1. Introduction
Soft corals of the genus
Sinularia, belonging to the phylum Cnidaria, class Alcyonaria, and family Alcyoniidae, are a prolific source of structurally diverse (such as diterpenoids, polyhydroxylated steroids, alkaloids, polyamines, etc.) and biologically active metabolites. These organisms have been the subjects of intensive research by natural product chemists and pharmacologists for their chemical diversities and broad pharmacological activity, such as cytotoxic, antiviral, and anti-inflammatory properties [
1,
2]. To date, more than 50 species of
Sinularia have been chemically investigated, with over 500 diverse and bioactive metabolites being elucidated [
3].
In the course of our ongoing project searching for novel and bioactive compounds from Chinese soft corals [
3,
4,
5,
6,
7,
8,
9], the title animal
Sinularia polydactyla was collected from the Xisha Islands in the South China Sea, Hainan Province, China. Among all of the
Sinularia species,
S. polydactyla in the Red Sea has been extensively investigated, while there is only one report regarding the chemical constituents of
S. polydactyla from the South China Sea, with only two sesquiterpenoids being characterized [
3]. Our previous chemical investigation of
S. polydactyla revealed the rich chemical diversity of this biological material and led to the discovery of three uncommon novel diterpenes with unprecedented carbon skeletons (
1–
3) [
3] displaying an interesting dose-dependent promotion effect on ConA-induced T lymphocyte proliferation. Stimulated by this discovery, and in order to find more chemically interesting and biologically active metabolites, especially trace components, we recently carried out a further chemical investigation of the Et
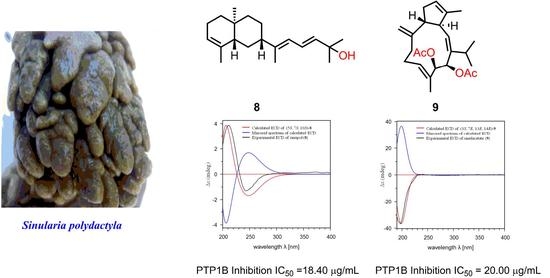
2O-soluble fraction of the title animal. This investigation resulted in the isolation and characterization of a new prenyleudesmane diterpene, sinupol (
8), and a new capnosane diterpene, sinulacetate (
9), together with five known related diterpenoids (
4–
7 and
10) (
Figure 1). The structure, including the stereochemistry of sinulacetate (
9), was determined by a comparison of the NMR data with those of the model compounds
11–
14. Described herein are the isolation, structure elucidation, and PTP1B inhibitory activity of these newly isolated compounds.
2. Results
On the basis of a detailed analysis of all of the isolated molecules and by comparison of their
1H NMR and
13C NMR spectral data and [α]
D values with those reported in the literature,
4–
7 and
10 were readily identified as a lobane-type diterpenoid, loba-8,10,13(15)-triene-17,18-diol (
4) [
10], lobatetraene (
5) [
10], fuscol (
6) [
9,
10],
7, an unnamed prenyleudesmane diterpene [
11], and sarcophytol B (
10) [
12], respectively.
Sinupol (
8) was obtained as a colorless oil. Its molecular formula was established as C
20H
32O by (+)-HR-EIMS, indicating five degrees of unsaturation in the molecule. The
13C NMR and HSQC spectra of
8 exhibited the presence of 20 carbon resonances, including five methyl groups, five sp
3 methylene, two sp
3 methine, two sp
3 quaternary, four sp
2 methine, and two sp
2 quaternary carbons. A detailed comparison of the NMR data of
8 with those of
7 suggested that
8 is a 1-dehydroxyl product of
7 for the identical chemical shifts in the side chains and significant carbon signal differences (
δC-1 76.9,
δC-2 33.0, and
δC-10 38.5 for
7,
δC-1 38.1,
δC-2 23.2, and
δC-10 32.4 for
8) around C-1. To fully elucidate the structure of
8, its two-dimensional (2D) NMR spectra were extensively analyzed. The
1H and
13C NMR resonances revealed the presences of one disubstituted and two trisubstituted double bonds, including a conjugated diene fragment (
Figure 2). Since the three double bonds accounted for three degrees of unsaturation, the remaining two degrees of unsaturation were attributed to a bicyclic ring system in
8. Further analysis of the
1H–
1H COSY spectrum revealed the presence of three coupling systems (
a–
c) from H
2-1 to H-3, H-5 to H
2-9, and H-12 to H-14 (
Figure 2).The key HMBC correlations from H
3-16 to C-3/C-4/C-5, from H
3-17 to C-1/C-5/C-10/C-9, and from H
2-6 to C-4/C-5/C-10/C-11 confirmed the presence of the bicyclic ring moiety. The side chain was connected to C-7 of the decalin ring moiety through C-11, and was supported by crosspeaks from H
3-18 to C-7/C-11/C-12 and from H
3-19/H
3-20 to C-14/C-15 in the HMBC spectrum. Thus, the planar structure of
8 was confirmed (
Figure 2).
