1. Introduction
Sea anemones have been understudied as a source of new pharmacological tools or therapeutic leads [
1]. Sea anemones belong to
Cnidaria that also includes corals, jellyfish and sponges. They have toxic peptides to incapacitate and immobilize prey and to defend from potential predators [
2]. Their toxin arsenal is complex, targeting a variety of pharmacological targets such as ionic channels, inflammatory receptors [
3] or pore forming protein in cellular membranes [
4]. Anti-hyperglycemic and anti-diabetic activities were also observed from sea anemone extract [
5].
Active principles from venomous species such as snakes or scorpions have a centralized venom system and have a structural homogeneity, which is not the case for sea anemones. Scorpion active principles for instance have a scaffold characterized by three beta sheets, an alpha helix and four disulfide bridges [
6]. In sea anemones, the venom system is decentralized in all parts of the animal body showing a higher diversity in size and scaffold of protein active principles. The active principles binding on receptors such as ionic channels are short size proteins between 3000 and 5000 Da cross linked with three disulfide bridges [
2]. They have different structural scaffolds regarding their pharmacological targets. Active principles binding on ionic channel are characterized by three beta strands [
7], while those binding on enzymes such as the family of Kunitz-type inhibitors have two beta strands and an alpha helix [
8]. Surprisingly, the sea anemone toxin Bg1, with a scaffold completely different, can compete at nanomolar concentrations with the Aah II scorpion toxin on the same pharmacological site on sodium channel [
9]. However, the superposition of the 3D structure of these two proteins shows that the lateral chains of four basic residues are in exactly the same positions [
9].
Cancer treatments are mainly based on anti-mitotic compounds that have side effects because they block the division of both cancer cells and healthy cells. Cancer cells need to be vascularized by endothelial cells, in a process called angiogenesis, to grow as a tumor and then to spread as metastases inducing the patient death. It was proposed almost 50 years ago to block angiogenesis to fight against cancer because angiogenesis is no longer important after embryogenesis [
10]. There are very few antiangiogenic compounds compared to antimitotic compounds and it has only been a decade since antiangiogenic compounds began being tested in clinical trials. Amazingly, antiangiogenic compounds showed a limited efficacy because they all bind on the Vascular Endothelial Growth Factor (VEGF) or the VEGF receptor and tumor cells can trigger different biological ways to have angiogenesis [
11]. Among antiangiogenic compounds binding on VEGF, the most used in clinical trial is Bevacizumab (known as Avastin), which is a monoclonal antibody [
11]. Bevacizumab is costly due to its size and difficulties to have germ free production as recombinant protein. Furthermore, resistance to Bevacizumab is observed due to the upregulation of other redundant angiogenic factors different from VEGF [
12]. Compounds binding on VEGF receptors that are not proteins have toxic effects that limit their use. Resistance towards VEGF-centered antiangiogenic therapy represents a substantial clinical challenge [
10]. There is therefore a need to have compounds blocking angiogenesis that do not bind on VEGF or VEGF receptor, are easy to produce and have no long-term toxicity.
Short size synthetic proteins (less than 50 residues) are suitable compounds for this goal.
Anemonia viridis (also called
Anemonia sulcata) is well studied and proven to be a remarkable source of low molecular proteins with different pharmacological binding sites on ionic channel receptors [
7]. Seven short size proteins (42–49 residues) binding on ionic channels were purified and characterized from
Anemonia viridis [
7]. Among them, AS2 is a 47-residue-long protein (4934 Da) having only beta structures [
13]. There is also BDS-1 that is a blood depressing protein of 43 residues (4708 Da) binding on a specific potassium channel [
14]. The main secondary structure of BDS-1 is a triple-stranded antiparallel beta-sheet without alpha helix [
7]. Recently, a partially purified extract of
Anemonia viridis was reported to affect the growth and viability of selected tumor cell lines [
15].
