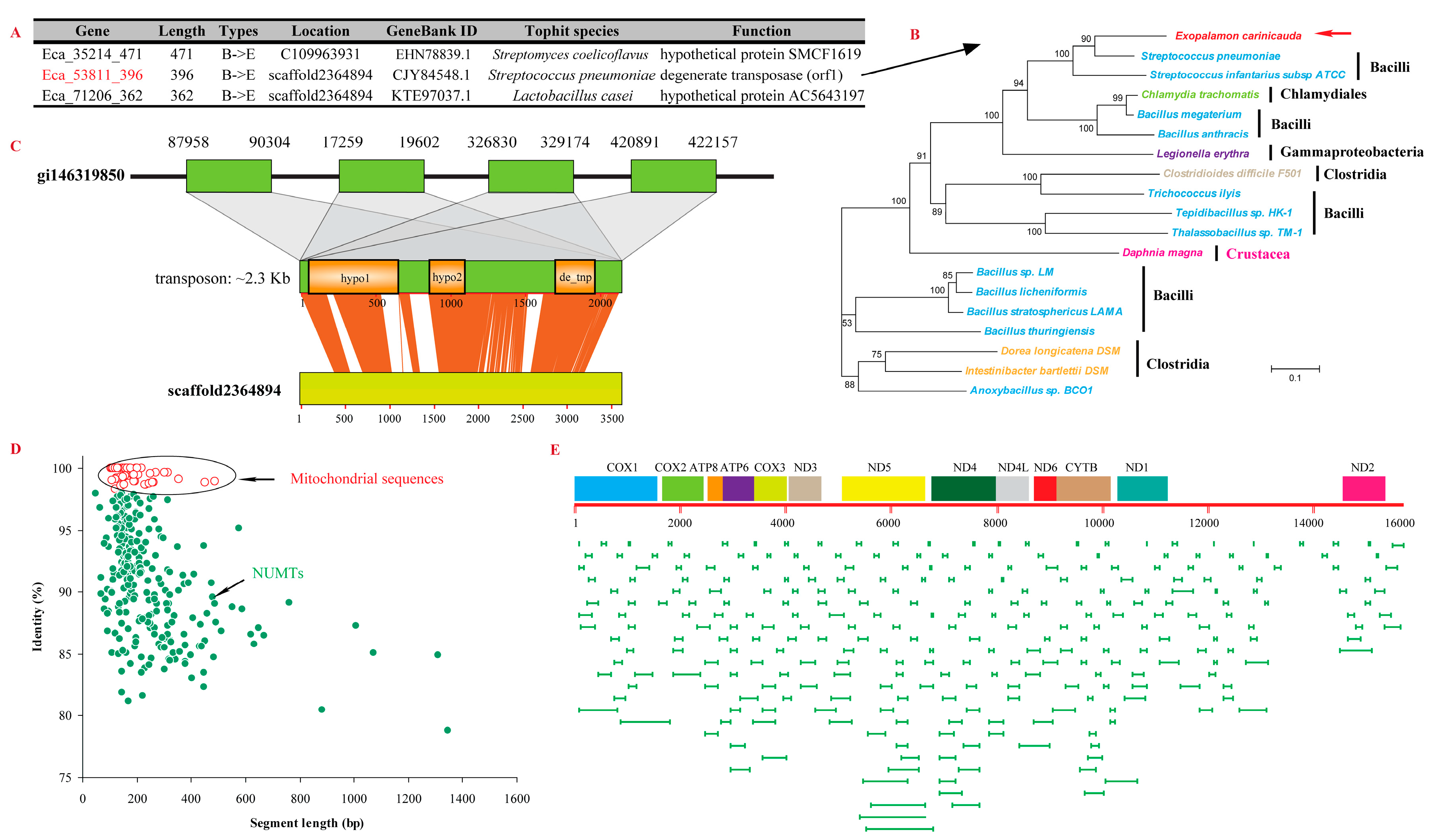
2.5. Horizontally Transferred Sequences
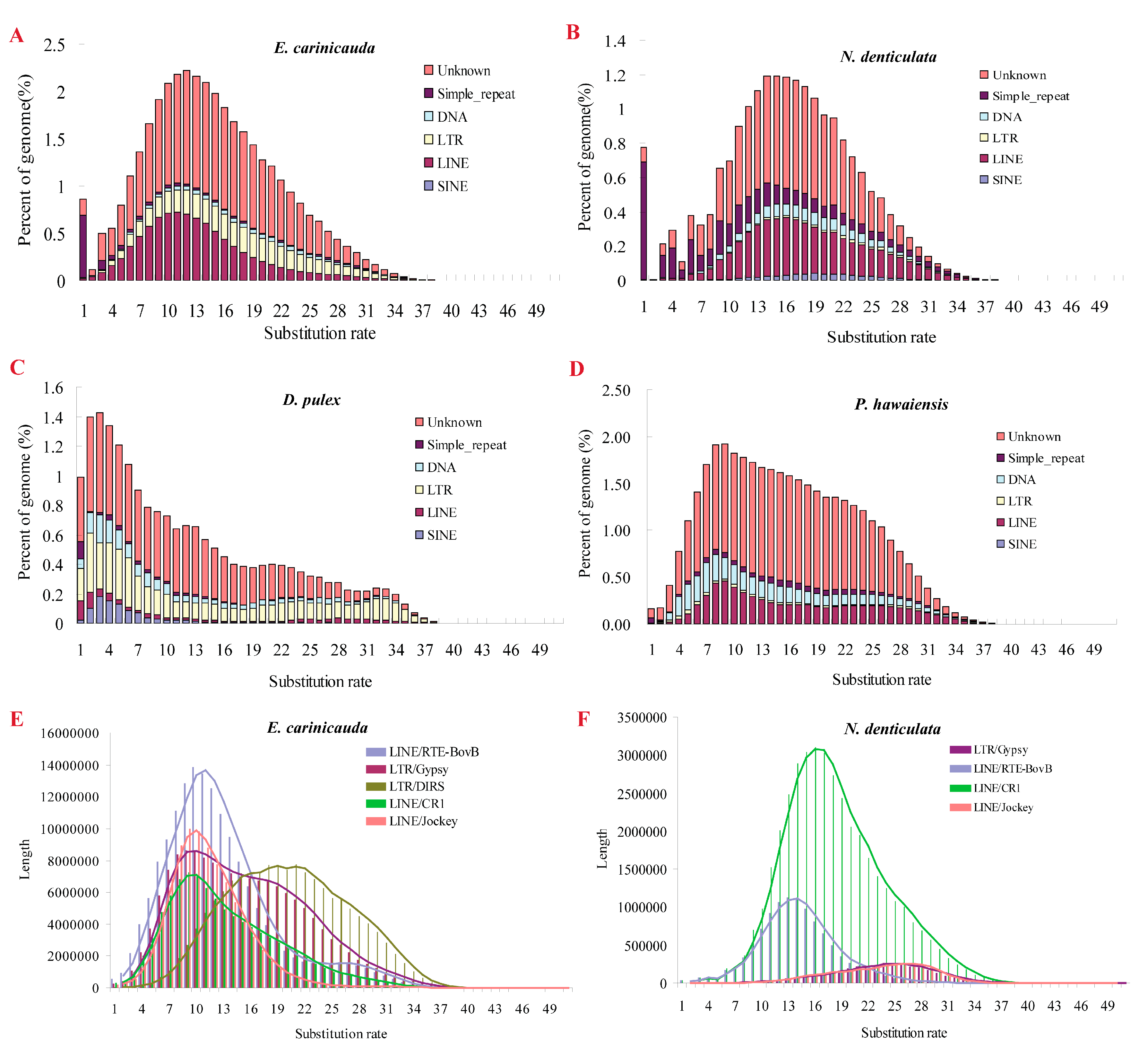
2.6. TE Expansion
3. Discussion
3.1. E. carinicauda Possesses a Large Complex Genome
3.2. Burst of TEs Responsible for Genome Expansion of E. carinicauda
4. Materials and Methods
4.1. Sample Preparation and Sequencing
4.2. Estimation of Genome Size, Heterozygosity, and Repetitiveness
4.3. Genome Assembly and Validating the Assembly
4.4. Phylogenetic Analysis
4.5. Allelic SNPs Analysis
4.6. Analysis of Hox Gene Cluster
4.7. Identification of HTGs and NUMTs
4.8. Repeats Annotation and Divergence Time Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Boore, J.L.; Lavrov, D.V.; Brown, W.M. Gene translocation links insects and crustaceans. Nature 1998, 392, 667–668. [Google Scholar] [CrossRef] [PubMed]
- Colbourne, J.K.; Pfrender, M.E.; Gilbert, D.; Thomas, W.K.; Tucker, A.; Oakley, T.H.; Tokishita, S.; Aerts, A.; Arnold, G.J.; Basu, M.K.; et al. The Ecoresponsive Genome of Daphnia pulex. Science 2011, 331, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Kao, D.; Lai, A.G.; Stamataki, E.; Rosic, S.; Konstantinides, N.; Jarvis, E.; Di Donfrancesco, A.; Pouchkina-Stancheva, N.; Semon, M.; Grillo, M.; et al. The genome of the crustacean Parhyale hawaiensis, a model for animal development, regeneration, immunity and lignocellulose digestion. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, X.; Yuan, J.; Li, F.; Chen, X.; Zhao, Y.; Huang, L.; Zheng, H.; Xiang, J. Genome survey and high-density genetic map construction provide genomic and genetic resources for the Pacific White Shrimp Litopenaeus vannamei. Sci. Rep. 2015, 5, 15612. [Google Scholar] [CrossRef] [PubMed]
- Xingqiang, W.; Binlun, Y.; Ma, S. Study on the biology and cultural ecology of Expopalaemon carinicauda. Shandong Fish. 2005, 22, 21–23. (In Chinese) [Google Scholar]
- Song, L.S.; Bian, C.; Luo, Y.J.; Wang, L.L.; You, X.X.; Li, J.; Qiu, Y.; Ma, X.Y.; Zhu, Z.F.; Ma, L.; et al. Draft genome of the Chinese mitten crab, Eriocheir sinensis. GigaScience 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Kenny, N.J.; Sin, Y.W.; Shen, X.; Zhe, Q.; Wang, W.; Chan, T.F.; Tobe, S.S.; Shimeld, S.M.; Chu, K.H.; Hui, J.H.L. Genomic Sequence and Experimental Tractability of a New Decapod Shrimp Model, Neocaridina denticulata. Mar. Drugs 2014, 12, 1419–1437. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ping, L.; Jian, L.; Jitao, L.; Peng, M.; Gao, B. Cloning and expression analysis of serine protease inhibitors gene of Exopalaemon carinicauda. J. Fish. Sci. China 2013, 20, 1166–1174. [Google Scholar]
