1. Introduction
Insulin resistance is one of the hallmarks of type 2 diabetes (T2DM) and obesity. Improvement of insulin sensitivity is an indispensable step to alleviate T2DM. The first phase of T2DM is characterized by pancreatic beta-cell compensation, displaying hyperinsulinemia in response to insulin resistance. Beta-cell overworking frequently end in dysfunction and cell death. At this point, decreased blood insulin levels exacerbate the onset of T2DM due to both insulin deficiency and resistance [
1].
To this day, pharmacological management of T2DM patients aims to achieve the best possible glycemic control, while avoiding hypoglycemia. However, the natural history of T2DM includes multiple dysfunctions affecting the α-cells, β-cells, liver, skeletal muscle, adipose tissue, the gastrointestinal tract, kidney and brain, what has been termed the ominous octet [
2]. This complex scenario of the pathophysiology of T2DM requires a shift in the current paradigm for the treatment of the disease.
A keystone in the management of diabetes is nutritional therapy along with regular physical activity. Considering that most people with T2DM are overweight or obese, weight loss is recommended to improve glycemic control [
3]. In addition, regular physical activity has demonstrated significant health benefits [
4]. When lifestyle interventions fail in the appropriate control of glucose homeostasis, T2DM is initially treated by monotherapy with oral agents, and eventually, it may require the combination of multiple drugs. In this way, insulin sensitizers and insulin secretagogues are safe therapies for T2DM treatment. Among the insulin sensitizers, metformin is successfully used as a first line pharmacotherapy for the treatment of T2DM patients. The major effect of this drug is the acute inhibition of hepatic gluconeogenesis through mechanisms involving the direct inhibition of the mitochondrial respiratory-chain complex I, leading to activation of AMP-activated protein kinase (AMPK) [
5]. The pleiotropic effects of metformin lead to lower fasting blood glucose and insulin levels with minimal risk of hypoglycemia [
6].
When glycemic control cannot be achieved using metformin as monotherapy, a second line of drugs is added to metformin treatment. Among these treatments, sulfonylureas improve insulin secretion by the regulation of the ATP-sensitive potassium channels in the plasma membrane of pancreatic β-cells [
7]. Thiazolidinediones, agonists of nuclear peroxisome proliferator-activated receptor gamma (PPARγ), increase insulin sensitivity in liver and skeletal muscle [
8]. Glinides stimulate insulin secretion rapidly and for a short period when needed [
9]. Incretins, such as glucagon-like peptide 1 (GLP-1) receptor agonists, gastric inhibitory polypeptide/glucose-dependent insulinotropic peptide (GIP) and dipeptidyl-peptidase-4 (DPP-4) inhibitors, increase insulin secretion and regulate glucose homeostasis [
10]. As the disease progresses, the β-cell function declines, and insulin therapy become necessary. The side effects of hypoglycemia and weight gain limit the efficacy of this therapy [
11]. However, novel insulin preparations and delivery systems are available to optimize insulin therapy [
12]. Regarding new glucose-lowering therapies, sodium glucose cotransporter 2 (SGLT2) receptor inhibitors lower blood glucose levels in an insulin-independent manner by increasing renal glucose excretion [
13]. Promising new pharmacological targets are currently under evaluation, such as sirtuin agonists, which enhance insulin secretion and/or insulin sensitivity; or the inhibitors of protein tyrosine phosphatase 1B, which prolong the action of insulin [
10].
Furanocembranolides are polyoxygenated diterpenoids, isolated from corals, in which a furanic ring and a β-lactone subunit are inserted in a cembrane skeleton [
14]. Leptolide is a member of the furanocembranolide family, which was isolated for the first time by Gutiérrez et al. in 2005 [
14] from the octocorals
Leptogorgia alba and
Leptogorgia rigida on the Pacific coast of Panama. It has been proposed that members of this family, such as pukalide, may function in nature as a defensive toxin against potential octocoral predators [
15,
16]. The potential pharmacological use of this family of compounds is largely unexplored. Although, few examples have been reported. Among them, lophotoxin is a neuromuscular toxin that binds selectively and irreversibly within the acetylcholine-recognition site of nicotinic acetylcholine receptors, thereby preventing acetylcholine from activating its receptor [
17,
18]. The antiproliferative and cytotoxic activities of some of these compounds have been studied against the cell lines L-929, K-562, HeLA, MDA-MB-231, A-549, HT-29 and P388 showing weak antiproliferative and cytotoxic properties [
19,
20,
21]. Furthermore, the antiplasmodial activity of six furanocembranolides and the irregular pseudopterolide isolated from specimens of
Leptogorgia alba and
L. rigida was evaluated, and among them, leptolide and pukalide showed no biological activity against the parasite [
14].