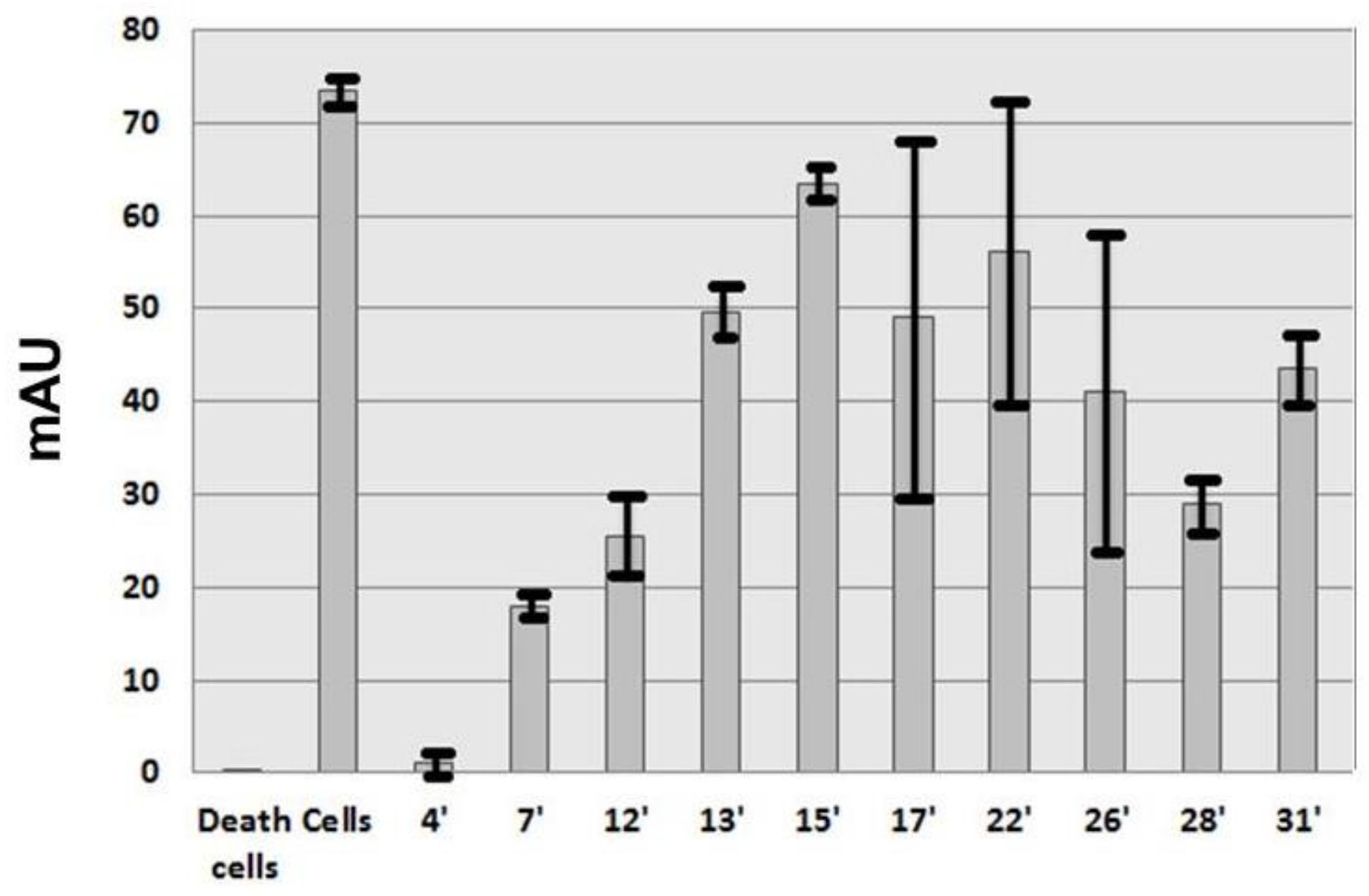
In this study, two cellular antiangiogenic screening tests were used with Human Microvascular Endothelial Cells (HMEC) having the Epithelial Growth Factor instead of VEGF [
16]. The first one measured the proliferation of HMEC and the second one the formation of HMEC capillary network. These screening tests made it possible to identify from
Anemonia viridis a protein fraction able to limit HMEC proliferation and angiogenesis at low concentration (IC
50 14 nM). Trypsin inhibition and the presence of a two turns alpha helix revealed by circular dichroism suggest that the active principle is a kunitz type inhibitor.