- Sun, Y.M.; Li, F.H.; Chi, Y.H.; Xiang, J.H. Enhanced resistance of marine shrimp Exopalamon carincauda Holthuis to WSSV by injecting live VP28-recombinant bacteria. Acta Oceanol. Sin. 2013, 32, 52–58. [Google Scholar] [CrossRef]
- Duan, Y.; Liu, P.; Li, J.; Chen, P. Immune gene discovery by expressed sequence tag (EST) analysis of hemocytes in the ridgetail white prawn Exopalaemon carinicauda. Fish Shellfish Immunol. 2013, 34, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Z.; Cheng, X.Q.; Yu, Y.X.; Shen, H.; Wan, X.H. Analysis of ITS1 sequences and genetic relationships between populations of ridgetail white prawn, Exopalaemon carinicauda, in the East China Sea. Genet. Mol. Res. 2015, 14, 12316–12322. [Google Scholar] [CrossRef] [PubMed]
- Gui, T.; Zhang, J.; Song, F.; Sun, Y.; Xie, S.; Yu, K.; Xiang, J. CRISPR/Cas9-mediated Genome Editing and Mutagenesis of EcChi4 in Exopalaemon carinicauda. G3 (Bethesda) 2016, 6, 3757–3764. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, N.W. Genome Size Diversity and Evolution in the Crustacea. In Theses & Dissertations; University of Guelph: Guelph, ON, Canada, 2015; Volume 174, pp. 1–262. [Google Scholar]
- Jeffery, N.W.; Hultgren, K.; Chak, S.T.; Gregory, T.R.; Rubenstein, D.R. Patterns of genome size variation in snapping shrimp. Genome 2016, 59, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.E.; Takebayashi, N.; Barker, M.S.; Mayrose, I.; Greenspoon, P.B.; Rieseberg, L.H. The frequency of polyploid speciation in vascular plants. Proc. Natl. Acad. Sci. USA 2009, 106, 13875–13879. [Google Scholar] [CrossRef] [PubMed]
- Piegu, B.; Guyot, R.; Picault, N.; Roulin, A.; Saniyal, A.; Kim, H.; Collura, K.; Brar, D.S.; Jackson, S.; Wing, R.A.; et al. Doubling genome size without polyploidization: Dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res. 2006, 16, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Kidwell, M.G. Transposable elements and the evolution of genome size in eukaryotes. Genetica 2002, 115, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Isambert, H.; Stein, R.R. On the need for widespread horizontal gene transfers under genome size constraint. Biol. Direct 2009, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Cordero, O.X.; Hogeweg, P. The impact of long-distance horizontal gene transfer on prokaryotic genome size. Proc. Natl. Acad. Sci. USA 2009, 106, 21748–21753. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.J.; Belzile, C.; Glemet, H.; Dufresne, F. Large genomes among caridean shrimp. Genome 2008, 51, 159–163. [Google Scholar] [PubMed]