As for the relative configuration of
8, the
E geometry of Δ
11,12 and Δ
13,14 was established by the NOE correlation from H-13 to CH
3-18 and the coupling constants between H-13 (
δH 6.96, dd,
J = 11.2, 15.3 Hz) and H-14 (
δH 6.13, d,
J = 15.3 Hz) (
Table 1). On the basis of the large coupling constant of H-5 (
δH 1.90, dd,
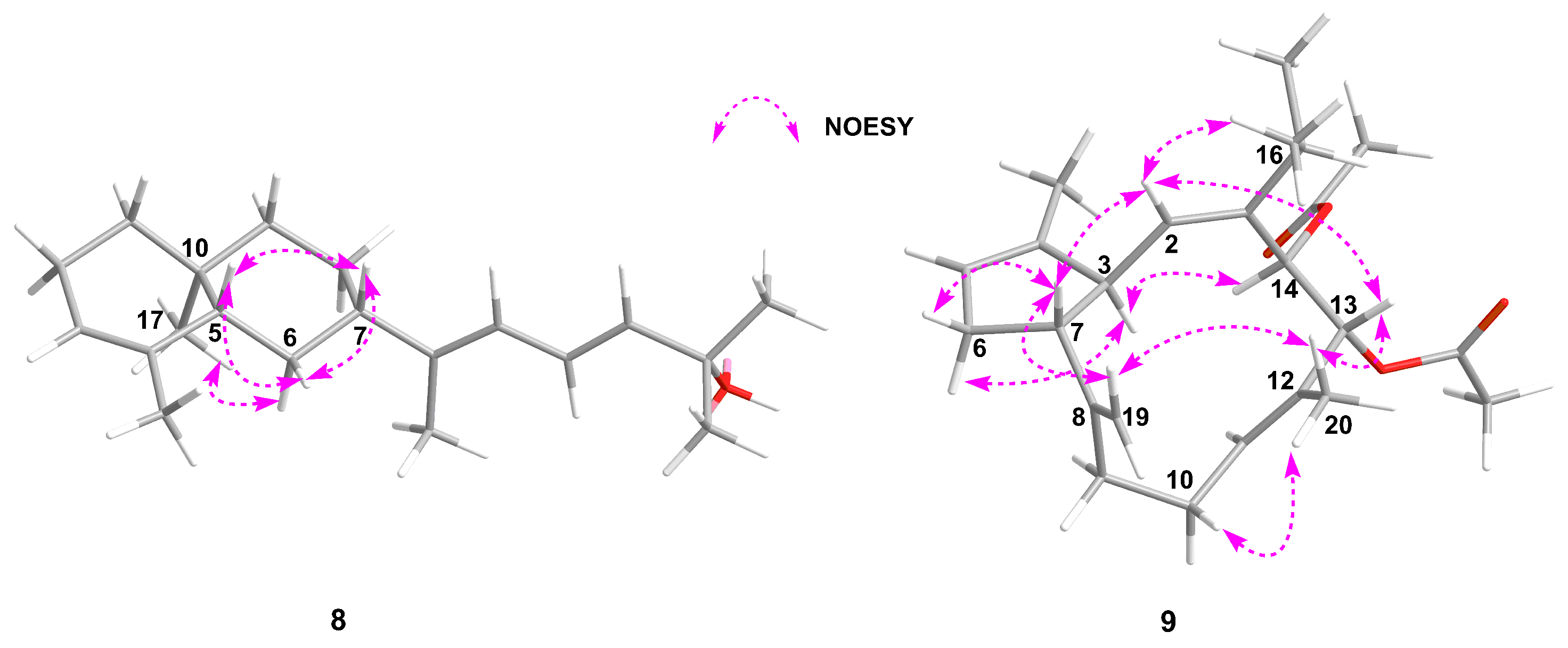
J = 12.7, 1.1 Hz), the proton of H-5 was determined as an axial orientation. The correlations from H-6β to H-5 and H-7 in the NOESY spectrum defined the
cis-configuration of H-5 and H-7 (
Figure 3). Additionally, the H
3-17 correlated with H-6α, which confirmed the
trans-configuration between CH
3-17 and H-5 (
Figure 3). The relative configuration of the C-5, C-7, and C-10 in
8 was thus confirmed as
S*,
S*,
S*, respectively.