3. Discussion
The purpose of this study was to identify and purify low molecular weight proteins from Anemonia viridis susceptible to have antiangiogenic activity not acting on the VEGF pathway. The purification protocol was set not to purify all compounds in Anemonia viridis having an antiangiogenic activity but only low molecular weight proteins. The reason is that synthetic proteins less than 50 residues can now be produced at low cost and have the great advantage to make possible a sterile production, which is not the case for monoclonal antibodies such as Avastin requiring a biological production. The high cost of Avastin is not related to the production as recombinant protein but to the purification process to have a germ free pharmaceutical production. A low molecular weight compound (<1000 Da) would be certainly less expensive to produce than a synthetic protein of 43 residues corresponding to BDS-5. However, a new chemical family of active principles requires now very expensive toxicological studies to have a Drug Master File suitable for clinical studies. Moreover, actual preclinical toxicity studies required for clinical studies are not sufficient to guarantee an absence of long term toxicities. These long-term toxicities are often due to accumulation in tissues of chemical compounds that cannot be or are insufficiently degraded. Therefore, a synthetic protein (with a molecular weight <5000 Da) represents a good compromise between cost of production and long-term safety issues.
This study shows that sea anemones are certainly a very interesting source of low molecular weight protein active principles, which have been understudied [
1]. Although most of these proteins have three disulfide bridges and share sequence homologies, point mutations can totally change their structures and their pharmacological properties [
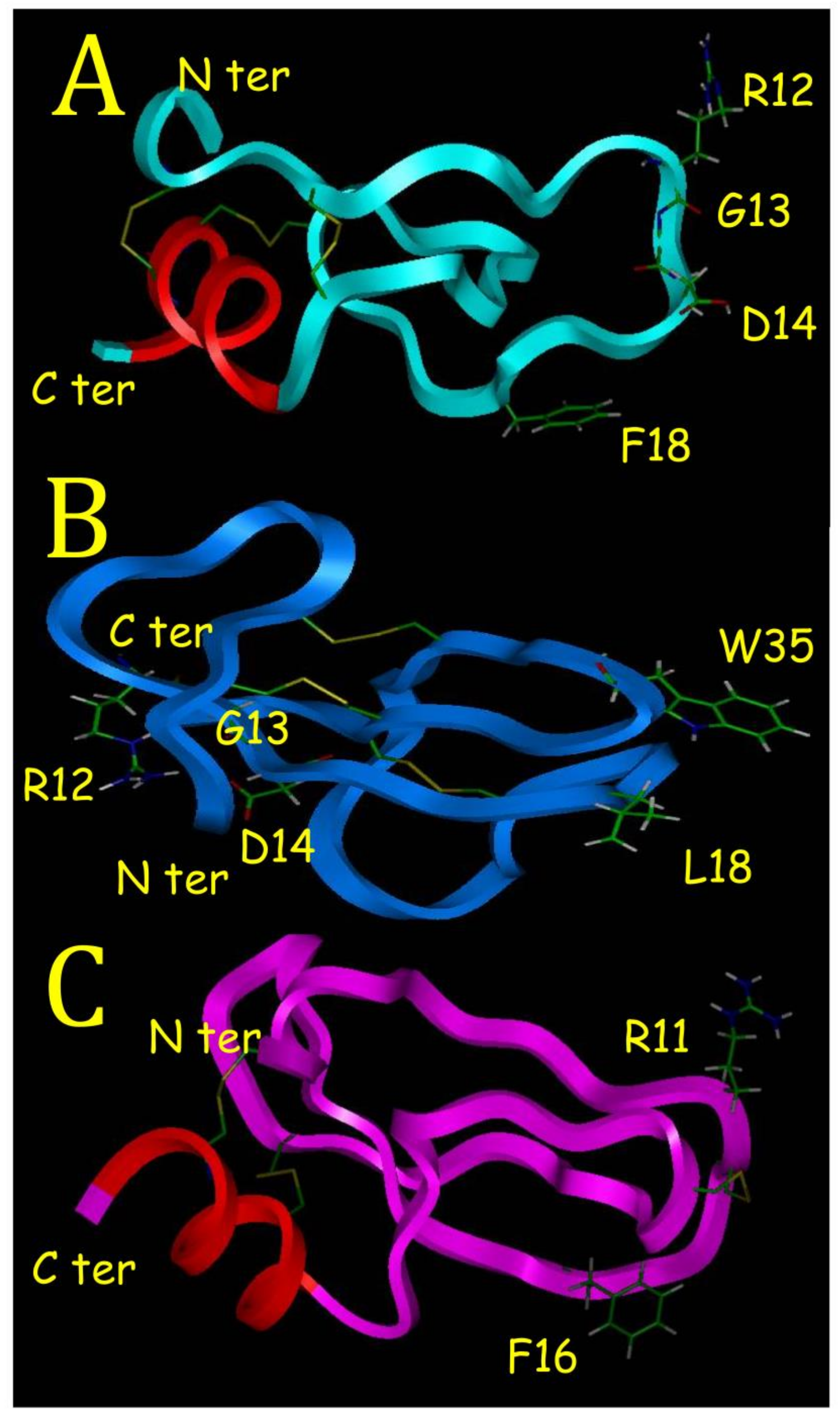
1]. This is well outlined in this study if we compare BDS-5 with BDS-1. With its C-terminal alpha helix, BDS-5 appears to have a 3D structure different from sea anemone protein binding on ionic channels such as BDS-1. Proteins binding on ionic channel identified in sea anemones or in scorpions are characterized by a hydrophobic surface, which helps for a correct positioning of basic residues located on the opposite side. These basic residues create ionic bonds with acidic residues located in their ionic channel binding sites [