- Bonnivard, E.; Catrice, O.; Ravaux, J.; Brown, S.C.; Higuet, D. Survey of genome size in 28 hydrothermal vent species covering 10 families. Genome 2009, 52, 524–536. [Google Scholar] [PubMed]
- Zhang, G.F.; Fang, X.D.; Guo, X.M.; Li, L.; Luo, R.B.; Xu, F.; Yang, P.C.; Zhang, L.L.; Wang, X.T.; Qi, H.G.; et al. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 2012, 490, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Chow, S.; Dougherty, W.J.; Sandifer, P.A. Meiotic Chromosome Complements and Nuclear-DNA Contents of 4 Species of Shrimps of the GenusPenaeus. J. Crustacean Biol. 1990, 10, 29–36. [Google Scholar] [CrossRef]
- Zhu, B.; Lou, M.M.; Xie, G.L.; Zhang, G.Q.; Zhou, X.P.; Li, B.; Jin, G.L. Horizontal gene transfer in silkworm, Bombyx mori. BMC Genom. 2011, 12, 248. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, N.W. The first genome size estimates for six species of krill (Malacostraca, Euphausiidae): Large genomes at the north and south poles. Polar Biol. 2012, 35, 959–962. [Google Scholar] [CrossRef]
- Bracken, D.H.; Toon, A.; Felder, D.L.; Martin, J.W.; Finley, M.; Rasmussen, J.; Palero, F.; Crandall, K.A. The Decapod Tree of Life: Compiling the Data and Moving toward a Consensus of Decapod Evolution. Arthropod Syst. Phylogeny 2009, 67, 99–116. [Google Scholar]
- Lin, F.J.; Liu, Y.; Sha, Z.L.; Tsang, L.M.; Chu, K.H.; Chan, T.Y.; Liu, R.Y.; Cui, Z.X. Evolution and phylogeny of the mud shrimps (Crustacea: Decapoda) revealed from complete mitochondrial genomes. BMC Genom. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- De Grave, S.; Chan, T.Y.; Chu, K.H.; Yang, C.H.; Landeira, J.M. Phylogenetics reveals the crustacean order Amphionidacea to be larval shrimps (Decapoda: Caridea). Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Bracken, H.D.; De Grave, S.; Toon, A.; Felder, D.L.; Crandall, K.A. Phylogenetic position, systematic status, and divergence time of the Procarididea (Crustacea: Decapoda). Zool. Scr. 2010, 39, 198–212. [Google Scholar] [CrossRef]
- Yang, L.; Ping, L.; Jian, L.; Jitao, L.; Gao, B. The chromosome preparetion and karyotype in ridgetail white prawn Exopalaemon carinicauda. J. Dalian Ocean Univ. 2012, 27, 453–456. [Google Scholar]
