Ascidian Toxins with Potential for Drug Development


Abstract
:1. Introduction
1.1. Overview of Ascidians
1.2. Ascidian Natural Products
1.3. Purpose of This Review
2. Symbiotic Organisms in the Biosynthesis of Ascidian Natural Products
2.1. Microbial Diversity
2.2. Prochloron
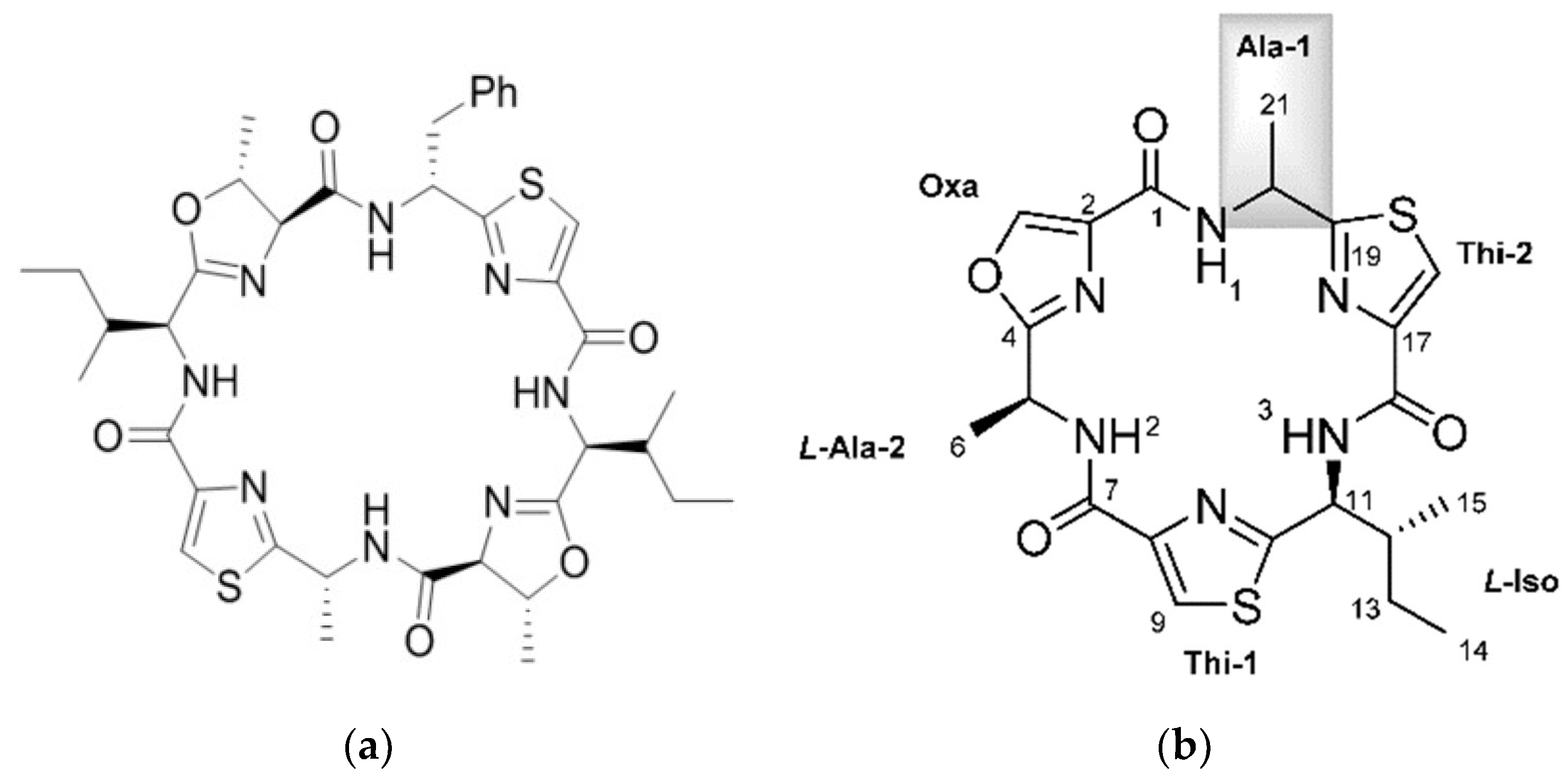
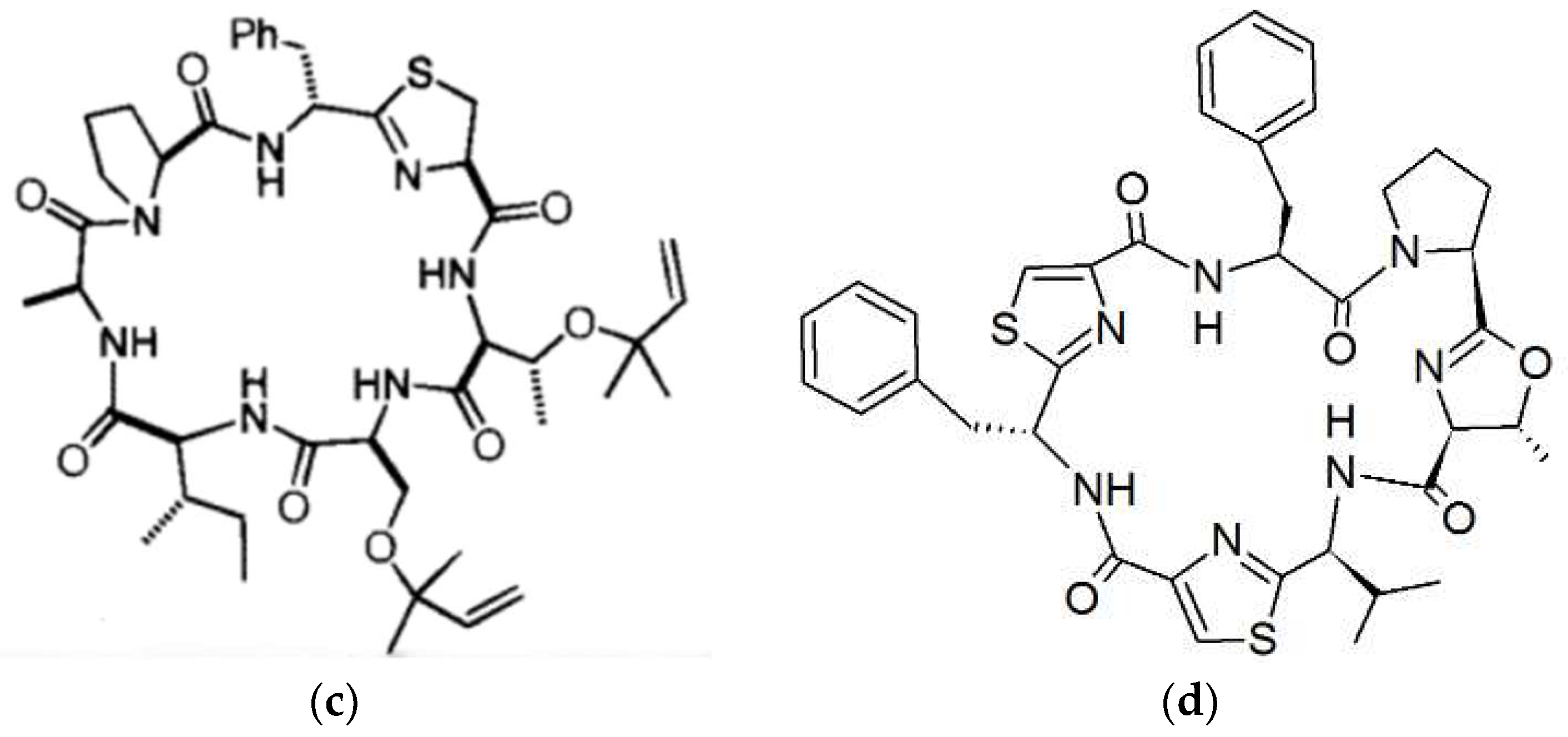
3. Cyanobactins
3.1. Biosynthesis of Cyanobactins
3.2. Biological Activity of Cyanobactins
4. Cyclic Depsipeptides and Polyketides
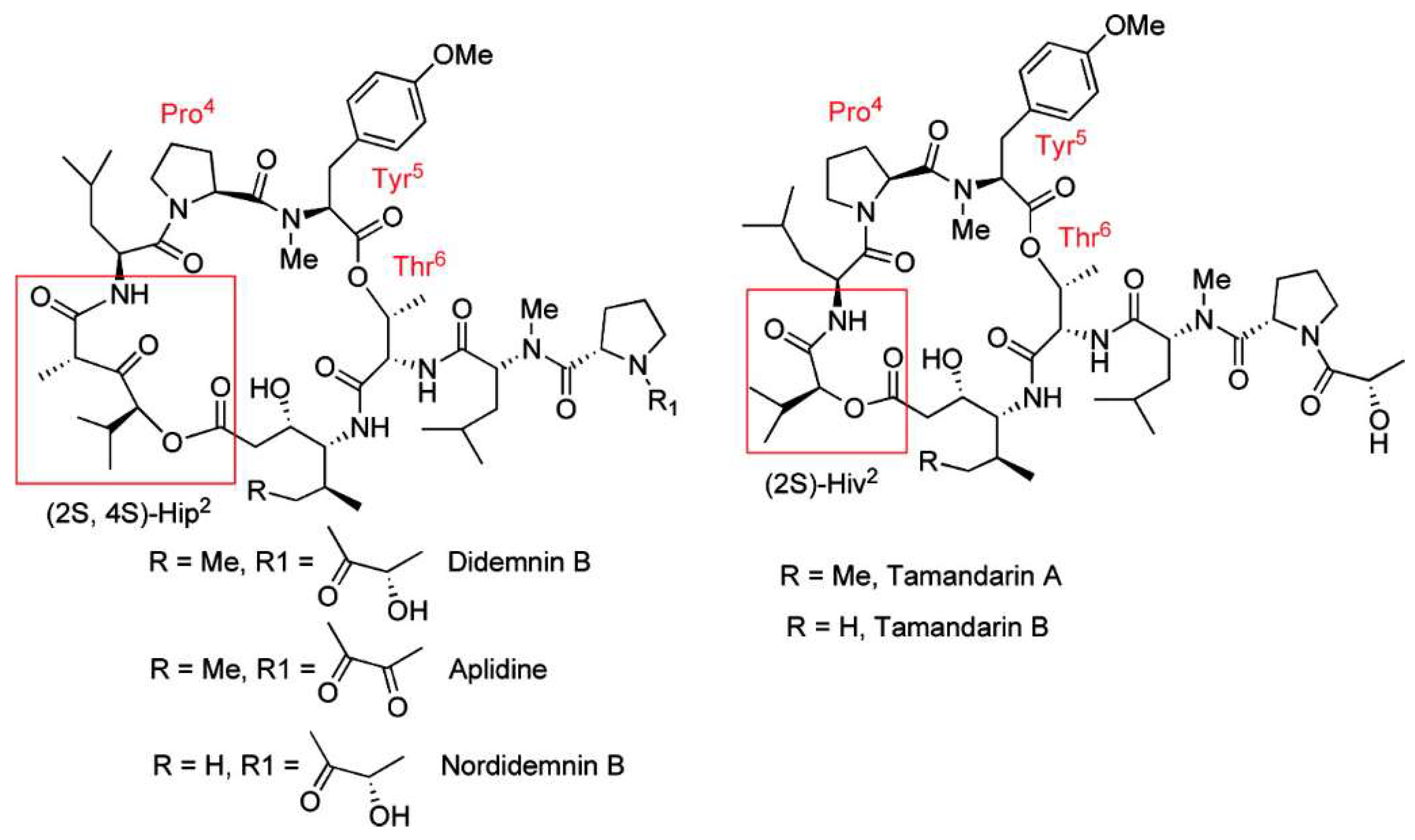
4.1. Didemnins
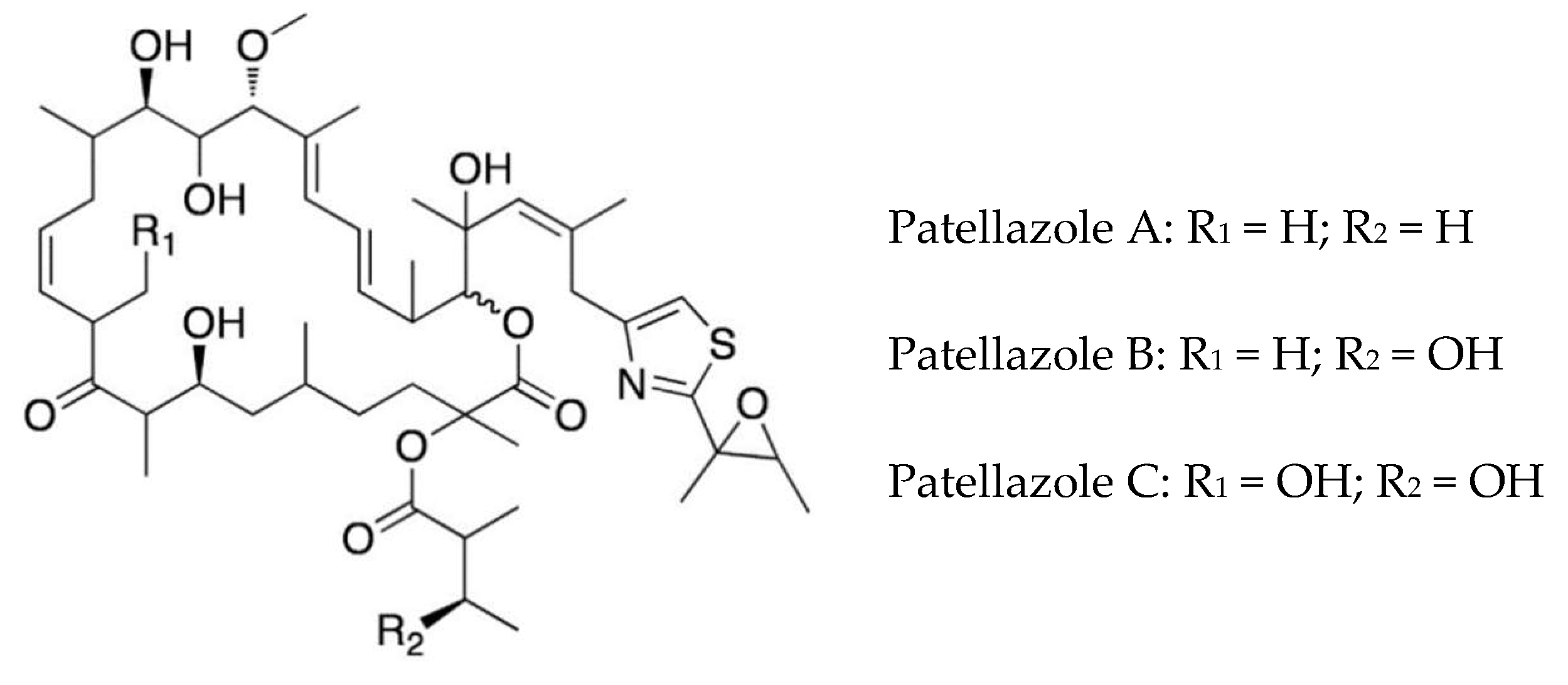
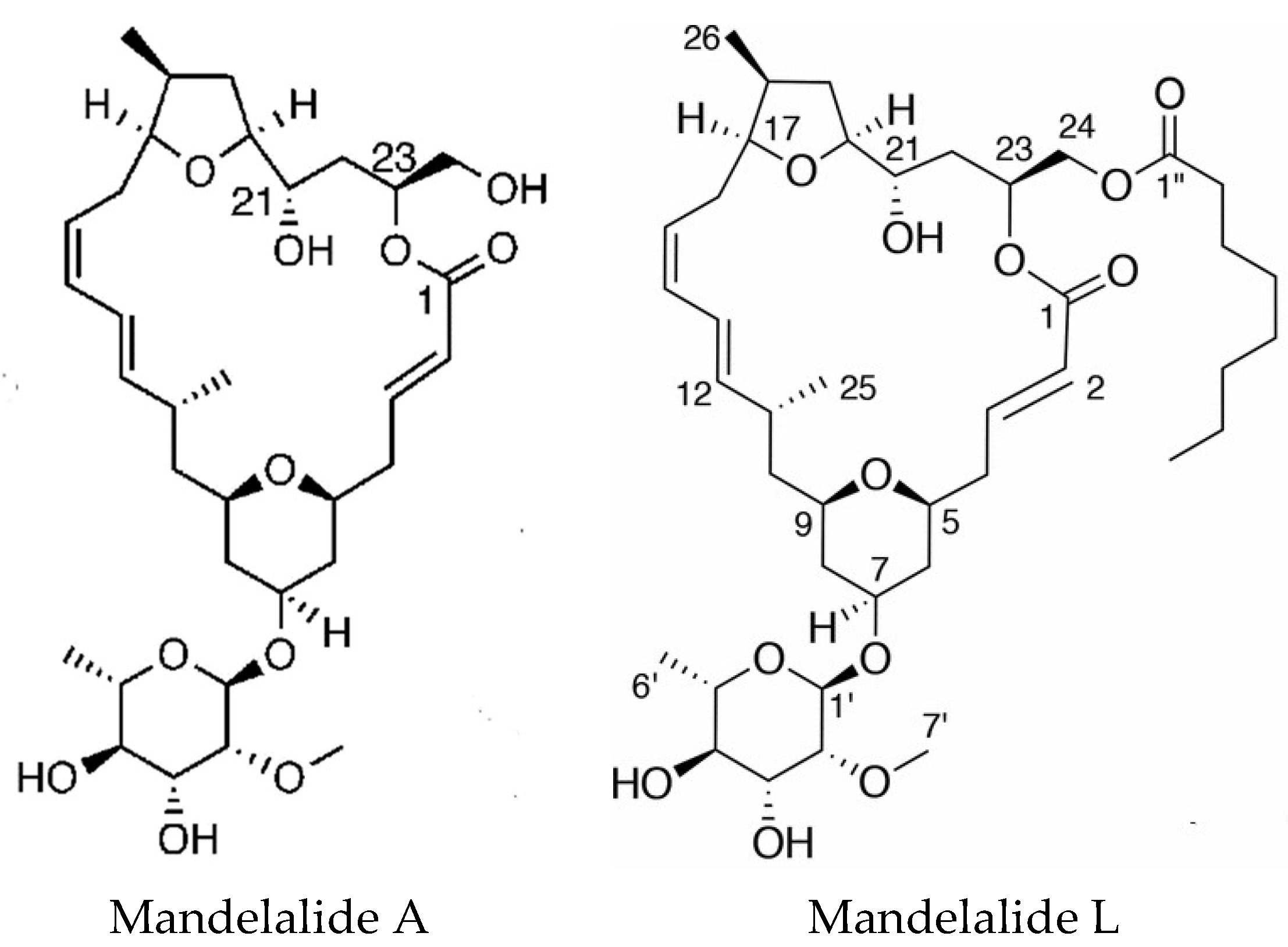
4.2. Polyketides
5. Alkaloids
5.1. Quinoline Alkaloids
5.2. Pyridoacridine Alkaloids
5.3. Beta-Carboline Alkaloids
5.4. Tyrosine and Phenylalanine Based Alkaloids
5.5. Indole Based Alkaloids
5.6. Other Alkaloids
6. Terpenoids and Quinones
6.1. Terpenoids
6.2. Quinones
7. Ascidian Compounds Affecting Signaling Pathways
7.1. Kinase Inhibitors
7.2. Acetylcholine Signaling Inhibitors
7.3. Phosphatase Inhibitors
8. Toxins Affecting the Cytoskeleton
8.1. Tubulin
8.2. Actin
9. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Lambert, C.C. Historical introduction, overview, and reproductive biology of the protochordates. Can. J. Zool. 2005, 83, 1–7. [Google Scholar] [CrossRef]
- Lambert, G. Ecology and natural history of the protochordates. Can. J. Zool. 2005, 83, 34–50. [Google Scholar] [CrossRef]
- Shenkar, N.; Swalla, B.J. Global diversity of Ascidiacea. PLoS ONE 2011, 6, e20657. [Google Scholar] [CrossRef] [PubMed]
- Shenkar, N.; Gittenberger, A.; Lambert, G.; Rius, M.; Moreira Da Rocha, R.; Swalla, B.J.; Turon, X. Ascidiacea World Database. 2018. Available online: http://www.marinespecies.org/ascidiacea (accessed on 10 January 2018).
- Bellante, A.; Piazzese, D.; Cataldo, S.; Parisi, M.G.; Cammarata, M. Evaluation and comparison of trace metal accumulation in different tissues of potential bioindicator organisms: Macrobenthic filter feeders Styela plicata, Sabella spallanzanii, and Mytilus galloprovincialis. Environ. Toxicol. Chem. 2016, 35, 3062–3070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumollard, R.; Gazo, I.D.L.; Gomes, I.; Besnardeau, L.; McDougall, A. Ascidians: An Emerging Marine Model for Drug Discovery and Screening. Curr. Top. Med. Chem. 2017, 17, 2056–2066. [Google Scholar] [CrossRef] [PubMed]
- Ueki, T.; Yamaguchi, N.; Isago, Y.; Tanahashi, H. Vanadium accumulation in ascidians: A system overview. Coord. Chem. Rev. 2015, 301, 300–308. [Google Scholar] [CrossRef]