Sinulacetate (
9) was obtained as a colorless oil. An (+)-HR-EIMS data analysis of
9 indicated the molecular formula C
24H
34O
4, which suggested the presence of eight degrees of unsaturation. By inspection of the
13C and
1H NMR spectra, the presence of two acetoxyl groups was immediately recognized (
δC 169.8 and 170.5, respectively). Eight carbon signals in the downfield implied the occurrence of four double bonds in the molecule, including one terminal sp
2 methylene (
δC 113.3). The remaining two degrees of unsaturation were due to the presence of two fused rings in
9. Detailed analysis of the
1H–
1H COSY spectrum of
9 readily characterized three coupling systems (
a–
c) (
Figure 2) by the strong correlations of H-2/H-3/H-7/H
2-6/H-5, H
2-9/H
2-10/H-11, and H-13/H-14, respectively. The key HMBC correlations from H
3-18 to C-3/C-5, H-7 to C-2/C-3/C-8/C-19, H
2-19 to C-8/C-9, H
2-10 to C-12, H-11 to C-20/C-13, H
3-20 to C-11/C-12/C-13, H-14 to C-1/C-13/C-12, H-15 to C-1/C-2/C-14, and H
3-16/H
3-17 to C-15/C-1 confirmed the connections of these coupling systems. In view of the above evidences,
9 belongs to a capnosane-type of diterpenoids. Further analysis of the HMBC crosspeaks from H-13 to the carbonyl group at
δC 169.8 and H-14 to the carbonyl at
δC 170.5 indicated that the acetoxyl acetate groups were on C-14 and C-13, respectively.
The relative stereochemistry of
9 was deduced by analysis of the NMR data and correlations from ROESY spectra. Correlations from H
2-10 to CH
3-20 and from H-2 to CH
3-16 in the ROESY spectrum confirmed the
E geometry of Δ
11,12 and the
Z geometry of Δ
1,2 in
9. A typical triplet with
JH–H values of 9.5 Hz of H-3 indicated that the dihedral angles between H-3 and H-7 and between H-3 and H-2 were approximately 0° or 180°. The ROESY correlations from H-6β to H-7 and from H-6α to H-3 implied the
trans-configuration of H-3 and H-7. The
thans-configuration of H-3 and H-7 in
9 was further confirmed by the similar multiplicity and coupling constants of H-3 and H-7 with those of sarcophytol L (
12) [
12], as well as the quite different coupling constants from those of trocheliophols H (
13) and I (
14) [
13] possessing the
cis-configuration of H-3 and H-7 (
Table 2). The absence of a crosspeak between H-3 and H-7 in the ROESY spectrum in
9 was also consistent with the above conclusion. As for the relative configurations at C-13 and C-14, the similar carbon chemical shift values between
9 (C-13 (
δC 77.1) and C-14 (
δC 73.6)) (
Table 1) and
11 (C-13 (
δC 77.8) and C-14 (
δC 74.2)) together with the similar coupling constants for protons of
9 (H-13, 4.95, d,
J = 10.2 Hz; H-14, 6.02, d,
J = 10.2 Hz) and
11 (H-13, 5.34, d,
J = 10.0 Hz; H-14, 6.15, d,
J = 10.0 Hz) [
14] indicated the
threo relative stereochemistry at C-13 and C-14 in
9. Key crosspeaks from H
2-19 to H-7/H
3-20 and from H-13 to H-2/H
3-20 in the ROESY spectrum indicated the same orientation (
β) of the protons at H-7, H
2-19, H
3-20, H-13, and H-2 in the three-dimensional (3D) model (
Figure 3). The
α-orientation of H-14 was determined by the strong ROESY correlation from H-3 to H-14 (
Figure 3). Thus, the relative configuration of the carbons in
9 was determined as 3
S*, 7
R*,
13R*, 14
R*.