6]. This is illustrated with BDS-1 that have a hydrophobic surface, with L18 and W35 (
Figure 7B), and on the opposite side R12, which is one of the basic residues interacting with the binding site on potassium channel [
7].
Although BDS-5 had never been purified and characterized, its sequence was previously known from a search for amino acid sequence motifs in sea anemone polypeptides, where 14 BDS-like sequences were identified in the
Anemonia viridis genome [
19]. This search was made in a cDNA library built from an Expressed Sequence Tag (EST) analysis performed on
Anemonia viridis revealing 14,504 unique protein sequences in its genome [
20]. Another EST analysis made it possible to discover BDS-15 [
21]. Interestingly this study showed that BDS-1, BDS-3, BDS-4, BDS-5 and BDS-6 were the most represented transcripts among BDS like proteins [
22]. The F18 mutation is found in half of the BDS like sequence. BDS-16 purified and sequenced in this study was never referenced before. Amazingly, the RGD motif is highly conserved in BDS sequences but no activity on integrins has been reported with BDS-1 or BDS-2. This is probably due to the N terminus in BDS-1 and BDS-2, which is masking a part of the RGD motif (
Figure 7B).
Kunitz-Type Inhibitors (KTI) have a conserved scaffold (a chain of 40–60 residues stabilized by three disulfide bridges, a two-beta strand sheet and a short C terminal alpha helix) and the most known is the Bovine Pancreatic Trypsin Inhibitor [
7]. Another property of KTI is to have an active site with amphoteric characteristic as described in the crystal structure of a Caribbean Sea anemone SHPI-1 in a complex with an Elastase enzyme used in this study [
18]. The amphoteric characteristic is due to hydrophobic and hydrophilic poles made of F16 and R11 for SHPI-1 (
Figure 7C), and F16 and R12 for BDS-5 (
Figure 7A), respectively. A KTI from a tick was able to inhibit Trypsin, Elastase and interfere with blood vessel formation [
22]. The RGD motif is recognized by Integrins that are membrane proteins playing a major role in cancer progression and metastasis [
23]. BDS-5 appears to be a KTI with a RGD motif that could block angiogenesis in binding on integrins.