- Palanisamy, S.K.; Thomas, O.P.; McCormack, G.P. Bio-invasive ascidians in Ireland: A threat for the shellfish industry but also a source of high added value products. Bioengineered 2018, 9, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Palanisamy, S.K.; Trisciuoglio, D.; Zwergel, C.; Del Bufalo, D.; Mai, A. Metabolite profiling of ascidian Styela plicata using LC–MS with multivariate statistical analysis and their antitumor activity. J. Enzyme Inhib. Med. Chem. 2017, 32, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Phat, C.; Hong, S.C. Structural diversity of marine cyclic peptides and their molecular mechanisms for anticancer, antibacterial, antifungal, and other clinical applications. Peptides 2017, 95, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Negi, B.; Kumar, D.S.; Rawat, D. Marine peptides as anticancer agents: A remedy to mankind by nature. Curr. Protein Pept. Sci. 2017, 18, 885–904. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.K.; Choi, M.C.; Seo, C.H.; Park, Y. Therapeutic Properties and Biological Benefits of Marine-Derived Anticancer Peptides. Int. J. Mol. Sci. 2018, 19, 919. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.Y.; Dahiya, R.; Qin, H.L.; Mourya, R.; Maharaj, S. Natural proline-rich cyclopolypeptides from marine organisms: Chemistry, synthetic methodologies and biological status. Mar. Drugs 2016, 14, 194. [Google Scholar] [CrossRef] [PubMed]
- Gogineni, V.; Hamann, M.T. Marine natural product peptides with therapeutic potential: Chemistry, biosynthesis, and pharmacology. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2018, 1862, 81–196. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Torres, V.; Encinar, J.A.; Herranz-López, M.; Pérez-Sánchez, A.; Galiano, V.; Barrajón-Catalán, E.; Mico, V. An updated review on marine anticancer compounds: The use of virtual screening for the discovery of small-molecule cancer drugs. Molecules 2017, 22, 1037. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.R.; Mohamed, G.A. Marine pyridoacridine alkaloids: Biosynthesis and biological activities. Chem. Biodivers. 2016, 13, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Imperatore, C.; Aiello, A.; D’Aniello, F.; Senese, M.; Menna, M. Alkaloids from marine invertebrates as important leads for anticancer drugs discovery and development. Molecules 2014, 19, 20391–20423. [Google Scholar] [CrossRef] [PubMed]
- Palanisamy, S.K.; Rajendran, N.M.; Marino, A. Natural Products Diversity of Marine Ascidians (Tunicates; Ascidiacea) and Successful Drugs in Clinical Development. Nat. Prod. Bioprospect. 2017, 7, 1–111. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, V.; Venkatesan, M.; Ramachandran, S.; Sundaresan, U. Bioactive Peptides from Marine Ascidians and Future Drug Development—A Review. Int. J. Pept. Res. Ther. 2018, 24, 13–18. [Google Scholar] [CrossRef]
- Cooper, E.L.; Albert, R. Tunicates: A vertebrate ancestral source of antitumor compounds. In Handbook of Anticancer Drugs from Marine Origin; Springer: Cham, Switzerland, 2015; pp. 383–395. [Google Scholar]
- Agrawal, S.; Adholeya, A.; Deshmukh, S.K. The Pharmacological Potential of Non-ribosomal Peptides from Marine Sponge and Tunicates. Front. Pharmacol. 2016, 7, 333. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.A. The Global Marine Pharmaceuticals Pipeline. Available online: http://marinepharmacology.midwestern.edu/clinPipeline.htm (accessed on 14 January 2018).