- Tianshu, Z.; Wang, Y. Studies on the chromosome of the Macrobrachium rosenbergii. J. Cent. China Norm. Univ. (Nat. Sci.) 2003, 37. [Google Scholar] [CrossRef]
- Bon, E.; Delaherche, A.; Bilhere, E.; De Daruvar, A.; Lonvaud-Funel, A.; Le Marrec, C. Oenococcus oeni Genome Plasticity Is Associated with Fitness. Appl. Environ. Microb. 2009, 75, 2079–2090. [Google Scholar] [CrossRef] [PubMed]
- Nikoh, N.; McCutcheon, J.P.; Kudo, T.; Miyagishima, S.Y.; Moran, N.A.; Nakabachi, A. Bacterial genes in the aphid genome: Absence of functional gene transfer from Buchnera to its host. PLoS Genet. 2010, 6, e1000827. [Google Scholar] [CrossRef] [PubMed]
- Park, H.K.; Myung, S.C.; Kim, W. Comparative transcriptomic analysis of Streptococcus pseudopneumoniae with viridans group streptococci. BMC Microbiol. 2012, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.V.; Yuhki, N.; Masuda, R.; Modi, W.; O’Brien, S.J. Numt, a recent transfer and tandem amplification of mitochondrial DNA to the nuclear genome of the domestic cat. J. Mol. Evol. 1994, 39, 174–190. [Google Scholar] [PubMed]
- Song, S.; Jiang, F.; Yuan, J.; Guo, W.; Miao, Y. Exceptionally high cumulative percentage of NUMTs originating from linear mitochondrial DNA molecules in the Hydra magnipapillata genome. BMC Genom. 2013, 14, 447. [Google Scholar] [CrossRef] [PubMed]
- Adelson, D.L.; Raison, J.M.; Edgar, R.C. Characterization and distribution of retrotransposons and simple sequence repeats in the bovine genome. Proc. Natl. Acad. Sci. USA 2009, 106, 12855–12860. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shi, X.; Hao, B.; Ge, S.; Luo, J. Duplication and DNA segmental loss in the rice genome: Implications for diploidization. New Phytol. 2005, 165, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Blanc, G.; Wolfe, K.H. Widespread paleopolyploidy in model plant species inferred from age distributions of duplicate genes. Plant Cell 2004, 16, 1667–1678. [Google Scholar] [CrossRef] [PubMed]
- Berthelot, C.; Brunet, F.; Chalopin, D.; Juanchich, A.; Bernard, M.; Noel, B.; Bento, P.; Da Silva, C.; Labadie, K.; Alberti, A.; et al. The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat. Commun. 2014, 5, 3657. [Google Scholar] [CrossRef] [PubMed]