With the establishment of the relative configurations of the chiral carbons of
8 and
9, the remaining task was to determine their absolute configurations (ACs). Due to the minute amount of the new compounds and unsuccessful efforts on crystallization, a reliable and powerful approach, the time-dependent density functional theory electronic circular dichroism (TDDFT ECD) calculation was applied to establish their absolute configurations. As shown in
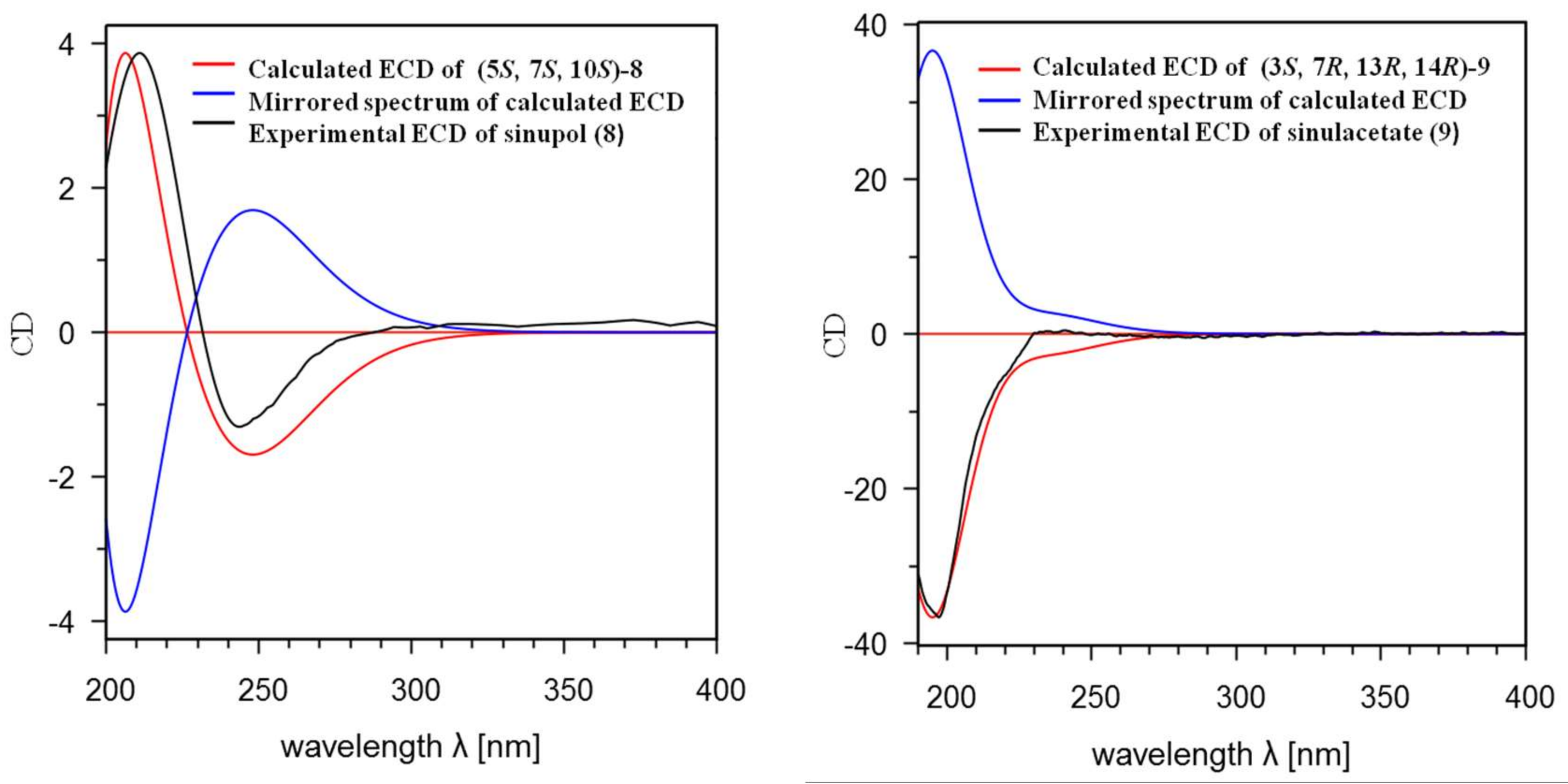
Figure 4, the experimental ECD spectrum (MeCN) of
8 showed a negative cotton effect (CE) at 234 nm (Δε −1.98) and a positive CE at 211 nm (Δε +4.83). The experimental ECD spectrum (MeCN) of
9 showed a negative cotton effect (CE) at 198 nm (Δε −2.56). The recognizable files for Schrödinger LLC, 2015-4 [
15] with the format of “mol2” were readily set out and then the initial torsional sampling (MCMM) and OPLS_2005 force field conformational searches of (5
S, 7
S, 10
S)-
8 and (3
S, 7
R,
13R, 14
R)-
9 were carried out by the Macromodel in Schrödinger LLC, 2015-4 [Ligprep, force field OPLS_2005; generate possible states at targets PH 7.0 ± 2.0; conformational search, force field OPLS_2005; dielectric constant, 1.0; method, torsional sampling (MCMM); customize the search with torsion sampling options: intermediate, with maximum number of steps: 1000, use 100 steps per rotatable bond; energy window, 21 KJ/mol; eliminate redundant conformers with maximum atom deviation cutoff: 0.5 Å; others were the default parameters in Schrödinger LLC, 2015-4], which afforded 67 and 15 conformers, respectively. The Boltzmann populations of the conformers were obtained based on the potential energy provided by the OPLS_2005 force field, leading to 20 conformers for
8 and eight conformers for
9 above 1% population for further optimization [
Supplementary Materials, Figures S1 and S3] with the output files format “sdf”. The above output files were transformed manually into the files with the format of “gjf” by text editor, which were recognizable for Gaussian 09 revision C.01 The re-optimization of the resulting geometries were all performed with Gaussian 09 revision C.01 [