Leptolide, among other members of its family, has already been shown to increase pancreatic beta-cell proliferation in vitro, in INS1cells (Insulin secreting beta cell derived line) and primary cultures of rodent pancreatic islets [
22]. In addition, epoxypukalide, another molecule of this family, has been shown to improve beta-cell protection in vitro and in vivo, in rodent islets and in a STZ (streptozotocin)-induced model of diabetes, respectively [
22,
23]. Interestingly, epoxypukalide also alleviates glucose intolerance in a preclinical model of type 1 diabetes [
23]. Thus, furanocembranolides appear to be attractive molecules to maintain functional beta-cell mass and glycemic control.
In this work, we have extended our initial findings and explored the capability of leptolide to improve insulin sensitivity. To this end, we have assessed the capacity of leptolide to enhance insulin signaling in insulin-resistant hepatocytes and in the liver and skeletal muscle of diet-induced obese mice.
2. Materials and Methods
Leptolide purification, characterization and molecular structure were described previously [
14]. Briefly, crude extracts from octocorals were subjected to fractionation. Leptolide was initially isolated as a novel compound with antiplasmodial activity, and its structure was determined by NMR and confirmed by single-crystal X-ray crystallography.
2.1. Cell Culture
HepG2 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA; #HB-8065). The cell line was originally isolated from a liver hepatocellular carcinoma of a 15-year-old Caucasian male. Cells were growth in DMEM (1X) supplemented with 4.5 g/L d-glucose, 0.6 g/L l-glutamine, 0.1 g/L sodium pyruvate and 10% fetal bovine serum.
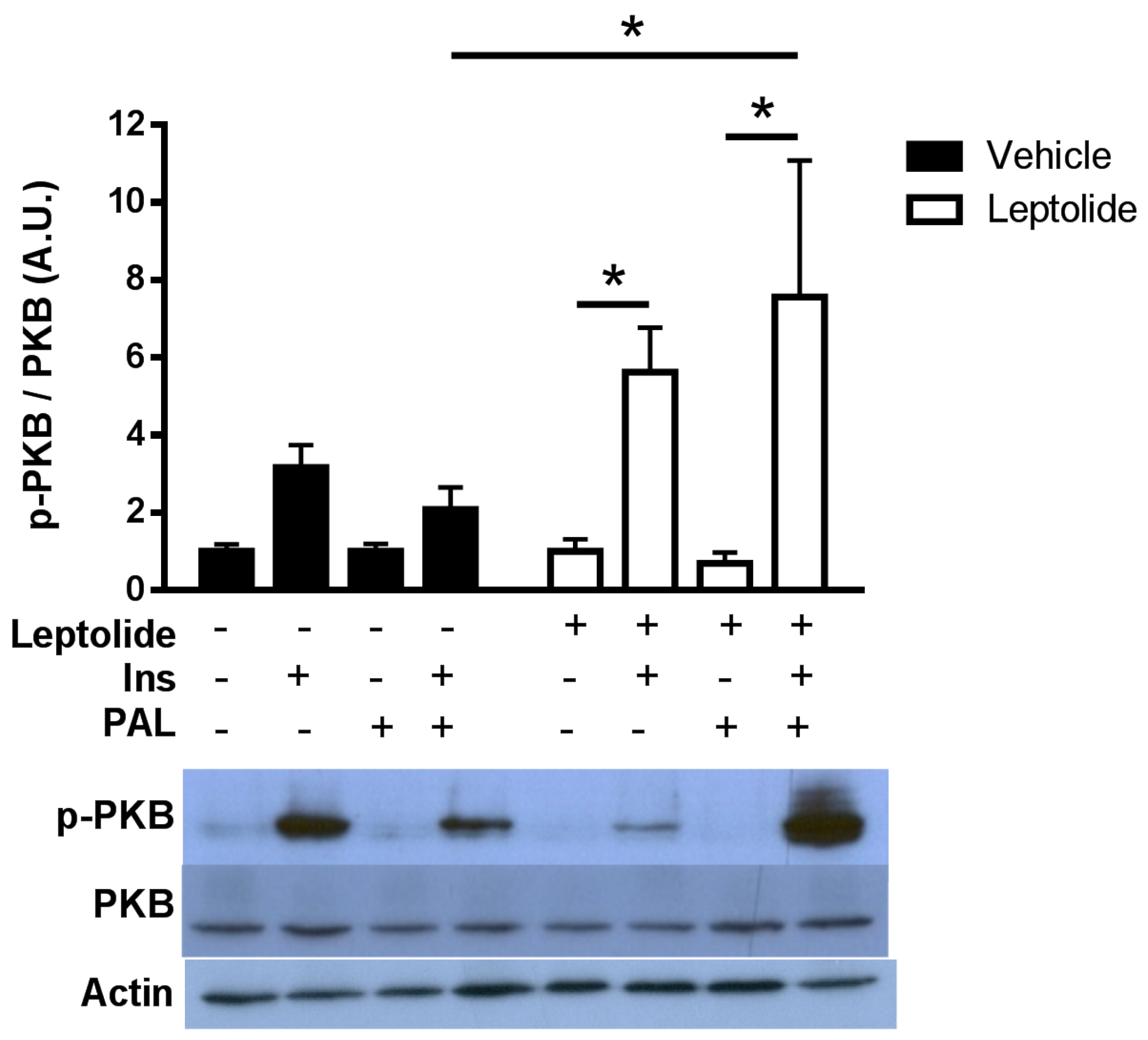
In order to analyze the effects of leptolide on the intracellular insulin signaling pathway, HepG2 cells were treated with 0.1 µM leptolide or vehicle (DMSO) during 24 h in medium without serum. Afterwards, 100 nM human insulin (Sigma, St. Louis, MO, USA) was added, and HepG2 cells were collected after 0, 5, 10, 15 and 30 min. To analyze the effects of leptolide in the setting of resistance, HepG2 cells were treated with 0.2 mM palmitate and 0.1 µM leptolide in serum-free medium for 24 h. Afterwards, 100 nM human insulin (Sigma, St. Louis, MO, USA) was added, and 15 min later, HepG2 cells were collected.