- Donia, M.S.; Fricke, W.F.; Partensky, F.; Cox, J.; Elshahawi, S.I.; White, J.R.; Phillippy, A.M.; Schatz, M.C.; Piel, J.; Haygood, M.G.; et al. Complex microbiome underlying secondary and primary metabolism in the tunicate-Prochloron symbiosis. Proc. Natl. Acad. of Sci. USA 2011, 108, E1423–E1432. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.W.; Donia, M.S.; McIntosh, J.A.; Fricke, W.F.; Ravel, J. Origin and variation of tunicate secondary metabolites. J. Nat. Prod. 2012, 75, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Fu, C.; Wang, G. Microbial diversity associated with ascidians: A review of research methods and application. Symbiosis 2017, 71, 19–26. [Google Scholar] [CrossRef]
- Schmidt, E.W. The secret to a successful relationship: Lasting chemistry between ascidians and their symbiotic bacteria. Invertebr. Biol. 2015, 134, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Tianero, M.D.; Kwan, J.C.; Wyche, T.P.; Presson, A.P.; Koch, M.; Barrows, L.R.; Bugni, T.S.; Schmidt, E.W. Species specificity of symbiosis and secondary metabolism in ascidians. ISME J. 2015, 9, 615–628. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.S.; Erwin, P.M.; Shenkar, N.; López-Legentil, S. Introduced ascidians harbor highly diverse and host-specific symbiotic microbial assemblages. Sci. Rep. 2017, 7, 11033. [Google Scholar] [CrossRef] [PubMed]
- Buedenbender, L.; Carroll, A.R.; Ekins, M.; Kurtböke, D.İ. Taxonomic and Metabolite Diversity of Actinomycetes Associated with Three Australian Ascidians. Diversity 2017, 9, 53. [Google Scholar] [CrossRef]
- Donia, M.S.; Ravel, J.; Schmidt, E.W. A global assembly line for cyanobactins. Nat. Chem. Biol. 2008, 4, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Sivonen, K.; Leikoski, N.; Fewer, D.P.; Jokela, J. Cyanobactins—Ribosomal cyclic peptides produced by cyanobacteria. Appl. Microbiol. Biotechnol. 2010, 86, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, L.; Raina, J.B.; Lutz, A.; Kot, W.; Albertsen, M.; Halkjær-Nielsen, P.; Sørensen, S.J.; Larkum, A.W.; Kühl, M. In situ metabolomic-and transcriptomic-profiling of the host-associated cyanobacteria Prochloron and Acaryochloris marina. ISME J. 2018, 12, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Hirose, E.; Neilan, B.A.; Schmidt, E.W.; Murakami, A.; Gault, P.M. Enigmatic life and evolution of Prochloron and related cyanobacteria inhabiting colonial ascidians. In Handbook on Cyanobacteria: Biochemistry, Biotechnology and Applications; Gault, P.M., Marler, H.J., Eds.; Nova Science Publishers, Inc.: New York, NY, USA, 2007; pp. 161–189. ISBN 978-1-60741-092-8. [Google Scholar]
- Hirose, E. Ascidian photosymbiosis: Diversity of cyanobacterial transmission during embryogenesis. Genesis 2015, 53, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.; Vasconcelos, V. Cyanobactins from cyanobacteria: Current genetic and chemical state of knowledge. Mar. Drugs 2015, 13, 6910–6946. [Google Scholar] [CrossRef] [PubMed]
- Morita, M.; Schmidt, E.W. Parallel lives of symbionts and hosts: Chemical mutualism in marine animals. Nat. Prod. Rep. 2018, 35, 357–378. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.W.; Nelson, J.T.; Rasko, D.A.; Sudek, S.; Eisen, J.A.; Haygood, M.G.; Ravel, J. Patellamide A and C biosynthesis by a microcin-like pathway in Prochloron didemni, the cyanobacterial symbiont of Lissoclinum patella. Proc. Natl. Acad. Sci. USA 2005, 102, 7315–7320. [Google Scholar] [CrossRef] [PubMed]
- Sardar, D.; Schmidt, E.W. Combinatorial biosynthesis of RiPPs: Docking with marine life. Curr. Opin. Chem. Biol. 2016, 31, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Czekster, C.M.; Ge, Y.; Naismith, J.H. Mechanisms of cyanobactin biosynthesis. Curr. Opin. Chem. Biol. 2016, 35, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Koehnke, J.; Bent, A.F.; Houssen, W.E.; Mann, G.; Jaspars, M.; Naismith, J.H. The structural biology of patellamide biosynthesis. Curr. Opin. Struct. Biol. 2014, 29, 112–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Torres, J.P.; Tianero, M.D.; Kwan, J.C.; Schmidt, E.W. Origin of chemical diversity in Prochloron-tunicate symbiosis. Appl. Environ. Microbiol. 2016, 82, 3450–3460. [Google Scholar] [CrossRef] [PubMed]
- Tianero, M.D.; Pierce, E.; Raghuraman, S.; Sardar, D.; McIntosh, J.A.; Heemstra, J.R.; Schonrock, Z.; Covington, B.C.; Maschek, J.A.; Cox, J.E.; et al. Metabolic model for diversity-generating biosynthesis. Proc. Natl. Acad. Sci. USA 2016, 113, 1772–1777. [Google Scholar] [CrossRef] [PubMed]
- Adaba, R.I.; Mann, G.; Raab, A.; Houssen, W.E.; McEwan, A.R.; Thomas, L.; Tabudravu, J.; Naismith, J.H.; Jaspars, M. Accurate quantification of modified cyclic peptides without the need for authentic standards. Tetrahedron 2016, 72, 8603–8609. [Google Scholar] [CrossRef]
- Bertram, A.; Pattenden, G. Marine metabolites: Metal binding and metal complexes of azole-based cyclic peptides of marine origin. Nat. Prod. Rep. 2007, 24, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Urda, C.; Fernández, R.; Rodríguez, J.; Pérez, M.; Jiménez, C.; Cuevas, C. Bistratamides M and N, oxazole-thiazole containing cyclic hexapeptides isolated from Lissoclinum bistratum interaction of zinc (II) with bistratamide K. Mar. Drugs 2017, 15, 209. [Google Scholar] [CrossRef] [PubMed]
- Comba, P.; Dovalil, N.; Gahan, L.R.; Hanson, G.R.; Westphal, M. Cyclic peptide marine metabolites and Cu II. Dalton Trans. 2014, 43, 1935–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comba, P.; Gahan, L.R.; Hanson, G.R.; Maeder, M.; Westphal, M. Carbonic anhydrase activity of dinuclear CuII complexes with patellamide model ligands. Dalton Trans. 2014, 43, 3144–3152. [Google Scholar] [CrossRef] [PubMed]
- Comba, P.; Eisenschmidt, A.; Gahan, L.R.; Herten, D.P.; Nette, G.; Schenk, G.; Seefeld, M. Is CuII Coordinated to Patellamides inside Prochloron Cells? Chem. Eur. J. 2017, 23, 12264–12274. [Google Scholar] [CrossRef] [PubMed]
- Lopez, D.; Martinez-Luis, S. Marine natural products with P-glycoprotein inhibitor properties. Mar. Drugs 2014, 12, 525–546. [Google Scholar] [CrossRef] [PubMed]
- Wipf, P.; Uto, Y. Total synthesis and revision of stereochemistry of the marine metabolite trunkamide A. J. Org. Chem. 2000, 65, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Savchenko, A.I.; Kerscher, M.; Grange, R.L.; Krenske, E.H.; Harmer, J.R.; Bauer, M.J.; Broit, N.; Watters, D.J.; Boyle, B.M.; et al. Heteratom-Interchanged isomers of Lissoclinamide 5: Copper(II) complexation, halide binding and biological activity. Eur. J. Org. Chem. 2018, 12, 1465–1476. [Google Scholar] [CrossRef]
- Salib, M.N.; Molinski, T.F. Cyclic Hexapeptide Dimers, Antatollamides A and B, from the Ascidian Didemnum molle. A Tryptophan-Derived Auxiliary for l-and d-Amino Acid Assignments. J. Org. Chem. 2017, 82, 10181–10187. [Google Scholar] [CrossRef] [PubMed]
- Taevernier, L.; Wynendaele, E.; Gevaert, B.; De Spiegeleer, B. Chemical classification of cyclic depsipeptides. Curr. Protein Peptide Sci. 2017, 18, 425–452. [Google Scholar] [CrossRef] [PubMed]
- Süssmuth, R.D.; Mainz, A. Nonribosomal Peptide Synthesis—Principles and Prospects. Angew. Chem. Int. Ed. 2017, 56, 3770–3821. [Google Scholar] [CrossRef] [PubMed]
- Amoutzias, G.D.; Chaliotis, A.; Mossialos, D. Discovery strategies of bioactive compounds synthesized by nonribosomal peptide synthetases and type-I polyketide synthases derived from marine microbiomes. Mar. Drugs 2016, 14, 80. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, K.L., Jr.; Gloer, J.B.; Cook, J.C., Jr.; Mizsak, S.A.; Scahill, T.A. Structures of the didemnins, antiviral and cytotoxic depsipeptides from a Caribbean tunicate. J. Am. Chem. Soc. 1981, 103, 1857–1859. [Google Scholar] [CrossRef]
- Marco, E.; Martín-Santamaría, S.; Cuevas, C.; Gago, F. Structural basis for the binding of didemnins to human elongation factor eEF1A and rationale for the potent antitumor activity of these marine natural products. J. Med. Chem. 2004, 47, 4439–4452. [Google Scholar] [CrossRef] [PubMed]
- Potts, M.B.; McMillan, E.A.; Rosales, T.I.; Kim, H.S.; Ou, Y.H.; Toombs, J.E.; Brekken, R.A.; Minden, M.D.; MacMillan, J.B.; White, M.A. Mode of action and pharmacogenomic biomarkers for exceptional responders to didemnin B. Nat. Chem. Biol. 2015, 11, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Thell, K.; Hellinger, R.; Schabbauer, G.; Gruber, C.W. Immunosuppressive peptides and their therapeutic applications. Drug Discov. Today 2014, 19, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Álvarez, S.; Pardal, E.; Sánchez-Nieto, D.; Navarro, M.; Caballero, M.D.; Mateos, M.V.; Martín, A. Plitidepsin: Design, development, and potential place in therapy. Drug Des. Dev. Ther. 2017, 11, 253–264. [Google Scholar] [CrossRef] [PubMed]
- US National Library of Medicine. Available online: www.clinicaltrials.gov (accessed on 15 January 2018).
- PharmaMar. PharmaMar Has Requested the Process of Re-Examination for Aplidin® from the EMA. Available online: https://www.pharmamar.com/wp-content/uploads/2018/01/PR_PharmaMar-has-requested-the-process-of-re-examination-for-Aplidin%C2%AE-from-the-EMA.pdf (accessed on 25 March 2018).