16] at the B3LYP/6-311G(d, p) level with IEFPCM (the Polarizable Continuum Model (PCM) using the integral equation formalism variant solvent model for MeCN # opt freq B3LYP/6-311g(d, p) scrf (solvent = acetonitrile), the input model used in the calculation was provided (Scheme S1)), and frequency analysis was performed as well to confirm that the re-optimized geometries were at the energy minima. Differences among all of the conformers for each compound were further illustrated through the figures with superimposed conformers (
Figures S2 and S4). The specific splitting patterns and
1H-
1H coupling constants for the most populated conformer of
9 were also compared with experimental ones and a high similarity was observed (
Table S2 and
Figure S5). The ECD spectra for all re-optimized geometries were then obtained by the TDDFT calculations with Gaussian 09 Revision C.01 ((B3LYP/6-311G (d, p) scrf (solvent = acetonitrile) td (nstates = 30, singlets), the input model used in the calculation was also provided (Scheme S2). Finally, SpecDis1.62 software was used to obtain the Boltzmann-averaged ECD spectra of (5
S, 7
S, 10
S)-
8 and (3
S, 7
R, 13
R, 14
R)-
9, which highly matched the experimental ones, whereas those of their enantiomers showed completely opposite curves (
Figure 4). The calculated ECD spectrum for (5
S, 7
S, 10
S)-
8 showed a positive and a negative CE at 208 nm and 243 nm, respectively, which were highly similar to the experimental results. Similarly, the calculated ECD curve for (3
S, 7
R, 13
R, 14
R)-
9 displayed a negative CE at 201 nm, indicating that had almost the same CE as the experimental result for
9. Consequently, the absolute configurations of sinupol (
8) and sinulacetate (
9) were determined to be 5
S, 7
S,10
S and 3
S, 7
R, 13
R, 14
R, respectively
.Although sinupol (
8) and sinulacetate (
9) formally displayed different carbon skeletons from the co-occurred fuscol (
6) and sarcophytrol B (
10), they were structurally related as they shared some common moieties, such as the conjugated diene chains (for
8 and
6) as well as isopropyl groups and 14-membered rings (for
9 and
10). To explain their possible biogenetic relationships, plausible biosynthetic pathways were proposed (
Scheme 1). Compound
8 was proposed to be a cyclization product of fuscol (
6). This hypothesis was also in agreement with the experimental results reported by Barry and co-workers [
17]. A capnosane diterpenoid, sinulacetate (
9), with 5:11-fused carbobicyclic skeleton, could be derived from the co-occurring cembranoid
10. As outlined in
Scheme 1, the formation of a C-C bond between C-3 and C-7 followed by acetylization at C-13 and C-14 generated
9.
