Benderamide A, a Cyclic Depsipeptide from a Singapore Collection of Marine Cyanobacterium cf. Lyngbya sp.

Abstract
:1. Introduction
2. Results
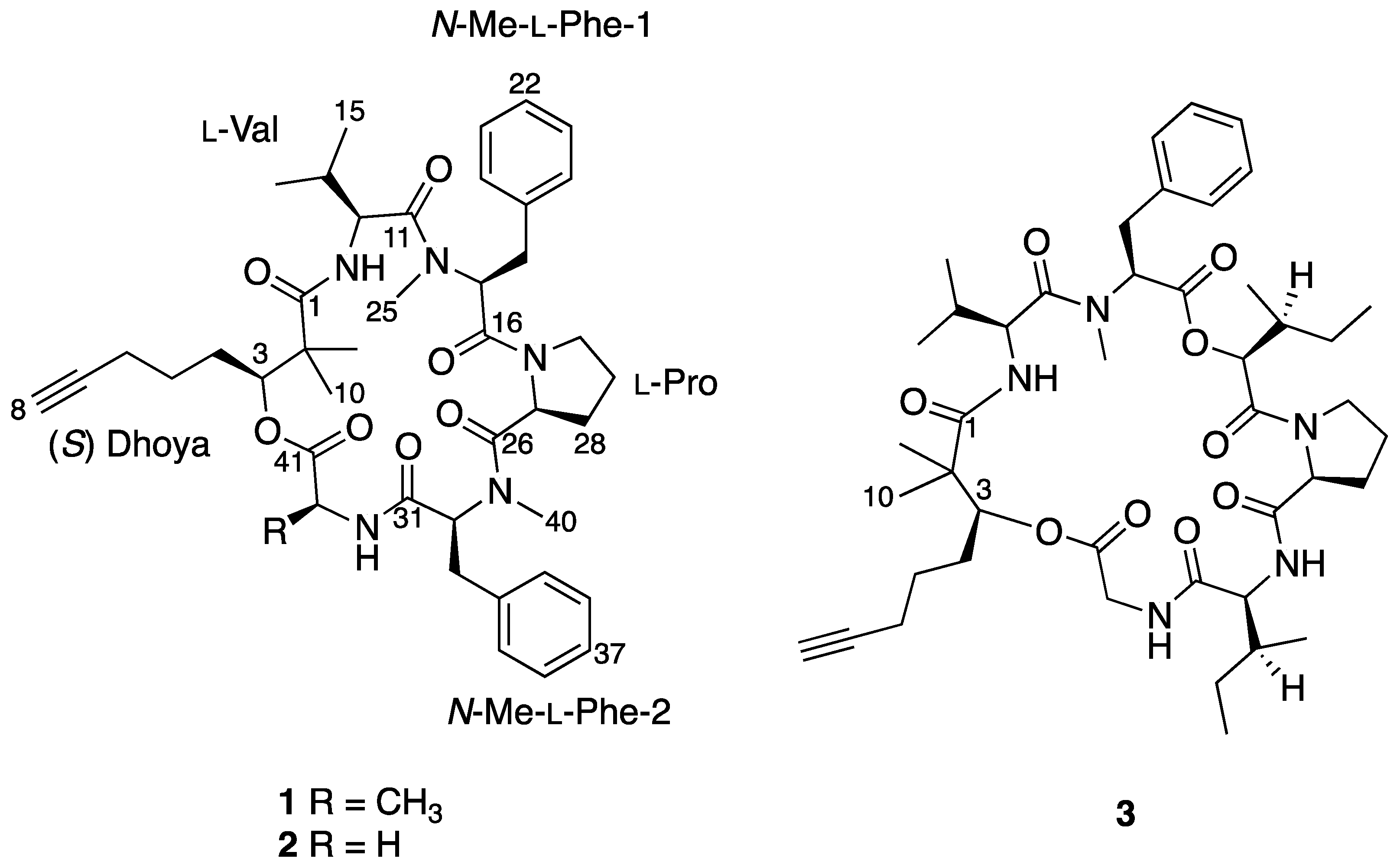
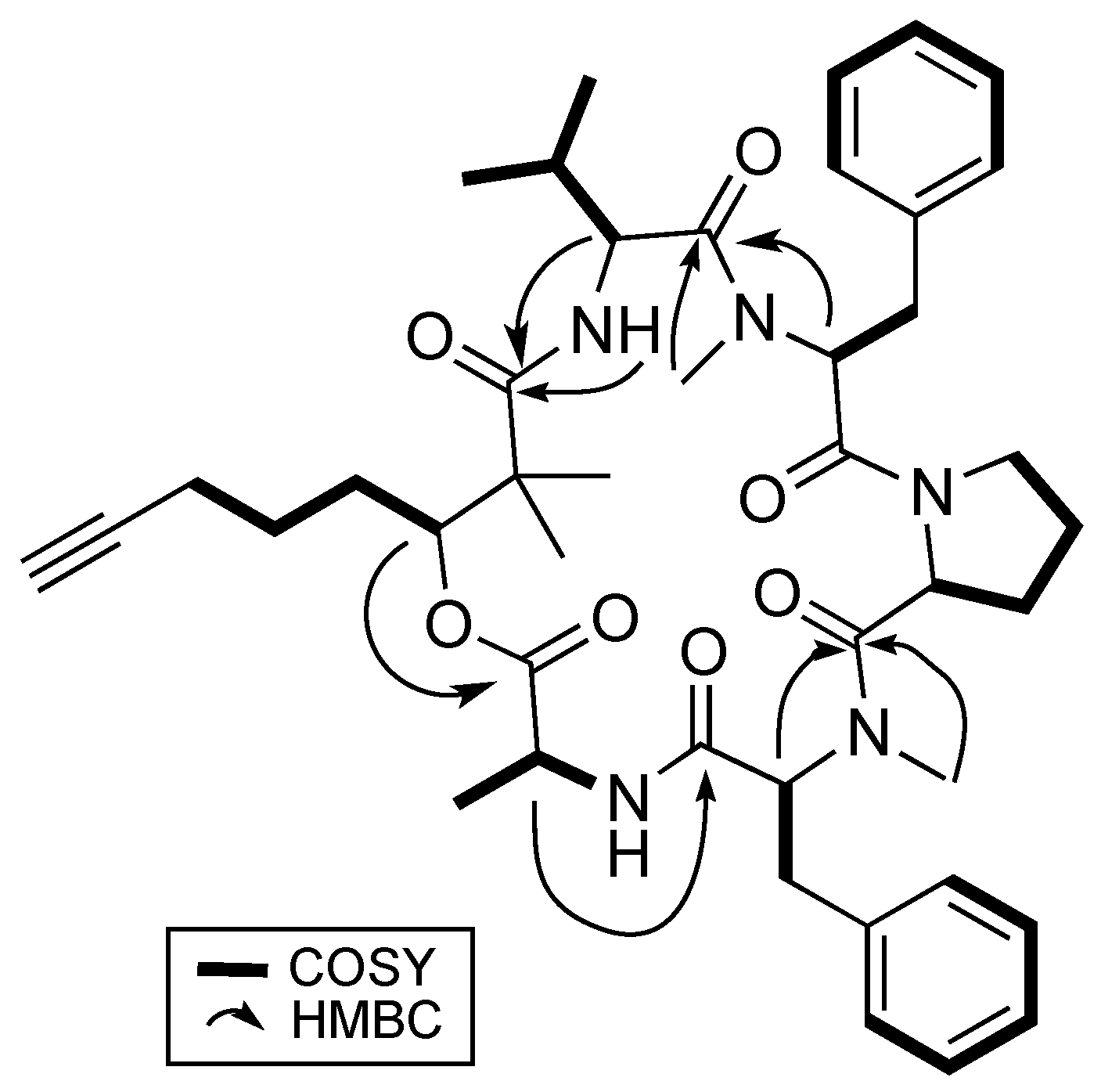
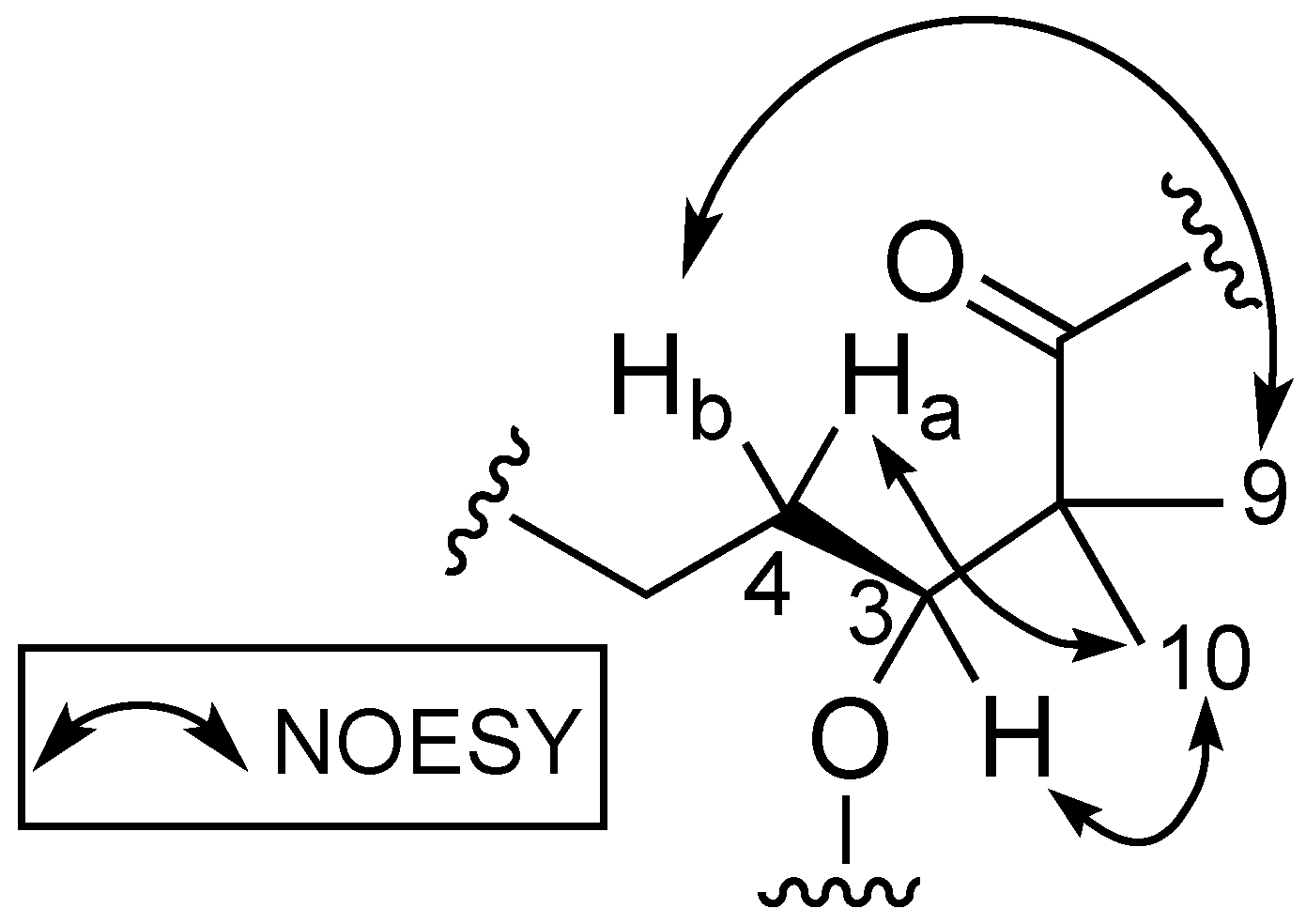
2.1. Structural Elucidation of 1
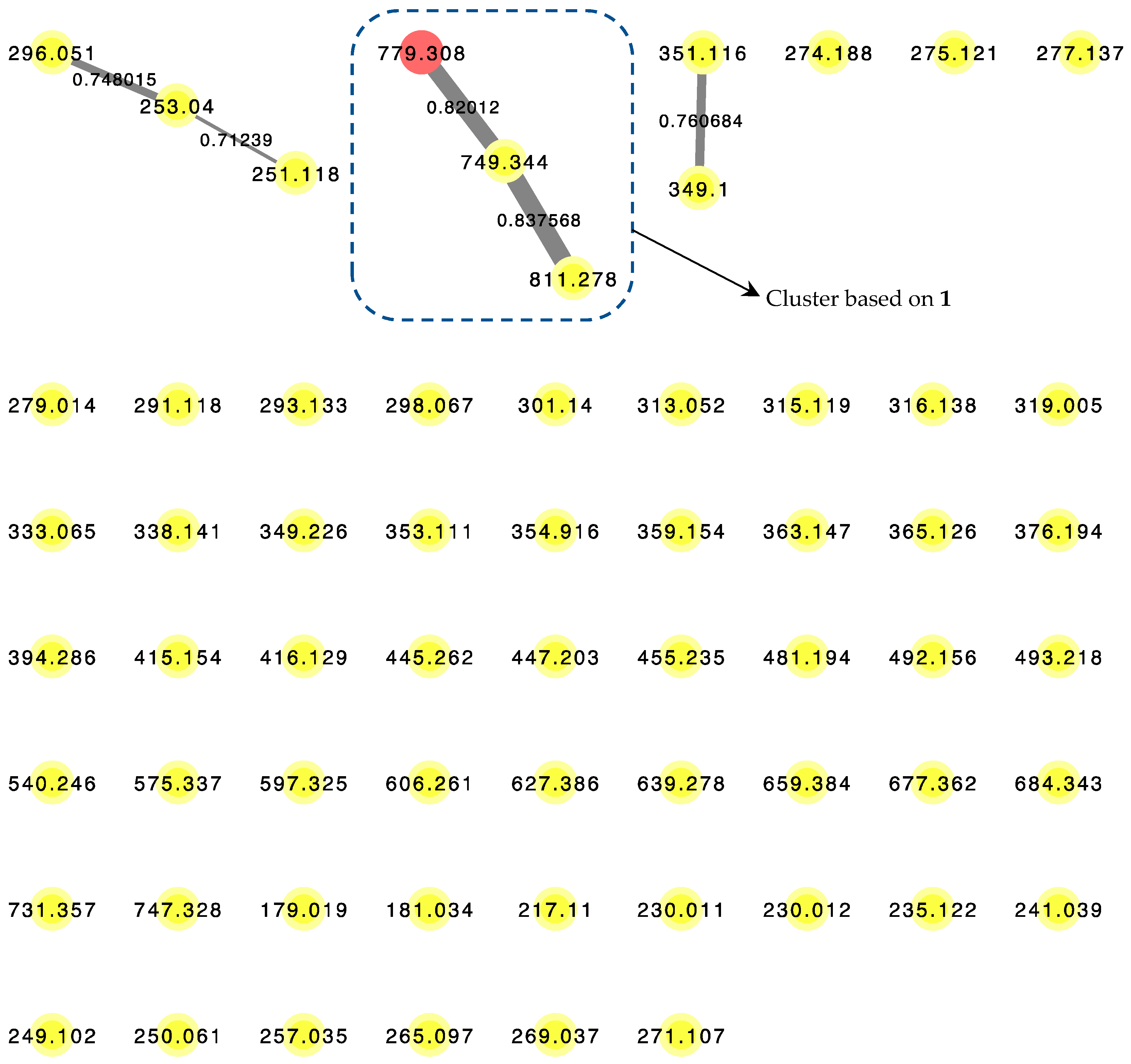
2.2. LC–MS/MS Molecular Networking of Benderamide A-Containing VFC Fraction
2.3. Biological Activity of Benderamide A (1)
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Biological Material
3.3. Extraction and Isolation
3.4. Advanced Marfey’s Analysis of Amino Acids
3.5. Molecular Networking
3.6. IncuCyte Live Cell Analysis Imaging System
3.7. Cell Titer-Glo and Caspase 3/7-Glo Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tan, L.T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry 2007, 68, 954–979. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, P.D.; Byrum, T.; Liu, W.T.; Dorrestein, P.C.; Gerwick, W.H. Viequeamide A, a cytotoxic member of the kulolide superfamily of cyclic depsipeptides from a marine button cyanobacterium. J. Nat. Prod. 2012, 75, 1560–1570. [Google Scholar] [CrossRef] [PubMed]