- Losada, A.; Muñoz-Alonso, M.J.; García, C.; Sánchez-Murcia, P.A.; Martínez-Leal, J.F.; Domínguez, J.M.; Lillo, M.P.; Gago, F.; Galmarini, C.M. Translation elongation factor eEF1A2 is a novel anticancer target for the marine natural product plitidepsin. Sci. Rep. 2016, 6, 35100. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Alonso, M.J.; González-Santiago, L.; Zarich, N.; Martínez, T.; Alvarez, E.; Rojas, J.M.; Muñoz, A. Plitidepsin has a dual effect inhibiting cell cycle and inducing apoptosis via Rac1/c-Jun NH2-terminal kinase activation in human melanoma cells. J. Pharmacol. Exp. Ther. 2008, 324, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- González-Santiago, L.; Suárez, Y.; Zarich, N.; Muñoz-Alonso, M.J.; Cuadrado, A.; Martinez, T.; Goya, L.; Iradi, A.; Sáez-Tormo, G.; Maier, J.V.; et al. Aplidin® induces JNK-dependent apoptosis in human breast cancer cells via alteration of glutathione homeostasis, Rac1 GTPase activation, and MKP-1 phosphatase downregulation. Cell Death Differ. 2006, 13, 1968–1981. [Google Scholar] [CrossRef] [PubMed]
- Broggini, M.; Marchini, S.V.; Galliera, E.; Borsotti, P.; Taraboletti, G.; Erba, E.; Sironi, M.; Jimeno, J.; Faircloth, G.T.; Giavazzi, R.; et al. Aplidine, a new anticancer agent of marine origin, inhibits vascular endothelial growth factor (VEGF) secretion and blocks VEGF-VEGFR-1 (flt-1) autocrine loop in human leukemia cells MOLT-4. Leukemia 2003, 17, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Borjan, B.; Steiner, N.; Karbon, S.; Kern, J.; Francesch, A.; Hermann, M.; Willenbacher, W.; Gunsilius, E.; Untergasser, G. The Aplidin analogs PM01215 and PM02781 inhibit angiogenesis in vitro and in vivo. BMC Cancer 2015, 15, 738. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Currano, J.N.; Carroll, P.J.; Joullié, M.M. Didemnins, tamandarins and related natural products. Nat. Prod. Rep. 2012, 29, 404–424. [Google Scholar] [CrossRef] [PubMed]
- Tsukimoto, M.; Nagaoka, M.; Shishido, Y.; Fujimoto, J.; Nishisaka, F.; Matsumoto, S.; Harunari, E.; Imada, C.; Matsuzaki, T. Bacterial production of the tunicate-derived antitumor cyclic depsipeptide didemnin B. J. Nat. Prod. 2011, 74, 2329–2331. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Kersten, R.D.; Nam, S.J.; Lu, L.; Al-Suwailem, A.M.; Zheng, H.; Fenical, W.; Dorrestein, P.C.; Moore, B.S.; Qian, P.Y. Bacterial biosynthesis and maturation of the didemnin anti-cancer agents. J. Am. Chem. Soc. 2012, 134, 8625–8632. [Google Scholar] [CrossRef] [PubMed]
- Adrio, J.; Cuevas, C.; Manzanares, I.; Joullié, M.M. Total synthesis and biological evaluation of tamandarin B analogues. J. Org. Chem. 2007, 72, 5129–5138. [Google Scholar] [CrossRef] [PubMed]
- Kwan, J.C.; Donia, M.S.; Han, A.W.; Hirose, E.; Haygood, M.G.; Schmidt, E.W. Genome streamlining and chemical defense in a coral reef symbiosis. Proc. Natl. Acad. Sci. USA 2012, 109, 20655–20660. [Google Scholar] [CrossRef] [PubMed]
- Kwan, J.C.; Schmidt, E.W. Bacterial endosymbiosis in a chordate host: Long-term co-evolution and conservation of secondary metabolism. PLoS ONE 2013, 8, e80822. [Google Scholar] [CrossRef] [PubMed]
- Helfrich, E.J.; Piel, J. Biosynthesis of polyketides by trans-AT polyketide synthases. Nat. Prod. Rep. 2016, 33, 231–316. [Google Scholar] [CrossRef] [PubMed]
- Sikorska, J.; Hau, A.M.; Anklin, C.; Parker-Nance, S.; Davies-Coleman, M.T.; Ishmael, J.E.; McPhail, K.L. Mandelalides A–D, cytotoxic macrolides from a new Lissoclinum species of South African tunicate. J. Org. Chem. 2012, 77, 6066–6075. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Yan, J.; Yu, J.; Liu, Y.; Wang, Z.; Xu, Z.; Ye, T. Total synthesis and stereochemical reassignment of mandelalide A. Angew. Chem. Int. Ed. 2014, 53, 6533–6537. [Google Scholar] [CrossRef] [PubMed]
- Veerasamy, N.; Ghosh, A.; Li, J.; Watanabe, K.; Serrill, J.D.; Ishmael, J.E.; McPhail, K.L.; Carter, R.G. Enantioselective total synthesis of mandelalide A and isomandelalide A: Discovery of a cytotoxic ring-expanded isomer. J. Am. Chem. Soc. 2016, 138, 770–773. [Google Scholar] [CrossRef] [PubMed]
- Nazari, M.; Serrill, J.D.; Sikorska, J.; Ye, T.; Ishmael, J.E.; McPhail, K.L. Discovery of mandelalide E and determinants of cytotoxicity for the mandelalide series. Org. Lett. 2016, 18, 1374–1377. [Google Scholar] [CrossRef] [PubMed]
- Nazari, M.; Serrill, J.D.; Wan, X.; Nguyen, M.H.; Anklin, C.; Gallegos, D.A.; Smith, A.B., III; Ishmael, J.E.; McPhail, K.L. New mandelalides expand a macrolide series of mitochondrial inhibitors. J. Med. Chem. 2017, 60, 7850–7862. [Google Scholar] [CrossRef] [PubMed]
- Issac, M.; Aknin, M.; Gauvin-Bialecki, A.; Pond, C.D.; Barrows, L.R.; Kashman, Y.; Carmeli, S. Mollecarbamates, Molleureas, and Molledihydroisoquinolone, o-Carboxyphenethylamide Metabolites of the Ascidian Didemnum molle Collected in Madagascar. J. Nat. Prod. 2017, 80, 1844–1852. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, I.; Suzuki, K.; Okamura, M.; Funakubo, S.; Onozaki, Y.; Kawamura, D.; Ohyoshi, T.; Kigoshi, H. Total Synthesis of Biselide E, a Marine Polyketide. Org. Lett. 2017, 19, 5713–5716. [Google Scholar] [CrossRef] [PubMed]
- Menna, M.; Fattorusso, E.; Imperatore, C. Alkaloids from marine ascidians. Molecules 2011, 16, 8694–8732. [Google Scholar] [CrossRef] [Green Version]
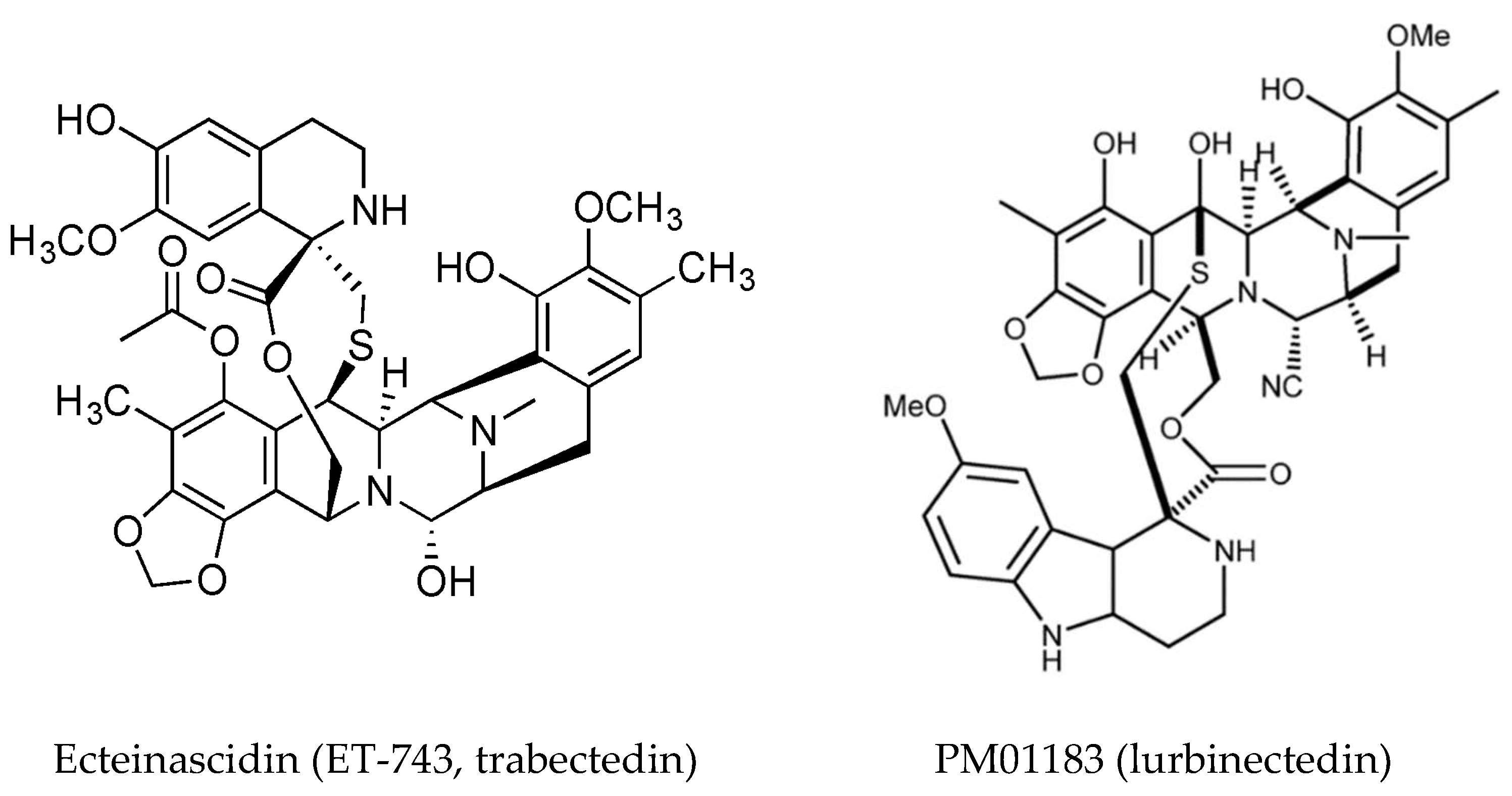
- Sakai, R.; Rinehart, K.L.; Guan, Y.; Wang, A.H. Additional antitumor ecteinascidins from a Caribbean tunicate: Crystal structures and activities in vivo. Proc. Natl. Acad. Sci. USA 1992, 89, 11456–11460. [Google Scholar] [CrossRef] [PubMed]
- Le, V.H.; Inai, M.; Williams, R.M.; Kan, T. Ecteinascidins. A review of the chemistry, biology and clinical utility of potent tetrahydroisoquinoline antitumor antibiotics. Nat. Prod. Rep. 2015, 32, 328–347. [Google Scholar] [CrossRef] [PubMed]