- Nossa, C.W.; Havlak, P.; Yue, J.X.; Lv, J.; Vincent, K.Y.; Brockmann, H.J.; Putnam, N.H. Joint assembly and genetic mapping of the Atlantic horseshoe crab genome reveals ancient whole genome duplication. GigaScience 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Kenny, N.J.; Chan, K.W.; Nong, W.; Qu, Z.; Maeso, I.; Yip, H.Y.; Chan, T.F.; Kwan, H.S.; Holland, P.W.; Chu, K.H.; et al. Ancestral whole-genome duplication in the marine chelicerate horseshoe crabs. Heredity 2016, 116, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Parikesit, A.A.; Steiner, L.; Stadler, P.F.; Prohaska, S.J. Pitfalls of Ascertainment Biases in Genome Annotations—Computing Comparable Protein Domain Distributions in Eukarya. Malays. J. Fundam. Appl. 2014, 10, 65–75. [Google Scholar]
- Bennetzen, J.L.; Ma, J.; Devos, K.M. Mechanisms of recent genome size variation in flowering plants. Ann. Bot. 2005, 95, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Biemont, C. Genome size evolution: Within-species variation in genome size. Heredity 2008, 101, 297–298. [Google Scholar] [CrossRef] [PubMed]
- Elliott, T.A.; Gregory, T.R. What’s in a genome? The C-value enigma and the evolution of eukaryotic genome content. Philos.Trans. R. Soc. Lond. Ser. B Biol. Sci. 2015, 370, 20140331. [Google Scholar] [CrossRef] [PubMed]
- Elliott, T.A.; Gregory, T.R. Do larger genomes contain more diverse transposable elements? BMC Evol. Biol. 2015, 15. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Bennetzen, J.L. Plant retrotransposons. Ann. Rev. Genet. 1999, 33, 479–532. [Google Scholar] [CrossRef] [PubMed]
- Jurka, J.; Kapitonov, V.V.; Kohany, O.; Jurka, M.V. Repetitive sequences in complex genomes: Structure and evolution. Ann. Rev. Genom. Hum. Genet. 2007, 8, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, T.J.; Poulter, R.T.; Lorenzen, M.D.; Beeman, R.W. DIRS retroelements in arthropods: Identification of the recently active TcDirs1 element in the red flour beetle Tribolium castaneum. Mol. Genet. Genom. 2004, 272, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Li, R.Q.; Fan, W.; Tian, G.; Zhu, H.M.; He, L.; Cai, J.; Huang, Q.F.; Cai, Q.L.; Li, B.; Bai, Y.Q.; et al. The sequence and de novo assembly of the giant panda genome. Nature 2010, 463, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Marcais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of K-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory—efficient short—read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Li, J.; Zhu, Y.; Hou, G.; Kong, X.; Kuang, Y.; Sun, X. L_RNA_scaffolder: Scaffolding genomes with transcripts. BMC Genom. 