- Reese, M.T.; Gulavita, N.K.; Nakao, Y.; Hamann, M.T.; Yoshida, W.Y.; Coval, S.J.; Scheuer, P.J. Kulolide: A cytotoxic depsipeptide from a cephalaspidean mollusk, Philinopsis speciosa. J. Am. Chem. Soc. 1996, 118, 11081–11084. [Google Scholar] [CrossRef]
- Nakao, Y.; Yoshida, W.Y.; Szabo, C.M.; Baker, B.J.; Scheuer, P.J. More peptides and other diverse constituents of the marine mollusk Philinopsis speciosa. J. Org. Chem. 1998, 63, 3272–3280. [Google Scholar] [CrossRef]
- Gunasekera, S.P.; Owle, C.S.; Montaser, R.; Luesch, H.; Paul, V.J. Malyngamide 3 and cocosamides A and B from the marine cyanobacterium Lyngbya majuscula from Cocos Lagoon, Guam. J. Nat. Prod. 2011, 74, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Almaliti, J.; Malloy, K.L.; Glukhov, E.; Spadafora, C.; Gutierrez, M.; Gerwick, W.H. Dudawalamides A-D, antiparasitic cyclic depsipeptides from the marine cyanobacterium Moorea producens. J. Nat. Prod. 2017, 80, 1827–1836. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Puddick, J.; Prinsep, M.R.; Lee, P.P.F.; Tan, L.T. Hantupeptin A, a cytotoxic cyclic depsipeptide from a Singapore collection of Lyngbya majuscula. J. Nat. Prod. 2009, 72, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Sitachitta, N.; Williamson, R.T.; Gerwick, W.H. Yanucamides A and