All of the isolates were evaluated for antibacterial and cytotoxic activities. Unfortunately, they were all inactive. Interestingly, in PTP1B (a recognized target for diabetes and obesity) inhibitory activity assay [
18], compounds
7,
8, and
9 exhibited inhibitory activity with IC
50 values of 75.5 μM, 63.9 μM, and 51.8 μM, respectively, as compared to the positive control of oleanolic acid (IC
50 = 2.56 μM).
4. Materials and Methods
4.1. General Experimental Procedures
All of the chemicals were obtained from commercial sources. Circular dichroism (CD) spectra were measured on a JASCO J-810 instrument (JASCO, Tokyo, Japan). Optical rotations were measured on Autopol VI Polarimeter (Rudolph, NJ, USA). NMR spectra were measured on a Bruker DRX-400 or Bruker DRX-500 spectrometer (BrukerBiospin AG, Fällanden, Germany). Chemical shifts (δ) are reported in ppm with reference to the solvent signals, and coupling constants (J) are in Hz. MS spectra were recorded on a Finnigan-MAT-95 mass spectrometer (FinniganMAT, San Jose, CA, USA). Commercial silica gel (Qingdao Haiyang Chemical Group Co., Ltd., Qingdao, China, 200–300 and 400–600 mesh), and Sephadex LH-20 gel (Amersham Biosciences, Little Chalfont, UK) were used for column chromatography, and precoated silica gel plates (Yan Tai Zi Fu Chemical Group Co., Yantai, China, G60 F-254) were used for analytical TLC (thin layer chromatography). Reversed-phase (RP) HPLC was performed on an Agilent 1260 series liquid chromatography equipped with a DAD G1315D detector at 210 and 254 nm. A semi-preparative ODS-HG-5 column (5 µm, 250 × 9.4 mm) was employed for the purifications. All solvents used for CC and HPLC were of analytical grade (Shanghai Chemical Reagents Co., Ltd., Shanghai, China) and chromatographic grade (Dikma Technologies Inc., Lake Forest, CA, USA), respectively.
4.2. Biological Materials
The biological samples of Sinularia polydactyla were collected by SCUBA diving at Xisha Islands, Hainan province, People’s Republic of China, in October 2013, and identified by Professor Hui Huang of the South China Sea Institute of Oceanology, Chinese Academy of Sciences. The biological material was frozen immediately after collection. A voucher specimen (NO. 13-XS-23) was deposited at the Shanghai Institute of Materia Medica, Chinese Academy of Sciences.
4.3. Isolation of Diterpenoids from Sinularia Polydactyla
Pieces of the freshly collected title animal (430 g, dry weight) were extracted exhaustively with acetone at room temperature (6 × 2.0 L). The organic extract was evaporated to give a residue, which was successively partitioned between Et2O and H2O, n-BuOH and H2O. The Et2O solution was concentrated under reduced pressure to give a dark brown residue (13.8 g), which was subjected to silica gel column chromatography and eluted with petroleum ether-Et2O (0–100% Et2O in petroleum ether (PE), gradient), yielding 11 fractions (Frs. 1–11). Fraction 6 (eluent: PE/Et2O, 9:1; components with an Rf from 0.3 to 0.5 (elution solvent for TLC, PE/Et2O = 1:1)) was then eluted with a gradient PE-Et2O (19:1 to 1:1, gradient) to yield four sub-fractions (6A–6D). Sub-fraction 6C (eluent: PE/Et2O, 7:3; Rf around 0.4 (elution solvent for TLC, PE/Et2O = 1:1)) was further purified using RP-HPLC (MeOH/H2O (90:10), 3.0 mL/min) to give compound 6 a (4.7 mg, tR 13.4 min). Fraction 7 (eluent: PE/Et2O, 6:4 to 4:6; components with an Rf value from 0.3–0.8 (elution solvent for TLC, PE/Et2O = 3:7)) was subjected to Sephadex LH-20 CC (PE/CH2Cl2/MeOH, 2:1:1) to give