4. Materials and Methods
4.1. Phase Extractions and Filtrations from Crude Anemonia viridis
Living animals were collected in the south bay at Marseille in a harbor called “Base nautique du Prado” where shallow and calm water made the development of an important colony of at least 1000 Anemonia viridis possible. No other sea anemone species were observed in this spot. They are spread on little stones and they were collected alive fixed on their stone. The very same day the anemones (n = 10) with an average size of 10 cm were transported in sea water to the lab at the faculty of Pharmacy and the tentacles were immediately cut and frozen at −20 °C. Tentacles were then lyophilized (Jouan, Nante, France) for 3 days. The dried tentacles were mashed and suspended in tert-butyl-methyl-Ether (Merck, Lyon, France). After a first centrifugation at 4500 rpm for 20 min (Hettich, Vlotho, Germany), the organic liquid phase containing hydrophobic compounds was eliminated and the dry pellet was suspended in water containing 0.1% Trifluoroatic acid (TFA) (Carlo Erba, Barcelona, Spain). A second centrifugation at 4500 rpm for 20 min was carried out and the pellet was eliminated. The liquid phase containing hydrophilic compounds was then centrifuged in a 30 kDa Centrifugal Filter Unit (Millipore, Burlington, MA, USA) at 4500 rpm for 20 min and then lyophilized. The liquid extract containing compounds <30,000 Da was filtered at 0.22 µm with a filter (Millipore, Guyancourt, France) fixed on a syringe and then lyophilized. UV spectrum of the filtered solution was carried out from 240 to 340 nm with a Beckman DU640.
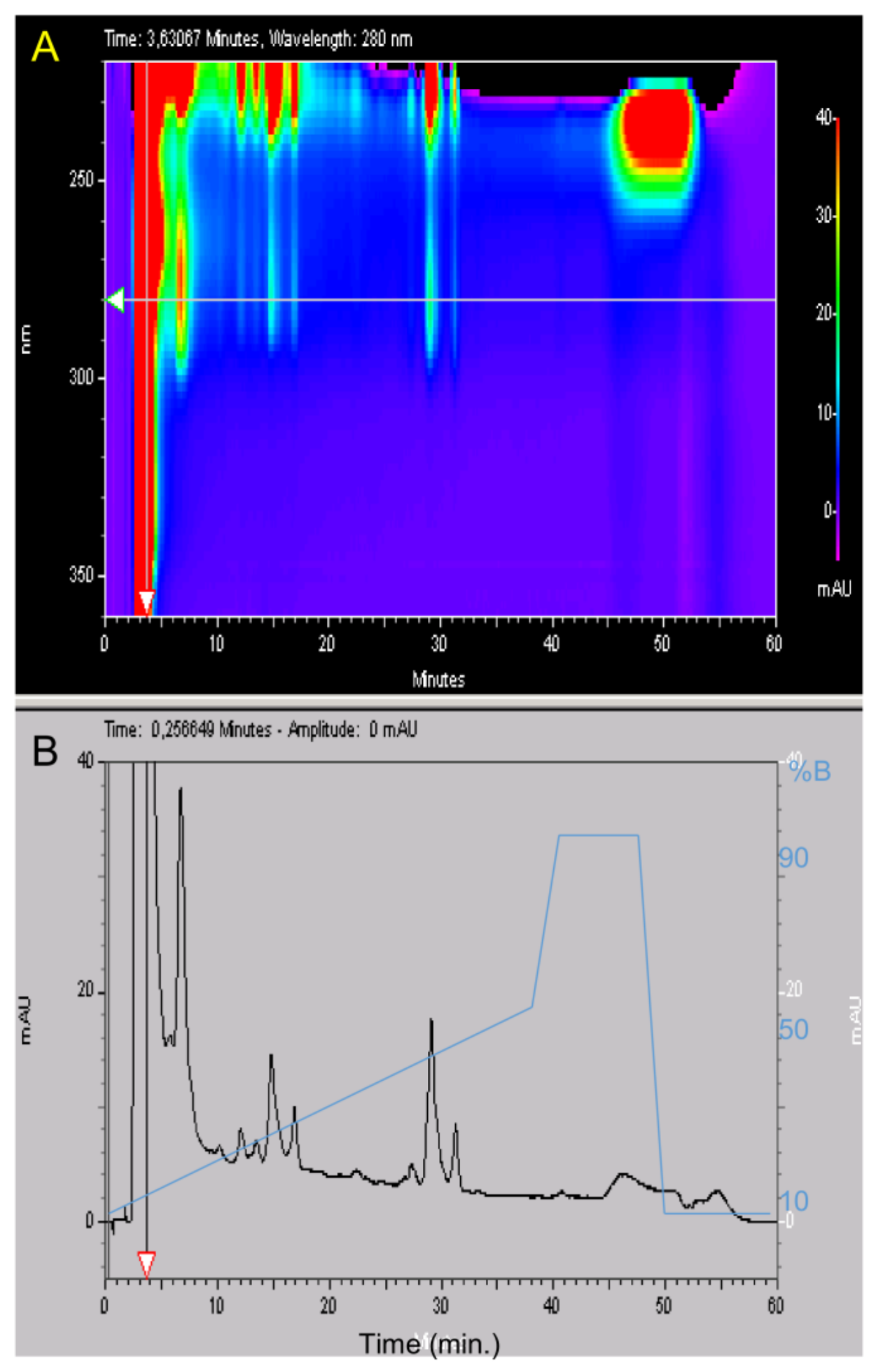
4.2. High Liquid Performance Chromatography (HPLC)
After filtration at 0.22 µm, the lyophilized material was suspended in water 0.1% TFA at 50 mg/mL. Purification was carried out using a Beckman HPLC System Gold 125 (Brea, CA, USA) apparatus with a Merck Lichrospher C8 reverse phase column (15 × 100 mm). Buffer A was water supplemented with 0.1% (v/v) TFA and buffer B was acetonitrile (Merck, Lyon, France) supplemented with 0.1% (v/v) TFA. Gradient was buffer B from 15% to 35% in 40 min, then 5 min at 90% B and a return to initial condition for 10 min at 15% B