B, two new depsipeptides from an assemblage of the marine cyanobacteria Lyngbya majuscula and Schizothrix species. J. Nat. Prod. 2000, 63, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2018, 35, 8–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mevers, E.; Liu, W.T.; Engene, N.; Mohimani, H.; Byrum, T.; Pevzner, P.A.; Dorrestein, P.C.; Spadafora, C.; Gerwick, W.H. Cytotoxic veraguamides, alkynyl bromide-containing cyclic depsipeptides from the marine cyanobacterium cf. Oscillatoria margaritifera. J. Nat. Prod. 2011, 74, 928–936. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Ikai, Y.; Oka, H.; Suzuki, M.; Harada, K.-I. A nonempirical method using LC/MS for determination of the absolute configuration of constituent amino acids in a peptide: Combination of Marfey’s method with mass spectrometry and its practical application. Anal. Chem. 1997, 69, 5146–5151. [Google Scholar] [CrossRef]
- Péter, A.; Olajos, E.; Casimir, R.; Tourwé, D.; Broxterman, Q.B.; Kaptein, B.; Armstrong, D.W. High-performance liquid chromatographic separation of the enantiomers of unusual α-amino acid analogues. J. Chromatogr. A 2000, 871, 105–113. [Google Scholar] [CrossRef]