2.2. Animal Procedures
C57Bl6J male mice were purchase from Charles River Laboratory (Écully, France). Male mice were chosen for metabolic phenotyping to avoid the potential variability related to estrous cycle. Experimental procedures were approved by the Animal Care and Use Committee of the University of Valladolid (UVa), Valladolid, Spain, in accordance with the European and Spanish Guidelines for the Care and Use of Mammals in Research. Mice were fed with standard rodent chow and water ad libitum in ventilated cages in a 12:12-h light/dark cycle.
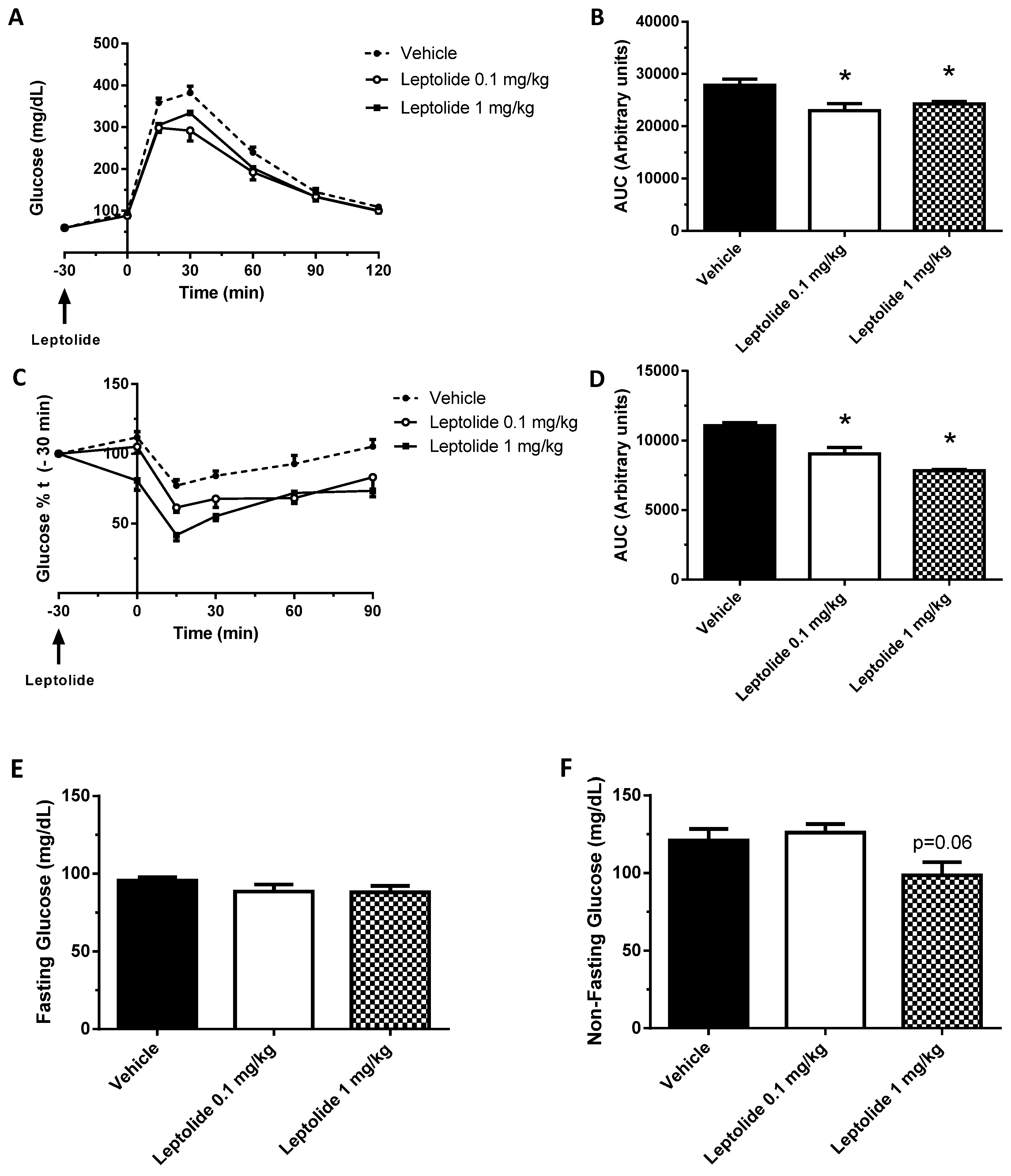
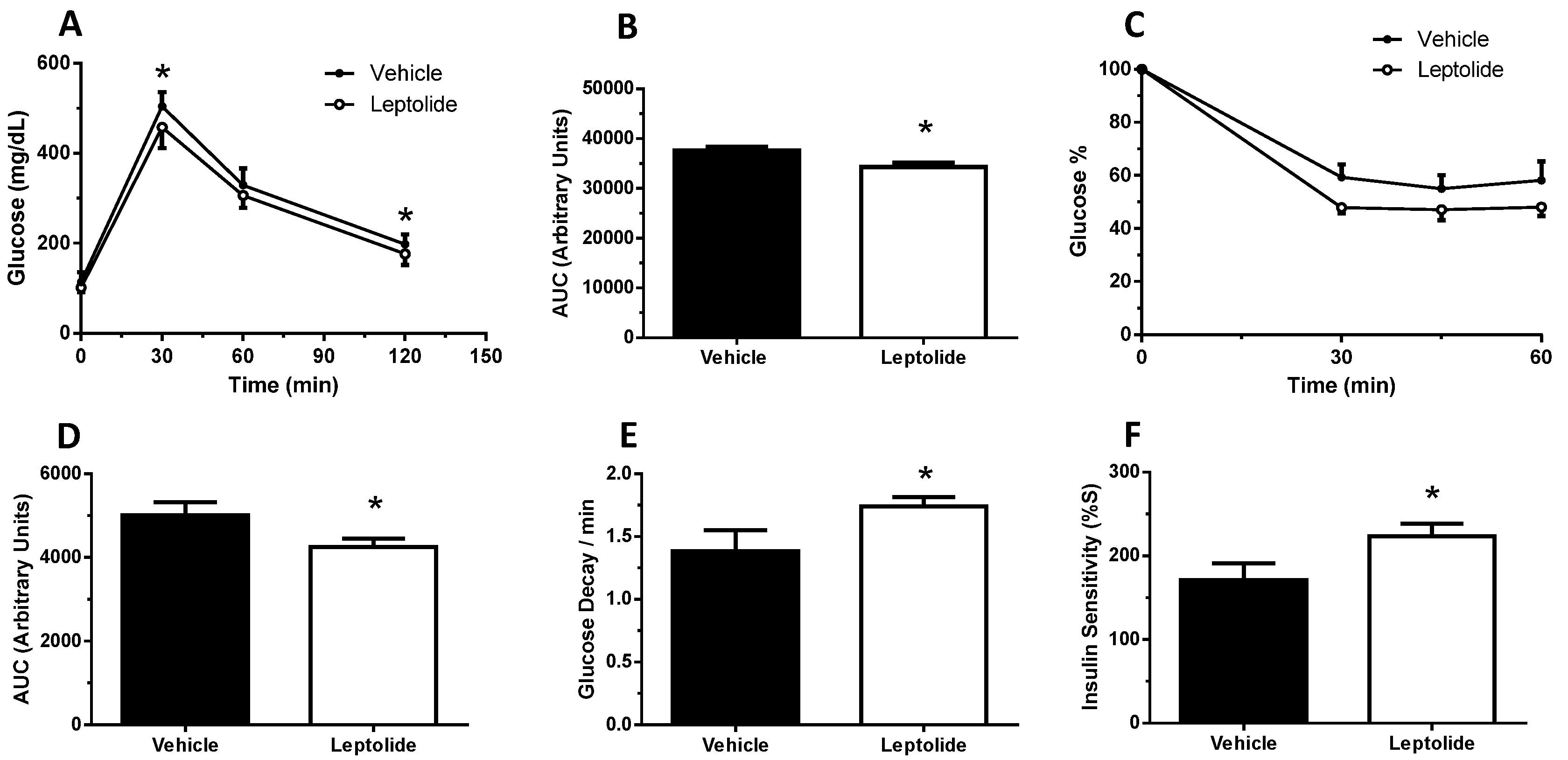
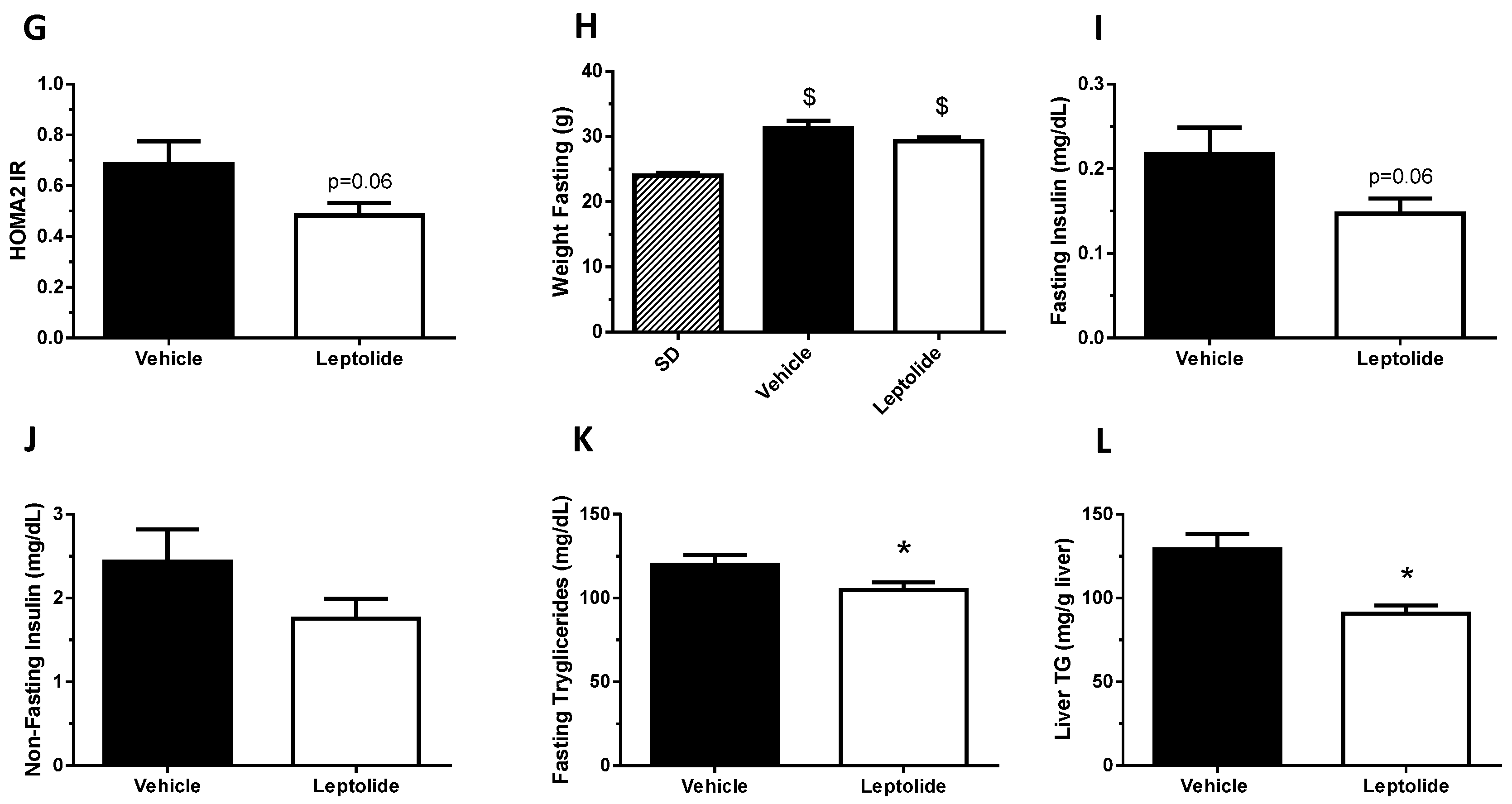
“Acute” administration of leptolide was performed in 12-week-old males fed a standard diet (SD) (33% protein; 58% carbohydrate; 9% fat) (#V1535, Ssniff, Soest, Germany) at the indicated doses (0.1 mg/kg and 1 mg/kg of body weight). “Chronic” administration of leptolide was performed in 6-week-old male mice fed a 60% kcal high fat diet (HFD) (20% protein; 20% carbohydrate; 60% fat) (#D12492, Research Diets, New Brunswick, NJ, USA) for 10 weeks. After 6 weeks of feeding with the HFD, mice were randomly divided into two groups, which were treated with once-daily ip injection of leptolide (0.1 mg/kg of body weight) or vehicle (DMSO) for another 4 weeks. All mice were maintained on HFD during the 4-week treatment. The day before sacrifice, mice were fasted overnight, followed by an ip insulin or saline injection, and 10 min later, mice were euthanized for liver and skeletal muscle tissues dissection as described previously [
24].