with a 2 mL/min flow rate. Fractions were collected manually and their absorptions were measured with a Beckman DU 640B before being frozen at −20 °C and lyophilized. HPLC analysis was carried out using a Merck Lichrospher RP-C8 column (4.6 × 100 mm) with similar buffers but a 0.8 mL/min flow rate using a linear gradient from 10% to 50% B in 40 min, then 50% to 90% B in 2 min, 5 min at 90% B, a return to initial condition in two min at 10% B and a final stationary phase of 10 min at 10% B. The total duration of the analytical run was 60 min. The UV absorbance of the effluent was measured from 220 to 330 nm with a diode ray Beckman 168 Gold detector and is expressed in milli Absorbance Unit (mAU).
4.3. VEGF Free HMEC Proliferation Assay
HMEC-1 cells were cultured in MCDB-131 medium containing 10% heat inactivated fetal bovine serum, 2 mmol/L glutamine, 100 units/mL penicillin and streptomycin, 1 mg/mL hydrocortisone and 10 ng/mL Epithelial Growth Factor. HMEC-1 cells were seeded on 0.1% gelatin-coated flasks and cultured at 37 °C with 5% CO2. HMEC-1 cells were transferred in 96-well plates at 5000 cells/well in culture medium. Twenty-four hours after seeding, cells were treated with extracts or the control vehicle solution. After 72 h of treatment, cells were exposed to 0.5 mg/mL of diMethyl Thiazol Tetrazolim (MTT) bromide for 3 h at 37 °C in culture medium. Cells were then washed with Phosphate Buffered Saline (PBS), formazan crystals were solubilized with 100 µL DMSO and absorbance was measured at 600 nm. MTT is degraded by a mitochondrial enzyme in living cells and forms a purple precipitate in mitochondria that is measured at 600 nm.
4.4. Tubulogenesis Assays
HMEC-1 cells were cultured as described in the proliferation inhibition assay. HMEC-1 capillary network formation on Matrigel were performed on Matrigel™ (Gibco, Beziers, France) added to 96-well plates and allowed to solidify for 30 min at 37 °C. HMEC-1 cells (10,000 cells/well) were added atop the Matrigel™, treated with fractions or a control vehicle solution and incubated for 5 h at 37 °C. Capillary-like structures formed in the gel were photographed and their length was quantified using the METAMORPH 7.6 software [
15].