- Luesch, H.; Pangilinan, R.; Yoshida, W.Y.; Moore, R.E.; Paul, V.J. Pitipeptolides A and B, new cyclodepsipeptides from the marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2001, 64, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Naman, C.B.; Rattan, R.; Nikoulina, S.E.; Lee, J.; Miller, B.W.; Moss, N.A.; Armstrong, L.; Boudreau, P.D.; Debonsi, H.M.; Valeriote, F.A.; et al. Integrating molecular networking and biological assays to target the isolation of a cytotoxic cyclic octapeptide, samoamide A, from an American Samoan marine cyanobacterium. J. Nat. Prod. 2017, 80, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Neta, P.; Farahani, M.; Simón-Manso, Y.; Liang, Y.; Yang, X.; Stein, S.E. Unexpected peaks in tandem mass spectra due to reaction of product ions with residual water in mass spectrometer collision cells. Rapid Commun. Mass Spectrom. 2014, 23, 2645–2660. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Shiota, I.; Sumimoto, S.; Matsubara, T.; Sato, T.; Suenaga, K. Kohamamides A, B, and C, cyclic depsipeptides from an Okeania sp. marine cyanobacterium. J. Nat. Prod. 2017, 80, 1948–1952. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Goeger, D.; Maier, C.S.; Gerwick, W.H. The Wewakpeptins, cyclic depsipeptides from a Papua New Guinea collection of the marine cyanobacterium Lyngbya semiplena. J. Org. Chem. 2005, 70, 3133–3139. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Unit | C/H No. | δC, Multi. | δH (J in Hz) | COSY | HMBC a |
|---|---|---|---|---|---|
| Dhoya | 1 | 176.6, C | |||
| 2 | 146.1, C | ||||
| 3 | 177.4, CH | 5.20, dd (11.0, 2.0) | H-4a, H-4b | 1, 2, 4, 5, 9, 41 | |
| 4a | 127.9, CH2 | 1.73–1.78, m | H-4b, H-5 | 2, 5, 6 | |
| 4b | 1.54, d (6.9) | H-3, H-4a | 5 | ||
| 5 | 124.7, CH2 | 1.49–1.44, m | H-4a, H-6 | 3, 4, 6, 7 | |
| 6 | 118.3, CH2 | 2.21, td (6.7, 2.6) | H-5 | 4, 5, 7, 8 | |
| 7 | 183.7, C | ||||
| 8 | 169.3, CH | 1.95, t (2.5) | 6 | ||
| 9 | 117.7, CH3 | 1.27, br. s | 1, 2, 3 | ||
| 10 | 123.6, CH3 | 1.19, br. s | 1, 2, 3 | ||
| Val | 11 | 172.3, C | |||
| 12 | 155.7, CH | 4.28, dd (9.5, 7.5) | NH, H-13 | 1, 11, 14, 15 | |
| 13 | 130.2, CH | 1.91–1.85, m | H-12, H-14, H-15 | 12, 15 | |
| 14 | 119.5, CH3 | 0.98, d (6.7) | H-13 | 12, 13, 15 | |
| 15 | 118.8, CH3 | 0.92, d (6.7) | H-13 | 12, 13, 14 | |
| NH | 5.70, d (7.2) | H-12 | 1, 12 | ||
| N-Me-Phe-1 | 16 | 169.2, C | |||
| 17 | 154.4, CH | 5.06, dd (12.1, 3.4) | H-18a, H-18b | 11, 16, 18, 19, 25 | |
| 18a | 138.0, CH2 | 3.17, s | H-17, H-18b | 16, 17, 19, 20, 24 | |