2013, 14, 604. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. BLAT—the BLAST—like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Koran, M.E.; Thornton-Wells, T.A.; Jahanshad, N.; Glahn, D.C.; Thompson, P.M.; Blangero, J.; Nichols, T.E.; Kochunov, P.; Landman, B.A. Impact of family structure and common environment on heritability estimation for neuroimaging genetics studies using Sequential Oligogenic Linkage Analysis Routines. J. Med. Imaging (Bellingham) 2014, 1, 014005. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B.; et al. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, F.; Patricio, M.; Muffato, M.; Pignatelli, M.; Bateman, A. TreeFam v9: A new website, more species and orthology-on-the-fly. Nucleic Acids Res. 2014, 42, D922–D925. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Coghlan, A.; Ruan, J.; Coin, L.J.; Heriche, J.K.; Osmotherly, L.; Li, R.; Liu, T.; Zhang, Z.; Bolund, L.; et al. TreeFam: A curated database of phylogenetic trees of animal gene families. Nucleic Acids Res. 2006, 34, D572–D580. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.W.; Shen, Y.H.; Xiang, Z.H.; Zhang, Z. Pathogen-origin horizontally transferred genes contribute to the evolution of Lepidopteran insects. BMC Evol. Biol. 2011, 11, 356. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Pelin, A.; Selman, M.; Aris-Brosou, S.; Farinelli, L.; Corradi, N. Genome analyses suggest the presence of polyploidy and recent human-driven expansions in eight global populations of the honeybee pathogen Nosema ceranae. Environ. Microbiol. 2015, 17, 4443–4458. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.B.; Zhang, X.J.; Liu, C.Z.; Wei, J.K.; Li, F.H.; Xiang, J.H. Horizontally transferred genes in the genome of Pacific white shrimp, Litopenaeus vannamei. BMC Evol. Biol. 2013, 13, 165. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Sun, M.; Wu, Z.; Tian, M.; Cheng, H.; Zhao, F.; Meng, X. The complete mitochondrial genome of the ridgetail white prawn Exopalaemon carinicauda Holthuis, 1950 (Crustacean: Decapoda: Palaemonidae) revealed a novel rearrangement of tRNA genes. Gene 2009, 437, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Criteria | Contig | Scaffold |
|---|---|---|
| Number | 20,407,032 | 13,897,062 |
| Total length (bp) | 4,865,350,937 | 5,567,872,237 |
| Longest (bp) | 13,513 | 553,834 |
| Shortest (bp) | 100 | 100 |
| N50 (bp) | 263 | 816 |
| N90 (bp) | 116 | 129 |