Fasting or non-fasting blood was collected from the tail vein into capillary tubes precoated with potassium-EDTA (Sarstedt, Nümbrecht, Germany) for the preparation of plasma or the determination of blood glucose levels using the Breeze 2 glucometer (Bayer, Leverkusen, Germany) as previously described [
24]. Insulin levels were measured using the ultrasensitive mouse ELISA assay (Mercodia, Uppsala, Sweden). Triglycerides were measured using a triglycerides kit (Biosystems, Barcelona, Spain). Cytokines and leptin were measured using Bio-Plex Luminex Immunoassays (Bio-Rad, Hercules, CA, USA). The detection limits of TNF-α, IL-1, IL6, insulin and leptin were 4.0 pg/mL, 1.6 pg/mL, 0.25 pg/mL, 0.025 pg/mL and 4.9 pg/mL respectively.
2.3. Glucose and Insulin Tolerance Tests, Glucose Decay and HOMA Indexes
The intraperitoneal glucose tolerance test (ip-GTT) was performed, 30 min after “acute” treatment or 4 weeks after “chronic” treatment, as previously described [
24]. Briefly, mice were fasted overnight (15 h) following the recommended standard operating procedure for phenotyping mice by the Eumorphia Consortium [
25]. Afterwards, mice were intraperitoneally injected with 2 g glucose/kg of body weight. Blood glucose levels were determined at 0, 15, 30, 60 and 120 min and plotted as a function of time. Likewise, the insulin tolerance test (ip-ITT) was performed, 30 min after “acute” treatment or 4 weeks after “chronic” treatment, as previously described [
24]. For ip-ITT, non-fasted mice were injected (1 U/kg of body weight) with insulin (Lilly, Indianapolils, IN, USA). Blood glucose levels were determined at 0, 15, 30, 60 and 90 min and plotted as a function of time. Ip-GTT and ip-ITT experiments were performed using the same group of mice. The timeline of the experiments was first the ip-GTT assays. Then, we let mice recover for three days, and ip-ITT experiments were performed. Glucose decay was calculated from the glucose measurements obtained during the insulin tolerance test. The measurements were converted into natural logarithm (Ln); the slope was calculated using linear regression (time × Ln[glucose]) and multiplied by 100 to obtain the glucose decay constant rate per minute (%/min).
The homeostasis model assessment (HOMA) estimates steady state beta cell function (%B), insulin sensitivity (%S), the inverse of %S and the insulin resistance (HOMA-IR). The HOMA Calculator software is freely available at the University of Oxford, United Kingdom, at the web page
www.dtu.ox.ac.uk/homacalculator.
2.4. Western-Blot Analysis
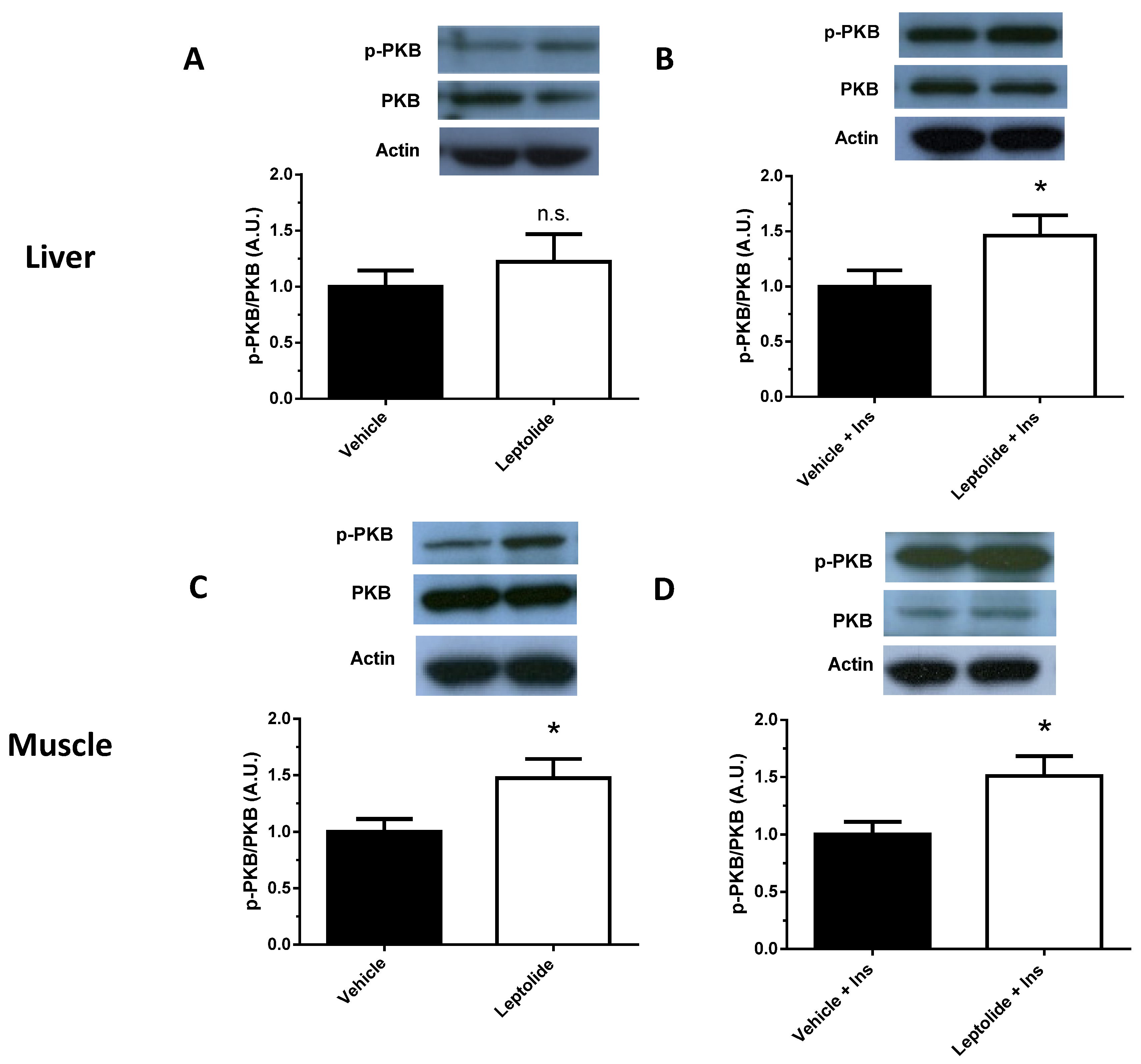