five sub-fractions (7A–7E). Sub-fraction 7C (with an Rf value from 0.5 to 0.7 (elution solvent for TLC, PE/Et2O = 3:7)) was separated by RP-HPLC (MeCN/H2O (85:15), 3.0 mL/min) to give compounds 5 a (2.1 mg, tR 17.6 min) and 8 a (2.4 mg, tR 14.5 min). Similarly, sub-fraction 7D (with an Rf value around 0.7 (elution solvent for TLC, PE/Et2O = 3:7)) was purified by RP-HPLC (MeOH/H2O (90:10), 3.0 mL/min) to give compound 9 b (0.9 mg), and sub-fraction 7E (with an Rf value from 0.3 to 0.5 (elution solvent for TLC, PE/Et2O = 3:7)) was chromatographed by RP-HPLC (MeOH/H2O (70:30), 3.0 mL/min) to give compounds 4 c (3.9mg, tR 15.6 min), 7 a (3.7 mg, tR 12.3 min), and 10 d (3.5mg, tR 18.3 min), respectively. (Note: a spots could be detected on the TLC under UV light (254 nm) and displayed blue through heating after spraying with anisaldehyde H2SO4 reagent; b spots displayed purple through heating after spraying with anisaldehyde H2SO4 reagent; c spots displayed blue through heating after spraying with anisaldehyde H2SO4 reagent; d spots could be detected on the TLC under UV light (254 nm) and displayed purple through heating after spraying with anisaldehyde H2SO4 reagent).
Compound
8: colorless oil; [α
+13.1 (
c 0.1, CH
3OH); IR (KBr): 3390, 1620, 834 cm
−1; UV λ
max (logε) 255 (3.5) nm;
1H and
13C NMR (C
5D
5N, 500 and 125 MHz), see
Table 1 and
Supplementary Materials; (+)-HREIMS
m/
z 288.2454 [M]
+ (calcd. for C
20H
32O,
m/
z 288.2453).
Compound
9: colorless oil; [α
−48.5 (
c 0.1, CHCl
3); IR (KBr): 1751, 1748, 883, 801 cm
−1;
1H and
13C NMR (CDCl
3, 500 and 125 MHz), see
Table 1 and
Supplementary Materials; (+)-HREIMS
m/
z 386.2449 [M]
+ (calcd. for C
24H
34O
4,
m/
z 386.2457).
4.4. Determination of the Absolute Configurations of Compounds 8 and 9 by TDDFT ECD Calculations
Conformational searches were carried out using the torsional sampling (MCMM) method and OPLS_2005 force field. Conformers above 1% population were re-optimized at the B3LYP/6-311G (d, p) level with IEFPCM (Polarizable Continuum Model using the Integral Equation Formalism variant) solvent model for acetonitrile [
15]. For the resulting geometries, ECD spectra were obtained by TDDFT calculations performed with the same functional basis set and solvent model as the energy optimization. Finally, the Boltzmann-averaged ECD spectra of the two compounds were obtained with SpecDis1.62 [
20].
4.5. Bioactivity Assay
Recombinant PTP1B catalytic domain was expressed and purified according to a previous report [
16]. The enzymatic activities of the PTP1B catalytic domain were determined at 30 °C by monitoring the hydrolysis of
pNPP. The dephosphorylation of
pNPP generated product
pNP, which was monitored at an absorbance of 405 nm by the EnVision multi label plate reader (Perkin Elmer Life Sciences, Boston, MA). In an atypical 100
µL assay mixture containing 50 mmol/L 3-[
N-morpholino] propanesulfonic acid (MOPs), pH 6.5, 2 mmol/L
pNPP, and 30 nmol/L recombinant PTP1B, activities were continuously monitored and the initial rate of hydrolysis was determined using the early linear region of the enzymatic reaction kinetic curve. The IC
50 was calculated with Prism 4 software (Graphpad, San Diego, CA). Compounds
5–
10 were tested in vitro and compounds
5,
6, and
10 were inactive. Interestingly,
7–
9 exhibited moderate inhibitory activity against PTP1B with IC
50 values 72.4 μM, 63.9 μM, and 51.8 μM, respectively, with the positive control of oleanolic acid (IC
50 = 2.56 μM).


 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}