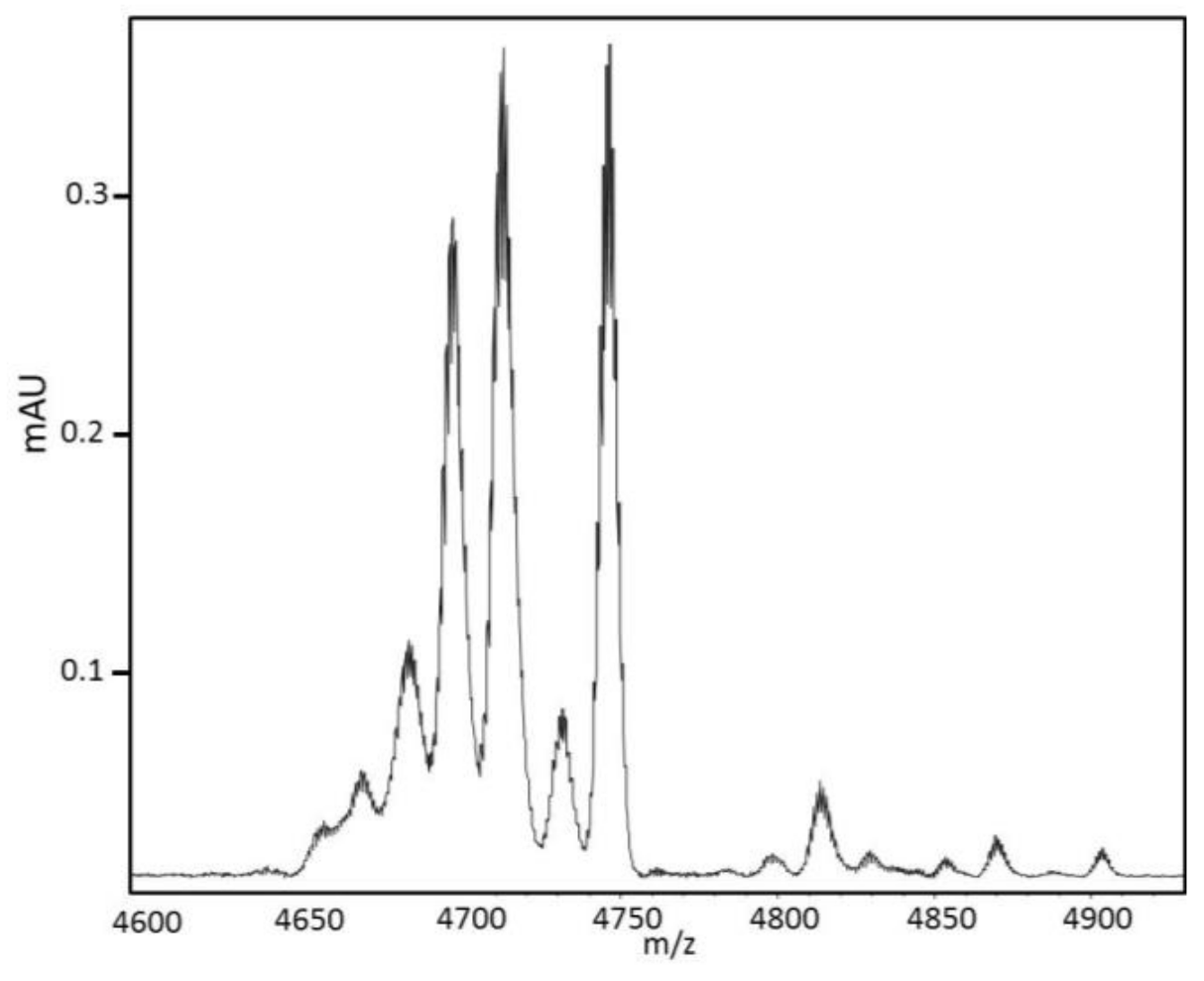
4.5. Mass Determination and Sequencing
Matrix-Assisted Laser Desorption Ionization–Mass Spectrometry (MALDI-MS) spectra were obtained on an UltraflXtreme apparatus (Bruker, Bremen, Germany) operating in positive linear mode with delayed extraction. The samples were co-crystallized with a 10 mg/mL solution of Hydroxy α-Cyano-4-Cinnamic Acid (HCCA) on the MALDI-MS target by the dry droplet method. MALDI-MS spectra were acquired with an accelerating potential of 20 KV and a laser power set to the minimum level necessary to get an efficient signal. Spectrum mass calibration was based on external calibration using an appropriate peptides standard mixture (Peptide Calibration Standard, Brüker, Bremen, Germany). Sequencing was carried out with the Edman degradation method on a PPSQ31 B Shimadzu sequencer. Enzymatic digestion was carried out on 50 µg of the 28-min fraction and 1 µg a Trypsin/Lys C mixture (Promega, Madison, WI, USA) in ammonium bicarbonate 100 mM pH 9 at 37 °C. Different digestions were carried out from one hour to three days. For Nano-HPLC-MS/MS mass spectrometry, the 28-min fraction was reduced with DTT (Sigma-Aldrich, St. Louis, MO, USA), and alkylated with iodoacetamide (Sigma-Aldrich) and incubated overnight at 37 °C in a solution of 12.5 ng/μL of trypsin (Sequencing grade, Roche, Basel, Switzerland) in 25 mM NH4HCO3. The protein digests were sequenced by Nano-LC–MS/MS (Dionex RSLC coupled to a hybrid Q orbitrap mass spectrometer equipped with a Nano-ESI source; Q exactive ThermoFisher scientific, Waltham, WA, USA) in the data-dependent acquisition mode (method top 10). Data were matched to the Uniprot protein database. MALDI MS In Source Decay (ISD) was performed using an Ultraflextreme TOF/TOF mass spectrometer controlled by the FlexControl 3.3 software (BrukerDaltonics, Bremen Germany). The laser was increased (20%) to fragment protein in the source of the mass spectrometer using a specific matrix enabling the generation of hydrogen radicals breaking the peptide backbone producing C ions and z ions