- Gordon, E.M.; Sankhala, K.K.; Chawla, N.; Chawla, S.P. Trabectedin for soft tissue sarcoma: Current status and future perspectives. Adv. Ther. 2016, 33, 1055–1071. [Google Scholar] [CrossRef] [PubMed]
- D’Incalci, M.; Galmarini, C.M. A review of trabectedin (ET-743): A unique mechanism of action. Mol. Cancer Ther. 2010, 9, 2157–2163. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Gorfajn, B.; Faircloth, G.; Scotto, K.W. Ecteinascidin 743, a transcription-targeted chemotherapeutic that inhibits MDR1 activation. Proc. Natl. Acad. Sci. USA 2000, 97, 6775–6779. [Google Scholar] [CrossRef] [PubMed]
- D’Incalci, M.; Badri, N.; Galmarini, C.M.; Allavena, P. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br. J. Cancer 2014, 111, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of macrophage targeting in the antitumor activity of trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Gomes, N.G.; Dasari, R.; Chandra, S.; Kiss, R.; Kornienko, A. Marine invertebrate metabolites with anticancer activities: Solutions to the “supply problem”. Mar. Drugs 2016, 14, 98. [Google Scholar] [CrossRef] [PubMed]
- Leal, J.F.; Martínez-Díez, M.; García-Hernández, V.; Moneo, V.; Domingo, A.; Bueren-Calabuig, J.A.; Negri, A.; Gago, F.; Guillén-Navarro, M.J.; Avilés, P.; et al. PM01183, a new DNA minor groove covalent binder with potent in vitro and in vivo anti-tumour activity. Brit. J. Pharmacol. 2010, 161, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, G.S.; Robles, C.M.; Giraudon, C.; Martínez-Leal, J.F.; Compe, E.; Coin, F.; Aviles, P.; Galmarini, C.M.; Egly, J.M. Lurbinectedin specifically triggers the degradation of phosphorylated RNA polymerase II and the formation of DNA breaks in cancer cells. Mol. Cancer Ther. 2016, 15, 2399–2412. [Google Scholar] [CrossRef] [PubMed]
- Soares, D.G.; Machado, M.S.; Rocca, C.J.; Poindessous, V.; Ouaret, D.; Sarasin, A.; Galmarini, C.M.; Henriques, J.A.; Escargueil, A.E.; Larsen, A.K. Trabectedin and its C subunit modified analogue PM01183 attenuate nucleotide excision repair and show activity toward platinum-resistant cells. Mol. Cancer Ther. 2011, 10, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Calvo, E.; Moreno, V.; Flynn, M.; Holgado, E.; Olmedo, M.E.; Lopez Criado, M.P.; Kahatt, C.; Lopez-Vilariño, A.; Siguero, M.; Fernandez-Teruel, C.; et al. Antitumor activity of lurbinectedin (PM01183) and doxorubicin in relapsed small-cell lung cancer: Results from a phase I study. Ann. Oncol. 2017, 28, 2559–2566. [Google Scholar] [CrossRef] [PubMed]
- Lima, M.; Bouzid, H.; Soares, D.G.; Selle, F.; Morel, C.; Galmarini, C.M.; Henriques, J.A.; Larsen, A.K.; Escargueil, A.E. Dual inhibition of ATR and ATM potentiates the activity of trabectedin and lurbinectedin by perturbing the DNA damage response and homologous recombination repair. Oncotarget 2016, 7, 25885. [Google Scholar] [CrossRef] [PubMed]
- Rath, C.M.; Janto, B.; Earl, J.; Ahmed, A.; Hu, F.Z.; Hiller, L.; Dahlgren, M.; Kreft, R.; Yu, F.; Wolff, J.J.; et al. Meta-omic characterization of the marine invertebrate microbial consortium that produces the chemotherapeutic natural product ET-743. ACS Chem. Biol. 2011, 6, 1244–1256. [Google Scholar] [CrossRef] [PubMed]
- Sherman, D.H.; Ehrlich, G.D.; Janto, B.; Williams, R.M.; Rath, C.M. Allegheny-Singer Research Institute, Colorado State University Research Foundation and University of Michigan. Biosynthetic Pathway for Heterologous Expression of a Nonribosomal Peptide Synthetase Drug and Analogs. U.S. Patent 9,611,491, 4 April 2017. [Google Scholar]
- Sandjo, L.P.; Kuete, V.; Biavatti, M.W. Pyridoacridine alkaloids of marine origin: NMR and MS spectral data, synthesis, biosynthesis and biological activity. Beilstein J. Org. Chem. 2015, 11, 1667–1699. [Google Scholar] [CrossRef] [PubMed]
- Kijjoa, A. Pyridoacridine Alkaloids from Marine Origin: Sources and Anticancer Activity. In Handbook of Anticancer Drugs from Marine Origin; Kim, S.-K., Ed.; Springer: Cham, Switzerland, 2015; pp. 771–802. ISBN 978-3-319-07145-9. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, K.; Xu, B. Telomerase Inhibitors from Natural Products and Their Anticancer Potential. Int. J. Mol. Sci. 2017, 19, E13. [Google Scholar] [CrossRef] [PubMed]
- Mengual Gomez, D.L.; Armando, R.G.; Cerrudo, C.S.; Ghiringhelli, P.D.; Gomez, D.E. Telomerase as a cancer target. Development of new molecules. Curr. Top. Med. Chem. 2016, 16, 2432–2440. [Google Scholar] [CrossRef]
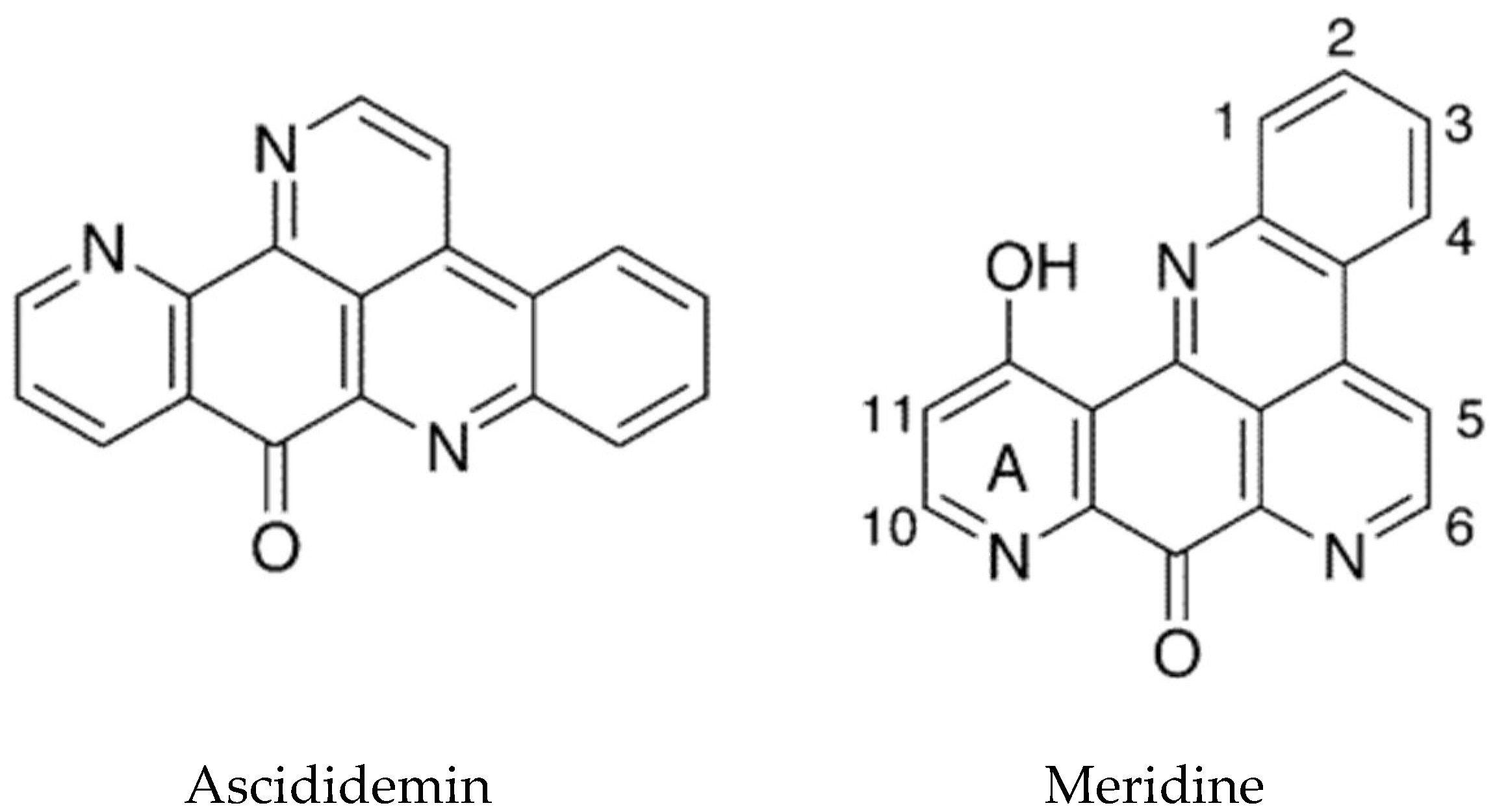
- Guittat, L.; De Cian, A.; Rosu, F.; Gabelica, V.; De Pauw, E.; Delfourne, E.; Mergny, J.L. Ascididemin and meridine stabilise G-quadruplexes and inhibit telomerase in vitro. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2005, 1724, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Delfourne, E.; Darro, F.; Bontemps-Subielos, N.; Decaestecker, C.; Bastide, J.; Frydman, A.; Kiss, R. Synthesis and characterization of the antitumor activities of analogues of meridine, a marine pyridoacridine alkaloid. J. Med. Chem. 2001, 44, 3275–3282. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Sharma, P.C.; Kumar, V. In Silico Molecular Docking Analysis of Natural Pyridoacridines as Anticancer Agents. Adv. Chem. 2016, 2016, 5409387. [Google Scholar] [CrossRef]
- Plodek, A.; Bracher, F. New Perspectives in the Chemistry of Marine Pyridoacridine Alkaloids. Mar. Drugs 2016, 14, 26. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, K.L.; Kobayashi, J.; Harbour, G.C.; Hughes, R.G., Jr.; Mizsak, S.A.; Scahill, T.A. Eudistomins C, E, K, and L, potent antiviral compounds containing a novel oxathiazepine ring from the Caribbean tunicate Eudistoma olivaceum. J. Amer. Chem. Soc. 1984, 106, 1524–1526. [Google Scholar] [CrossRef]