| 18b | 3.00–2.95, m | H-17 | 16, 17, 19, 20, 24 | ||
| 19 | 137.8, C | ||||
| 20/24 | 130.1, CH | 7.42, m | H-21/23 | 18, 21, 22, 23 | |
| 21/23 | 129.0, CH | 7.22–7.19, m | H-20/24, H-22 | 19, 20, 21, 23, 24 | |
| 22 | 127.2, CH | 7.19–7.14, m | H-21/23 | 20, 21, 23, 24 | |
| 25 | 132.4, CH3 | 3.59, s | 11, 17 | ||
| Pro | 26 | 171.7, C | |||
| 27 | 156.4, CH | 3.15–3.09, m | H-28a | 28, 29 | |
| 28a | 129.9, CH2 | −0.02–(−0.10), m | H-28a, H-29a | 26, 27 | |
| 28b | 0.55–0.47, m | H-28b | 26, 27 | ||
| 29a | 122.3, CH2 | 0.87–0.83, m | H-29a | 27, 28 | |
| 29b | 1.19, br. s | H-27, H-28a, H-28b, H-30a, H-30b | 30 | ||
| 30a | 146.4, CH2 | 3.23–3.19, m | H-29a | 29 | |
| 30b | 3.40–3.33, m | H-29a | 28, 29 | ||
| N-Me-Phe-2 | 31 | 169.9, C | |||
| 32 | 163.4, CH | 3.95, dd (9.0, 4.4) | H-33a, H-33b | 26, 31, 33, 34, 40 | |
| 33a | 135.4, CH2 | 2.75–2.68, m | H-32, H-33a | 31, 32, 34, 35, 39 | |
| 33b | 3.63, d (4.0) | H-32 | 31, 32, 34, 35, 39 | ||
| 34 | 138.4, C | ||||
| 35/39 | 129.8, CH | 7.06, m | H-36/38 | 33, 36, 37, 38 | |
| 36/38 | 128.7, CH | 7.22–7.19, m | H-35/39 | 34, 36, 38 | |
| 37 | 127.2, CH | 7.19–7.14, m | H-37 | 35, 36, 38, 39 | |
| 40 | 131.1, CH3 | 2.79, s | 26, 32 | ||