| >2 kb | 28,741 | 286,753 |
| Criteria | Unigenes |
|---|---|
| Unigene num | 81,135 |
| Match unigene num | 77,374 |
| Match unigene num (%) | 95.36% |
| 90% in one scaf | 40,002 |
| 90% in one scaf (%) | 49.30% |
| 50% in one scaf | 68,128 |
| 50% in one scaf (%) | 83.96% |
| Repeats | E. carinicauda | N. denticulata | P. hawaiensis | D. pulex |
|---|---|---|---|---|
| Total length | 5.57 Gb | 1.72 Gb | 4.02 Gb | 197 Mb |
| GC level | 37.47% | 35.11% | 40.84% | 40.77% |
| Bases masked | 1.99 Gb | 3.79 Gb | 1.49 Gb | 40 Mb |
| Repeat percent | 36.37% | 22.03% | 37.17% | 20.45% |
| SINEs: | 0.01% | 0.51% | 0.03% | 0.98% |
| LINEs: | 8.86% | 5.07% | 6.43% | 0.90% |
| RTE-BovB | 3.36% | 0.63% | 0.19% | 0.24% |
| Jockey | 2.12% | 0.20% | 0.15% | 0.05% |
| L3/CR1 | 1.65% | 2.31% | 3.31% | 0.00% |
| LTR elements | 5.41% | 0.26% | 0.58% | 5.48% |
| Gypsy | 2.41% | 0.21% | 0.00% | 2.77% |
| DIRS | 2.87% | 0.00% | 0.00% | 0.28% |
| DNA elements | 0.90% | 1.14% | 4.49% | 1.75% |
| Charlie | 0.02% | 0.10% | 0.13% | 0.00% |
| Tigger | 0.48% | 0.33% | 0.05% | 0.02% |
| Unclassified | 19.28% | 9.54% | 24.31% | 10.22% |
| Total TEs | 34.47% | 16.52% | 35.84% | 19.33% |
| Satellites | 0.01% | 0.09% | 0.04% | 0.00% |
| Simple repeats | 1.39% | 3.47% | 1.27% | 0.44% |
| Low complexity | 0.64% | 2.00% | 0.13% | 0.67% |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, J.; Gao, Y.; Zhang, X.; Wei, J.; Liu, C.; Li, F.; Xiang, J. Genome Sequences of Marine Shrimp Exopalaemon carinicauda Holthuis Provide Insights into Genome Size Evolution of Caridea. Mar. Drugs 2017, 15, 213. https://doi.org/10.3390/md15070213
Yuan J, Gao Y, Zhang X, Wei J, Liu C, Li F, Xiang J. Genome Sequences of Marine Shrimp Exopalaemon carinicauda Holthuis Provide Insights into Genome Size Evolution of Caridea. Marine Drugs. 2017; 15(7):213. https://doi.org/10.3390/md15070213
Chicago/Turabian StyleYuan, Jianbo, Yi Gao, Xiaojun Zhang, Jiankai Wei, Chengzhang Liu, Fuhua Li, and Jianhai Xiang. 2017. "Genome Sequences of Marine Shrimp Exopalaemon carinicauda Holthuis Provide Insights into Genome Size Evolution of Caridea" Marine Drugs 15, no. 7: 213. https://doi.org/10.3390/md15070213
APA StyleYuan, J., Gao, Y., Zhang, X., Wei, J., Liu, C., Li, F., & Xiang, J. (2017). Genome Sequences of Marine Shrimp Exopalaemon carinicauda Holthuis Provide Insights into Genome Size Evolution of Caridea. Marine Drugs, 15(7), 213. https://doi.org/10.3390/md15070213