- Giulietti, J.M.; Tate, P.M.; Cai, A.; Cho, B.; Mulcahy, S.P. DNA-binding studies of the natural β-carboline eudistomin U. Bioorg. Med. Chem. Lett. 2016, 26, 4705–4708. [Google Scholar] [CrossRef] [PubMed]
- Ota, Y.; Chinen, T.; Yoshida, K.; Kudo, S.; Nagumo, Y.; Shiwa, Y.; Yamada, R.; Umihara, H.; Iwasaki, K.; Masumoto, H.; et al. Eudistomin C, an antitumor and antiviral natural product, targets 40S ribosome and inhibits protein translation. ChemBioChem 2016, 17, 1616–1620. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Singh, A.; Kumar, K.; Kumar, V. Recent insights into synthetic β-carbolines with anti-cancer activities. Eur. J. Med. Chem. 2017, 142, 48–73. [Google Scholar] [CrossRef] [PubMed]
- Laine, A.E.; Lood, C.; Koskinen, A.M. Pharmacological importance of optically active tetrahydro-β-carbolines and synthetic approaches to create the C1 stereocenter. Molecules 2014, 19, 1544–1567. [Google Scholar] [CrossRef] [PubMed]
- Panarese, J.D.; Waters, S.P. Room-temperature aromatization of tetrahydro-β-carbolines by 2-iodoxybenzoic acid: Utility in a total synthesis of Eudistomin U. Org. Lett. 2010, 12, 4086–4089. [Google Scholar] [CrossRef] [PubMed]
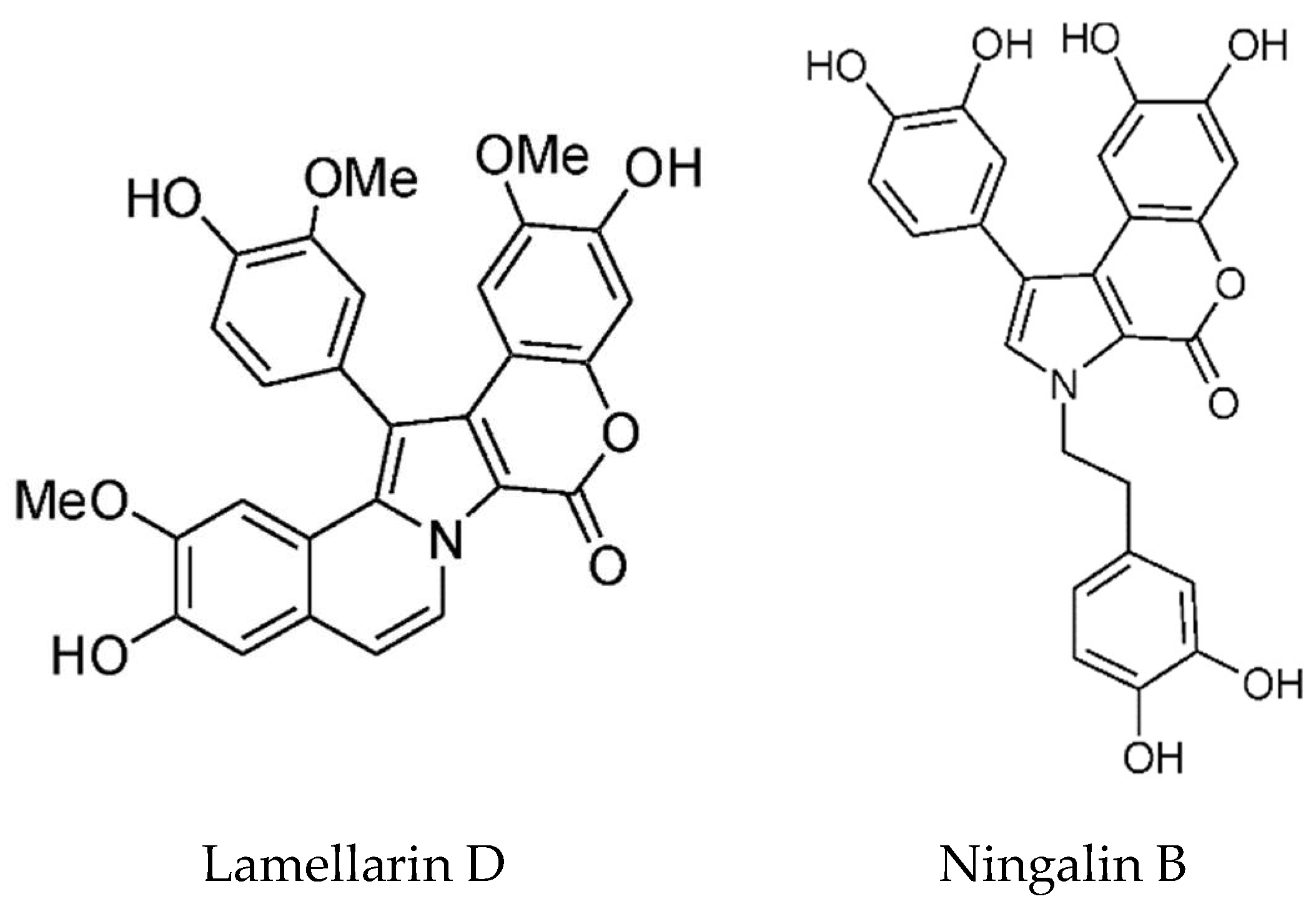
- Fan, H.; Peng, J.; Hamann, M.T.; Hu, J.F. Lamellarins and related pyrrole-derived alkaloids from marine organisms. Chem. Rev. 2008, 108, 264–287. [Google Scholar] [CrossRef] [PubMed]
- Marco, E.; Laine, W.; Tardy, C.; Lansiaux, A.; Iwao, M.; Ishibashi, F.; Bailly, C.; Gago, F. Molecular Determinants of Topoisomerase I Poisoning by Lamellarins: Comparison with Camptothecin and Structure–Activity Relationships. J. Med. Chem. 2005, 48, 3796–3807. [Google Scholar] [CrossRef] [PubMed]
- Bailly, C. Anticancer properties of lamellarins. Mar. Drugs 2015, 13, 1105–1123. [Google Scholar] [CrossRef] [PubMed]
- Khiati, S.; Seol, Y.; Agama, K.; Dalla Rosa, I.; Agrawal, S.; Fesen, K.; Zhang, H.; Neuman, K.C.; Pommier, Y. Poisoning of mitochondrial topoisomerase I by lamellarin D. Mol. Pharmacol. 2014, 86, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Ballot, C.; Martoriati, A.; Jendoubi, M.; Buche, S.; Formstecher, P.; Mortier, L.; Kluza, J.; Marchetti, P. Another facet to the anticancer response to lamellarin D: Induction of cellular senescence through inhibition of topoisomerase I and intracellular ROS production. Mar. Drugs 2014, 12, 779–798. [Google Scholar] [CrossRef] [PubMed]
- Long, S.; Sousa, E.; Kijjoa, A.; Pinto, M.M. Marine natural products as models to circumvent multidrug resistance. Molecules 2016, 21, 892. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Kumar, P.; Anreddy, N.; Xiao, X.; Yang, D.H.; Chen, Z.S. P-gp Inhibitory Activity from Marine Sponges, Tunicates and Algae. In Handbook of Anticancer Drugs from Marine Origin; Kim, S.-K., Ed.; Springer: Cham, Switzerland, 2015; pp. 593–619. ISBN 978-3-319-07145-9. [Google Scholar]
- Plisson, F.; Huang, X.C.; Zhang, H.; Khalil, Z.; Capon, R.J. Lamellarins as Inhibitors of P-Glycoprotein-Mediated Multidrug Resistance in a Human Colon Cancer Cell Line. Chem. Asian J. 2012, 7, 1616–1623. [Google Scholar] [CrossRef] [PubMed]
- Imbri, D.; Tauber, J.; Opatz, T. Synthetic approaches to the lamellarins—A comprehensive review. Mar. Drugs 2014, 12, 6142–6177. [Google Scholar] [CrossRef] [PubMed]
- Lade, D.M.; Pawar, A.B.; Mainkar, P.S.; Chandrasekhar, S. Total Synthesis of Lamellarin D Trimethyl Ether, Lamellarin D, and Lamellarin H. J. Org. Chem. 2017, 82, 4998–5004. [Google Scholar] [CrossRef] [PubMed]
- Manjappa, K.B.; Lin, J.M.; Yang, D.Y. Construction of Pentacyclic Lamellarin Skeleton via Grob Reaction: Application to Total Synthesis of Lamellarins H and D. J. Org. Chem. 2017, 82, 7648–7656. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wong, I.L.; Peng, K.; Liu, Z.; Wang, P.; Jiang, T.; Jiang, T.; Chow, L.M.; Wan, S.B. Extending the structure–activity relationship study of marine natural ningalin B analogues as P-glycoprotein inhibitors. Eur. J. Med. Chem. 2017, 125, 795–806. [Google Scholar] [CrossRef] [PubMed]
- Plisson, F.; Conte, M.; Khalil, Z.; Huang, X.C.; Piggott, A.M.; Capon, R.J. Kinase inhibitor scaffolds against neurodegenerative diseases from a Southern Australian ascidian, Didemnum sp. ChemMedChem 2012, 7, 983–990. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kim, D.H.; Jeon, T.H.; Kim, W.H.; Cho, C.G. Total Syntheses of Ningalins D and G. Org. Lett. 2017, 19, 4688–4691. [Google Scholar] [CrossRef] [PubMed]
- Cherigo, L.; Lopez, D.; Martinez-Luis, S. Marine natural products as breast cancer resistance protein inhibitors. Mar. Drugs 2015, 13, 2010–2029. [Google Scholar] [CrossRef] [PubMed]
- Takada, K.; Imamura, N.; Gustafson, K.R.; Henrich, C.J. Synthesis and structure–activity relationship of botryllamides that block the ABCG2 multidrug transporter. Bioorg. Med. Chem. Lett. 2010, 20, 1330–1333. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.R.; Sharma, S.; Joshi, P.; Wani, A.; Vishwakarma, R.A.; Kumar, A.; Bharate, S.B. Meridianin derivatives as potent Dyrk1A inhibitors and neuroprotective agents. Bioorg. Med. Chem. Lett. 2015, 25, 2948–2952. [Google Scholar] [CrossRef] [PubMed]
- Bharate, S.B.; Yadav, R.R.; Battula, S.; Vishwakarma, A.R. Meridianins: Marine-derived potent kinase inhibitors. Mini Rev. Med. Chem. 2012, 12, 618–631. [Google Scholar] [CrossRef] [PubMed]
- More, K.N.; Jang, H.W.; Hong, V.S.; Lee, J. Pim kinase inhibitory and antiproliferative activity of a novel series of meridianin C derivatives. Bioorg. Med. Chem. Lett. 2014, 24, 2424–2428. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.K.; Lee, T.Y.; Choi, J.S.; Hong, V.S.; Lee, J.; Park, J.W.; Jang, B.C. Inhibition of adipogenesis and leptin production in 3T3-L1 adipocytes by a derivative of meridianin C. Biochem. Biophys. Res. Commun. 2014, 452, 1078–1083. [Google Scholar] [CrossRef] [PubMed]