| Ala | 41 | 168.9, C | |||
| 42 | 148.8, CH | 4.99, dd (9.0, 7.0) | NH, H-43 | 31, 41, 43 | |
| 43 | 118.4, CH3 | 1.54, d (6.9) | H-42 | 41, 42 | |
| NH | 9.03, d (9.1) | H-42 | 31, 32, 41, 42, 43 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, C.Y.G.; Ong, J.F.M.; Goh, H.C.; Coffill, C.R.; Tan, L.T. Benderamide A, a Cyclic Depsipeptide from a Singapore Collection of Marine Cyanobacterium cf. Lyngbya sp. Mar. Drugs 2018, 16, 409. https://doi.org/10.3390/md16110409
Ding CYG, Ong JFM, Goh HC, Coffill CR, Tan LT. Benderamide A, a Cyclic Depsipeptide from a Singapore Collection of Marine Cyanobacterium cf. Lyngbya sp. Marine Drugs. 2018; 16(11):409. https://doi.org/10.3390/md16110409
Chicago/Turabian StyleDing, Chi Ying Gary, Ji Fa Marshall Ong, Hui Chin Goh, Cynthia R. Coffill, and Lik Tong Tan. 2018. "Benderamide A, a Cyclic Depsipeptide from a Singapore Collection of Marine Cyanobacterium cf. Lyngbya sp." Marine Drugs 16, no. 11: 409. https://doi.org/10.3390/md16110409
APA StyleDing, C. Y. G., Ong, J. F. M., Goh, H. C., Coffill, C. R., & Tan, L. T. (2018). Benderamide A, a Cyclic Depsipeptide from a Singapore Collection of Marine Cyanobacterium cf. Lyngbya sp. Marine Drugs, 16(11), 409. https://doi.org/10.3390/md16110409