from 1000 to 5000 Da generating tag masses that could be used to search proteins in databases. Spectra were acquired in positive reflectron ion mode with 2000 laser shots accumulated and the laser power was set with an increase of 20% of fluensce with a frequency of 1000 Hz. The mass spectrometer parameters were set according manufacturer’s settings for optimal acquisition performance. Sequences were analyzed on Flex Analysis 3.0 software, (BrukerDaltonics, Bremen, Germany).
4.6. Far UV Absorption and Circular Dichroism (CD) Spectra
Spectra were measured with a 100 µm path length from 260 to 178 nm at 20 °C on a JASCO J-810 spectropolarimeter. Data were collected at 0.5 nm intervals using a step auto response procedure (JASCO, Oklahoma City, OK, USA) and the buffer background was subtracted. Protein concentrations were 1 mg/mL in 20 mM pH 7 phosphate buffer. Absorption spectra were measured to verify both concentration and light transmission. CD spectra are presented as ∆ε per amide rather than in ∆A unit to have a measure independent of mass and concentration according to the Beer Lambert law. Furthermore, the signal was divided by the number of amino acid residues of the protein minus one to have ∆ε per amide [
17]. The instrument was calibrated with (+)-10-camphorsulfonic acid, a ratio of 2:1 was found between the positive CD band at 290.5 nm and the negative band at 192.5 nm.
4.7. Molecular Modelling
BDS-5 model was built with the Insight II software from MSI Technologies, Inc. (San Diego, CA, USA). The model was optimized with the Consistent Valence Force Field (CVFF) in term of the internal energies, using the van der Waals energy to monitor each step of the model. Model of BDS-5 was built with the InsightII Homology pulldown from the crystal structure of SHPI 1 in a complex with Elastase [
19] available in the Brookhaven data bank (PDB code 1SHP). Minimization was performed with steepest descent and conjugate gradient algorithms. Dynamic was performed at 300 K for 1.1 ps using 1000 steps.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