- Núñez-Pons, L.; Nieto, R.M.; Avila, C.; Jiménez, C.; Rodríguez, J. Mass spectrometry detection of minor new meridianins from the antarctic colonial ascidians Aplidium falklandicum and Aplidium meridianum. J. Mass Spectrom. 2015, 50, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Sandtorv, A.H. Chemical Synthesis of Meridianins and Related Derivatives. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, Netherlands, 2017; Volume 53, pp. 143–166. [Google Scholar]
- Jarry, M.; Lecointre, C.; Malleval, C.; Desrues, L.; Schouft, M.T.; Lejoncour, V.; Liger, F.; Lyvinec, G.; Joseph, B.; Loaëc, N.; et al. Impact of meriolins, a new class of cyclin-dependent kinase inhibitors, on malignant glioma proliferation and neo-angiogenesis. Neuro-Oncol. 2014, 16, 1484–1498. [Google Scholar] [CrossRef] [PubMed]
- Liberio, M.S.; Sadowski, M.C.; Davis, R.A.; Rockstroh, A.; Vasireddy, R.; Lehman, M.L.; Nelson, C.C. The ascidian natural product eusynstyelamide B is a novel topoisomerase II poison that induces DNA damage and growth arrest in prostate and breast cancer cells. Oncotarget 2015, 6, 43944. [Google Scholar] [CrossRef] [PubMed]
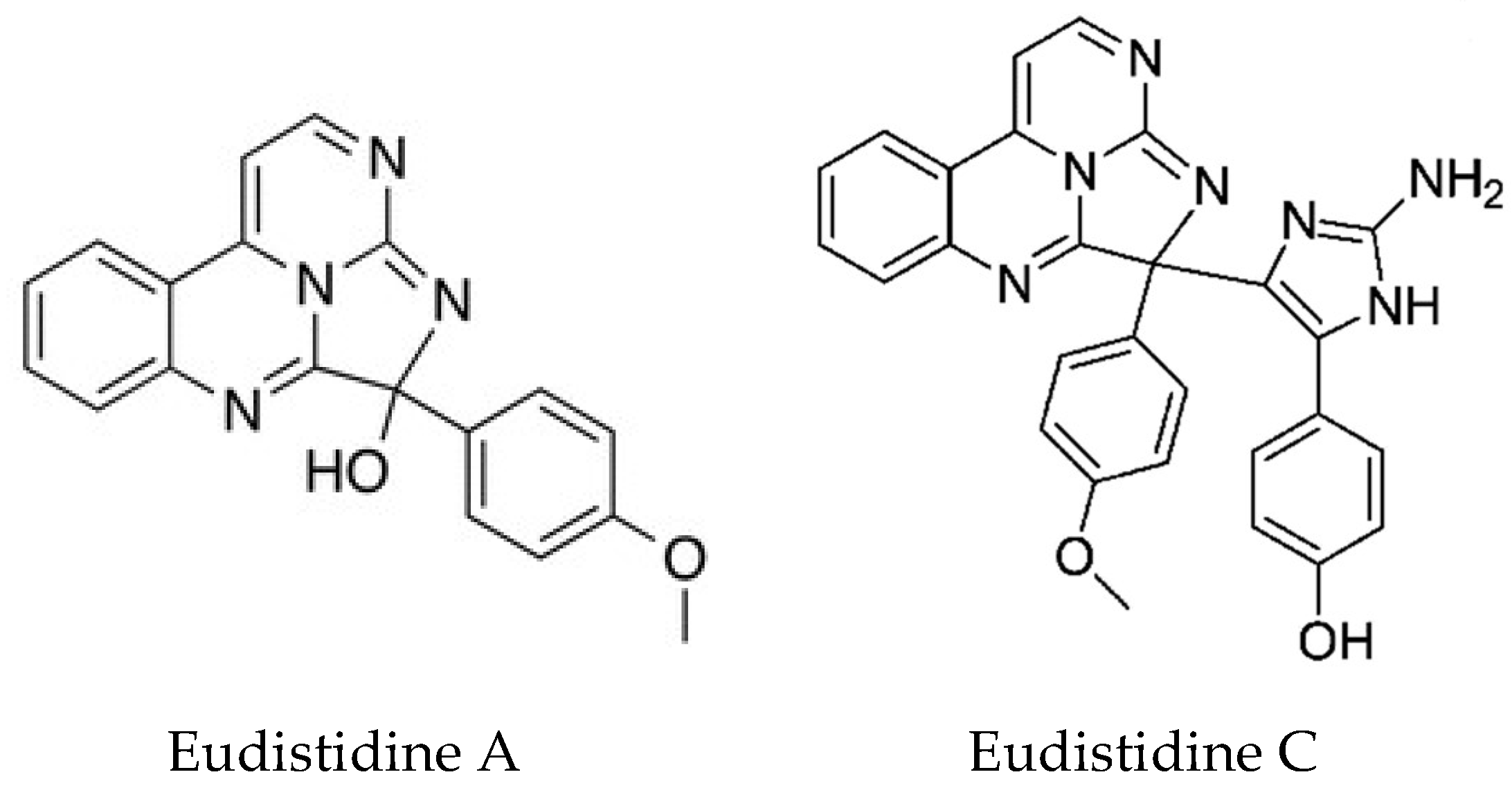
- Chan, S.T.; Patel, P.R.; Ransom, T.R.; Henrich, C.J.; McKee, T.C.; Goey, A.K.; Cook, K.M.; Figg, W.D.; McMahon, J.B.; Schnermann, M.J.; et al. Structural Elucidation and Synthesis of Eudistidine A: An Unusual Polycyclic Marine Alkaloid that Blocks Interaction of the Protein Binding Domains of p300 and HIF-1α. J. Am. Chem. Soc. 2015, 137, 5569–5575. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.T.; Nani, R.R.; Schauer, E.A.; Martin, G.E.; Williamson, R.T.; Saurí, J.; Buevich, A.V.; Schafer, W.A.; Joyce, L.A.; Goey, A.K.; et al. Characterization and Synthesis of Eudistidine C, a Bioactive Marine Alkaloid with an Intriguing Molecular Scaffold. J. Org. Chem. 2016, 81, 10631–10640. [Google Scholar] [CrossRef] [PubMed]
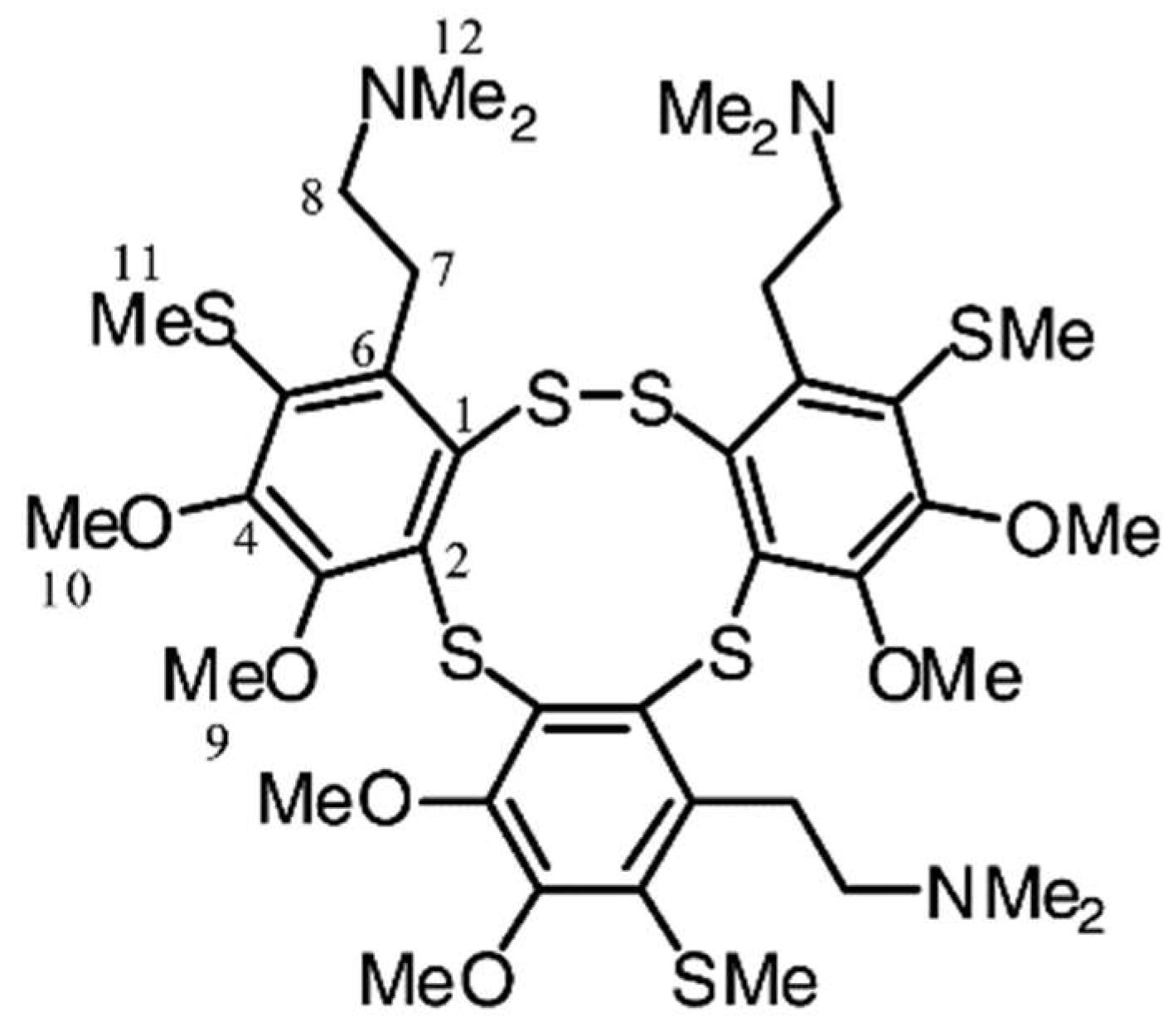
- Tatsuta, T.; Hosono, M.; Rotinsulu, H.; Wewengkang, D.S.; Sumilat, D.A.; Namikoshi, M.; Yamazaki, H. Lissoclibadin 1, a Polysulfur Aromatic Alkaloid from the Indonesian Ascidian Lissoclinum cf. badium, induces Caspase-Dependent Apoptosis in Human Colon Cancer Cells and Suppresses Tumor Growth in Nude Mice. J. Nat. Prod. 2017, 80, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Oda, T.; Fujiwara, T.; Liu, H.; Ukai, K.; Mangindaan, R.E.; Mochizuki, M.; Namikoshi, M. Effects of Lissoclibadins and Lissoclinotoxins, Isolated from a Tropical Ascidian Lissoclinum cf. badium, on IL-8 production in a PMA-stimulated Promyelocytic Leukemia Cell Line. Mar. Drugs 2006, 4, 15–21. [Google Scholar] [CrossRef]
- Fukuzawa, S.; Matsunaga, S.; Fusetani, N. Isolation of 13 New Ritterazines from the Tunicate Ritterella tokioka and Chemical Transformation of Ritterazine B1. J. Org. Chem. 1997, 62, 4484–4491. [Google Scholar] [CrossRef] [PubMed]
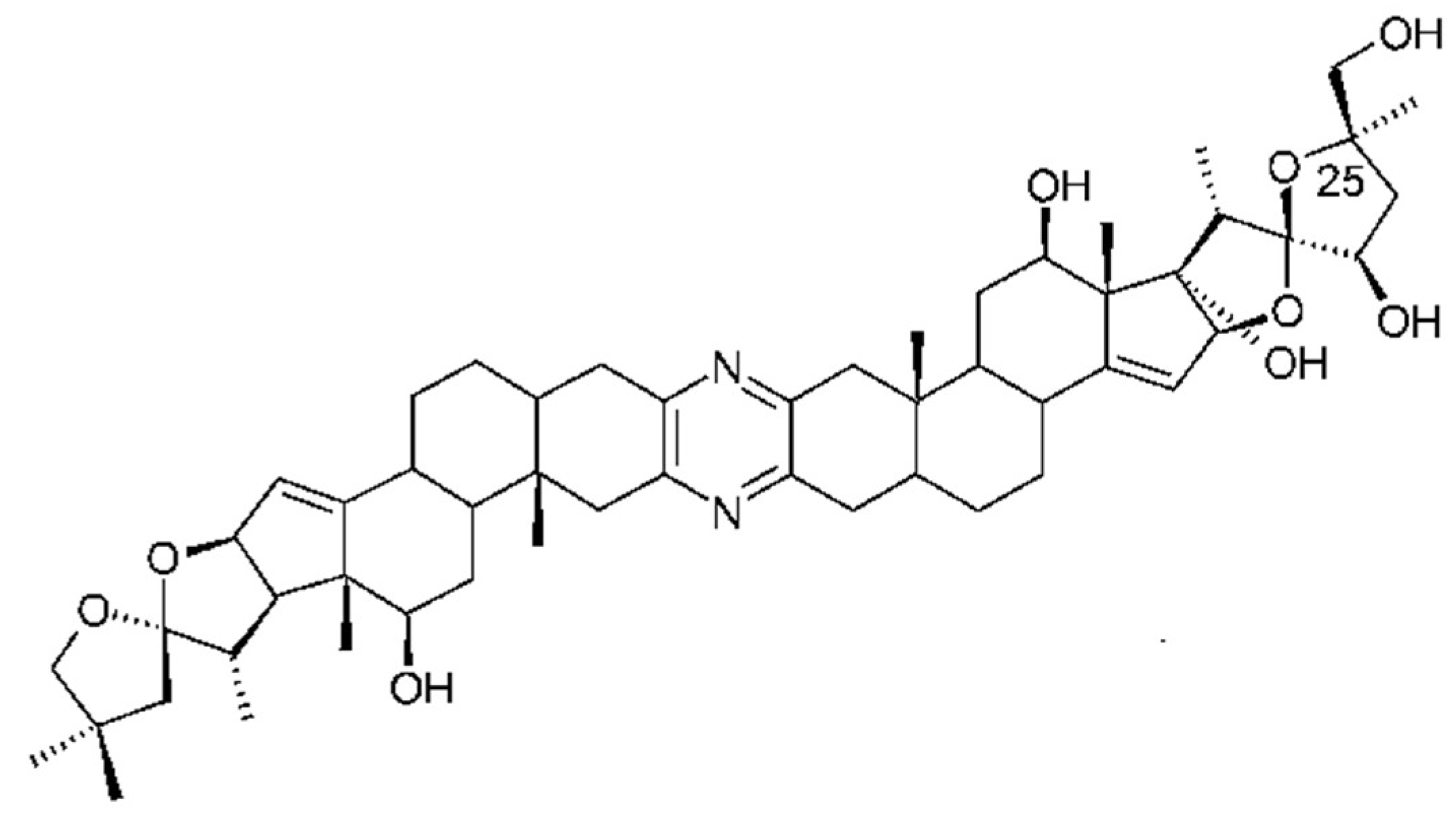
- Lee, S.; LaCour, T.G.; Fuchs, P.L. Chemistry of trisdecacyclic pyrazine antineoplastics: The cephalostatins and ritterazines. Chem. Rev. 2009, 109, 2275–2314. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, A.J.; Santos, E.A.; Jimenez, P.C.; Rocha, D.D.; Wilke, D.V.; Beuzer, P.; Axelrod, J.; Kanduluru, A.K.; Fuchs, P.L.; Cang, H.; et al. Ritterostatin GN1N, a Cephalostatin–Ritterazine Bis-steroidal Pyrazine Hybrid, Selectively Targets GRP78. ChemBioChem 2017, 18, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Tahtamouni, L.H.; Nawasreh, M.M.; Al-Mazaydeh, Z.A.; Al-Khateeb, R.A.; Abdellatif, R.N.; Bawadi, R.M.; Bamburg, J.R.; Yasin, S.R. Cephalostatin 1 analogues activate apoptosis via the endoplasmic reticulum stress signaling pathway. Eur. J. Pharmacol. 2018, 818, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Jia, L.; Xiao, Q.; Lan, Q.; Tang, X.; Wang, D.; Li, M.; Ji, Y.; Zhou, T.; Tian, W. A practical synthesis of cephalostatin 1. Chem. Asian J. 2011, 6, 786–790. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Jiang, X.L.; Tian, W.S. Synthesis of 12,12′-azo-13,13′-diepi-Ritterazine N. J. Org. Chem. 2017, 82, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Piccialli, V. Ruthenium tetroxide and perruthenate chemistry. Recent advances and related transformations mediated by other transition metal oxo-species. Molecules 2014, 19, 6534–6582. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.D.; Pham, N.B.; Ekins, M.; Hooper, J.N.; Quinn, R.J. Isolation and total synthesis of stolonines A–C, unique taurine amides from the Australian marine tunicate Cnemidocarpa stolonifera. Mar. Drugs 2015, 13, 4556–4575. [Google Scholar] [CrossRef] [PubMed]
- Won, T.H.; Kim, C.K.; Lee, S.H.; Rho, B.J.; Lee, S.K.; Oh, D.C.; Oh, K.B.; Shin, J. Amino Acid-Derived Metabolites from the Ascidian Aplidium sp. Mar. Drugs 2015, 13, 3836–3848. [Google Scholar] [CrossRef] [PubMed]
- Biard, J.F.; Malochet-Grivois, C.; Roussakis, C.; Cotelle, P.; Hénichart, J.P.; Débitus, C.; Verbist, J.F. Lissoclimides, cytotoxic diterpenes from Lissoclinum voeltzkowi Michaelsen. Nat. Prod. Lett. 1994, 4, 43–50. [Google Scholar] [CrossRef]
- Könst, Z.A.; Szklarski, A.R.; Pellegrino, S.; Michalak, S.E.; Meyer, M.; Zanette, C.; Cencic, R.; Nam, S.; Voora, V.K.; Horne, D.A.; et al. Synthesis facilitates an understanding of the structural basis for translation inhibition by the lissoclimides. Nat. Chem. 2017, 9, 1140–1149. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, V.; Plunkett, W.; Cortes, J.E. Omacetaxine: A protein translation inhibitor for treatment of chronic myelogenous leukemia. Clin. Cancer Res. 2014, 20, 1735–1740. [Google Scholar] [CrossRef] [PubMed]