HepG2 cells were preincubated in the presence or absence of leptolide at the above indicated times and doses. At the end of the incubation period, culture media were discarded and cells collected and washed with ice-cold PBS, followed by homogenization in lysis buffer (20 mmol/L Tris·HCl, pH 7.5, 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% (vol/vol) Triton X-100, 2.5 mmol/L sodium pyrophosphate, 1 mmol/L µ-glycerophosphate, 1 mmol/L Na3VO4, 1 µg/mL leupeptin and 1 mmol/L phenylmethylsulfonyl fluoride) plus protease inhibitors (Protease Inhibitor Cocktail; Sigma). After 10 min on ice, extracts were sonicated and centrifugated at 18,000× g for 10 min at 4 °C. Pellets were discarded, and solubilized proteins (~20–40 µg/sample) were resolved by 10% SDS-PAGE for anti-p-PKB (Ser473) (1:1000; Cell Signaling, Danvers, MA, USA) and electrotransferred onto polyvinylidene difluoride filters (PDVF Immobilon-P membrane (Millipore, Billerica, MA, USA)) for immunoblotting by conventional means. After probing with specific antibodies, the membranes were stripped and reprobed with antibody against actin (1:3000; Sigma) and PKB (1:1000; Cell Signaling). Signals were detected by chemiluminescence (Immun-Start Western Chemiluminescence Kit; Bio-Rad, Madrid, Spain), and band densitometry was quantified with Image J software (National Institutes of Health, Bethesda, MD, USA).
For animal tissues, liver and skeletal muscle from mice, stimulated or not with insulin, were homogenized with a polytron (OMNI, Kennesaw, GA, USA) in cell lysis buffer (Cell Signaling, USA) in the presence of protease/phosphatase inhibitors. As described for HepG2 cells, ~40–60 µg/sample were resolved in 10%-SDS PAGE for anti-pPKB, PKB and actin.
2.5. Statistical Analysis
Statistical analysis of data was performed using the GraphPad Prism Software 6.0 (La Jolla, CA, USA). Distributions were checked with the Kolmogorov–Smirnov test. Data are presented as the means ± S.E.M. Homogeneity of variance was performed using the Levene test. Comparisons between two groups were done using the unpaired Student’s t-test (if homogeneity of variance) or the Welch test (if heterogeneity of variance) when a variable was distributed normally; in the case of a non-parametric variable, the Mann–Whitney U-test was used. Comparisons between more than two groups were done using the one-way ANOVA or the Kruskal–Wallis test when a variable was distributed normally or non-normally, respectively. For post-hoc analyses, the Bonferroni test or Dunnett’s test was used if there was homogeneity or heterogeneity of variance, respectively. Differences were considered significant at p < 0.05.
4. Discussion
T2DM is a metabolic disease characterized by insulin resistance, which may be joined with reduced insulin production and secretion. New drugs for T2DM treatment should include the capability of improving insulin sensitivity and protect functional beta-cell mass. We have demonstrated that furanocembranolides are promising molecules in the treatment of T2DM. Thus, we have shown that furanocembranolides induce beta-cell proliferation and protection, maintaining functional beta-cell mass and insulin production in type 1 diabetes [
22,
23]. In this work, we have shown that leptolide acts as an insulin sensitizer, improving insulin sensitivity and intracellular insulin signaling in the liver and muscle of obese mice.
Obesity is associated with insulin resistance [
26]. In this work, we have shown that leptolide reduced weight gain, which was parallel with lower pro-inflammatory circulating cytokines and leptin. These effects may mediate one of the molecular mechanisms by which leptolide improved insulin sensitivity in skeletal muscle and liver tissues. In this line of thinking, we have previously demonstrated that epoxypukalide, a member of the furanocembranolide family, protected primary rat β-cell cultures from a cocktail of pro-inflammatory cytokines including IL-1β, IFN-γ and TNF-α [
22].
T2DM is a complex disease that hardly can be managed using mono-therapeutic approaches and often requires double or triple therapeutic combinations depending on the disease progression [
1,
27]. In addition, the comorbidities associated with T2DM, such as cardiovascular disease, require specific medication. Thus, polymedicated T2DM patients are more susceptible to lower adherence to treatment, increased risk of harmful drug interactions and increased healthcare spending [
28]. In this line of argumentation, the discovery and development of a new class of drugs with pleiotropic effects are highly relevant for T2DM management.
Leptolide is an exceptional drug that can enhance the post-receptor intracellular insulin signaling cascade and increases beta-cell proliferation [
22]. These characteristics make leptolide an attractive molecule to explore the possibility of overcoming polymedication in T2DM patients. Further studies are warranted for the determination of the optimum drug dosage, timing of dosages, systemic bioavailability and the route of administration to enhance leptolide activity in vivo.
The functional activity of this family of natural compounds on mammalian cells is mostly unknown. Our data suggest that one potential mechanism of action of these compounds is through the activation of the insulin signaling pathway in multiple tissues. Upon binding of insulin, the kinase domains of the insulin receptor (IR) are activated by autophosphorylation on tyrosine residues, resulting in tyrosine phosphorylation of insulin receptor substrate (IRS) proteins [
29]. In the liver, IRS2 is important for the integration of the insulin signal and the metabolic control of the hepatocytes [
30]. Phosphorylated-IRS proteins allow the association and activation of phosphatidylinositol 3-kinase (PI3K), leading to the production of phosphatidylinositol-3,4,5-triphosphate (PIP3), a lipid second messenger located on the plasma membrane. PIP3 allows the recruitment and activation of 3-phosphoinositide-dependent protein kinase 1 (PDK1) and serine/threonine protein kinase AKT (also known as PKB) [
29]. These proteins (IRS, PI3K and PKB) are considered three critical nodes of the canonical insulin receptor signal transduction network [
31]. In the liver, AKT2 is one of the major mediators of the metabolic effects of insulin [
32]. For this reason, we evaluated the phosphorylation levels of PKB in obese mice.
Here, we show that leptolide enhances the phosphorylation of PKB in liver and skeletal muscle tissues of a preclinical model of insulin resistance. However, the insulin signaling pathway also regulates cell growth and differentiation, emanating from the IRS node. The regulation of these processes is mediated by the Raf/Ras/MEK/MAPK (mitogen-activated protein kinase, also known as ERK or extracellular signal regulated kinase) pathway [
31]. Interestingly, we have previously demonstrated that epoxypukalide is able to induce the ERK1/2 pathway, but not PKB in pancreatic beta-cells [
22].
Leptolide and epoxypukalide possess the same carbon skeleton and contain the same macrocycle. The only structural difference between them lies at C-18, which is oxidized to a methyl ester in epoxypukalide and to an aldehyde in leptolide [
22,
33]. Our results suggest that this difference is responsible for their ability to activate different pathways depending on cell type and cell environment. Slight variations in functional groups of furanocembranolides have been reported to make a difference in their activities [
14,
17]. The way these molecules enter the cell or activate signaling pathways is not known yet. Further research is necessary to address these open questions.
In conclusion, our findings demonstrate the feasibility of furanocembranolides as a new therapeutic strategy to treat T2DM.

 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