- Quinn, R.K.; Könst, Z.A.; Michalak, S.E.; Schmidt, Y.; Szklarski, A.R.; Flores, A.R.; Nam, S.; Horne, D.A.; Vanderwal, C.D.; Alexanian, E.J. Site-selective aliphatic C–H chlorination using N-chloroamides enables a synthesis of chlorolissoclimide. J. Am. Chem. Soc. 2016, 138, 696–702. [Google Scholar] [CrossRef] [PubMed]
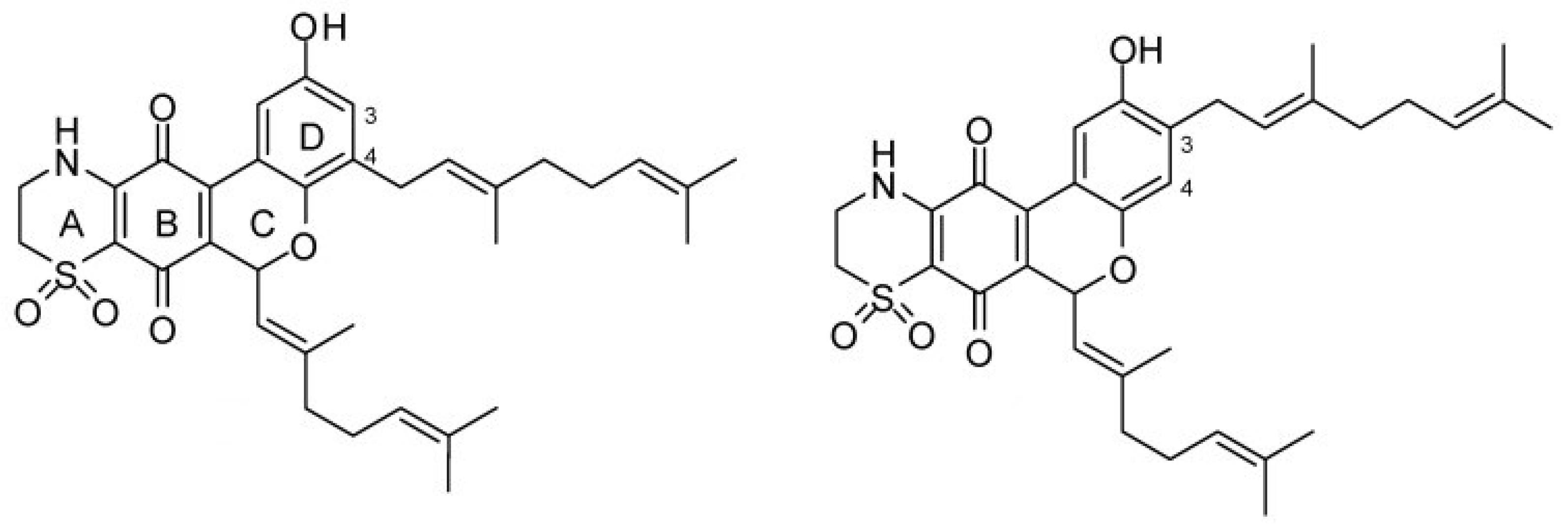
- Imperatore, C.; Cimino, P.; Cebrián-Torrejón, G.; Persico, M.; Aiello, A.; Senese, M.; Fattorusso, C.; Menna, M.; Doménech-Carbó, A. Insight into the Mechanism of Action of Marine Cytotoxic Thiazinoquinones. Mar. Drugs 2017, 15, 335. [Google Scholar] [CrossRef] [PubMed]
- Aiello, A.; Fattorusso, E.; Luciano, P.; Macho, A.; Menna, M.; Muñoz, E. Antitumor effects of two novel naturally occurring terpene quinones isolated from the Mediterranean ascidian Aplidium conicum. J. Med. Chem. 2005, 48, 3410–3416. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.L.; Khalil, I.M.; Shaw, L.; Bourguet-Kondracki, M.L.; Dubois, J.; Valentin, A.; Barker, D.; Copp, B.R. Structure–activity relationships of the bioactive thiazinoquinone marine natural products thiaplidiaquinones A and B. Mar. Drugs 2015, 13, 5102–5110. [Google Scholar] [CrossRef] [PubMed]
- Cadelis, M.M.; Bourguet-Kondracki, M.L.; Dubois, J.; Kaiser, M.; Brunel, J.M.; Barker, D.; Copp, B.R. Structure–activity relationship studies on thiaplidiaquinones A and B as novel inhibitors of Plasmodium falciparum and farnesyltransferase. Bioorg. Med. Chem. 2017, 25, 4433–4443. [Google Scholar] [CrossRef] [PubMed]
- Possner, S.T.; Schroeder, F.C.; Rapp, H.T.; Sinnwell, V.; Franke, S.; Francke, W. 3, 7-Isoquinoline quinones from the ascidian tunicate Ascidia virginea. Z. Naturforsch. C. 2017, 72, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Lien, W.C.; Chen, T.Y.; Sheu, S.Y.; Lin, T.C.; Kang, F.C.; Yu, C.H.; Kuan, T.S.; Huang, B.M.; Wang, C.Y. 7-hydroxy-staurosporine, UCN-01, induces DNA damage response and autophagy in human osteosarcoma U2-OS cells. J. Cell. Biochem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhao, B.; Britton, R.; Lim, L.Y.; Leong, D.; Sanghera, J.S.; Zhou, B.B.; Piers, E.; Andersen, R.J.; Roberge, M. Inhibition of Chk1 by the G2 DNA damage checkpoint inhibitor isogranulatimide. Mol. Cancer Ther. 2004, 3, 1221–1227. [Google Scholar] [PubMed]
- Lavrard, H.; Rodriguez, F.; Delfourne, E. Design of granulatimide and isogranulatimide analogues as potential Chk1 inhibitors: Study of amino-platforms for their synthesis. Bioorg. Med. Chem. 2014, 22, 4961–4967. [Google Scholar] [CrossRef] [PubMed]
- Lavrard, H.; Salvetti, B.; Mathieu, V.; Rodriguez, F.; Kiss, R.; Delfourne, E. Synthesis and in vitro antiproliferative activity of amido and amino analogues of the marine alkaloid isogranulatimide. ChemMedChem 2015, 10, 607–609. [Google Scholar] [CrossRef] [PubMed]
- Deslandes, S.; Chassaing, S.; Delfourne, E. Marine pyrrolocarbazoles and analogues: Synthesis and kinase inhibition. Mar. Drugs 2009, 7, 754–786. [Google Scholar] [CrossRef] [PubMed]
- Loaëc, N.; Attanasio, E.; Villiers, B.; Durieu, E.; Tahtouh, T.; Cam, M.; Davis, R.A.; Alencar, A.; Roué, M.; Bourguet-Kondracki, M.L.; et al. Marine-Derived 2-Aminoimidazolone Alkaloids. Leucettamine B-Related Polyandrocarpamines Inhibit Mammalian and Protozoan DYRK & CLK Kinases. Mar. Drugs 2017, 15, 316. [Google Scholar] [CrossRef]
- Oddo, S.; LaFerla, F.M. The role of nicotinic acetylcholine receptors in Alzheimer’s disease. J. Physiol.-Paris 2006, 99, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Llorach-Pares, L.; Nonell-Canals, A.; Sanchez-Martinez, M.; Avila, C. Computer-Aided Drug Design Applied to Marine Drug Discovery: Meridianins as Alzheimer’s Disease Therapeutic Agents. Mar. Drugs 2017, 15, 366. [Google Scholar] [CrossRef] [PubMed]
- Dev, K.; Maurya, R. Marine-Derived Anti-Alzheimer’s Agents of Promise. In Neuroprotective Natural Products: Clinical Aspects and Mode of Action, 1st ed.; Brahmachari, G., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2017; Chapter 7; pp. 153–184. ISBN 978–3-527-80379-8. [Google Scholar]
- Tadesse, M.; Svenson, J.; Sepčić, K.; Trembleau, L.; Engqvist, M.; Andersen, J.H.; Jaspars, M.; Stensvåg, K.; Haug, T. Isolation and synthesis of pulmonarins A and B, acetylcholinesterase inhibitors from the colonial ascidian Synoicum pulmonaria. J. Nat. Prod. 2014, 77, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Tadokoro, Y.; Nishikawa, T.; Ichimori, T.; Matsunaga, S.; Fujita, M.J.; Sakai, R. N-Methyl-β-carbolinium Salts and an N-Methylated 8-Oxoisoguanine as Acetylcholinesterase Inhibitors from a Solitary Ascidian, Cnemidocarpa irene. ACS Omega 2017, 2, 1074–1080. [Google Scholar] [CrossRef]
- Kudryavtsev, D.; Makarieva, T.; Utkina, N.; Santalova, E.; Kryukova, E.; Methfessel, C.; Tsetlin, V.; Stonik, V.; Kasheverov, I. Marine natural products acting on the acetylcholine-binding protein and nicotinic receptors: From computer modeling to binding studies and electrophysiology. Mar. Drugs 2014, 12, 1859–1875. [Google Scholar] [CrossRef] [PubMed]
- Tsuneki, H.; You, Y.; Toyooka, N.; Sasaoka, T.; Nemoto, H.; Dani, J.A.; Kimura, I. Marine alkaloids (−)-pictamine and (−)-lepadin B block neuronal nicotinic acetylcholine receptors. Biol. Pharm. Bull. 2005, 28, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hu, L.; Jia, J.; Gu, H.; Jia, Y.; Chen, X. A Stereoselective Approach toward (−)-Lepadins A–C. Org. Lett. 2017, 19, 5372–5375. [Google Scholar] [CrossRef] [PubMed]
- Lazo, J.S.; McQueeney, K.E.; Burnett, J.C.; Wipf, P.; Sharlow, E.R. Small molecule targeting of PTPs in cancer. Int. J. Biochem. Cell Biol. 2018, 96, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, W.; Liu, X.; Yu, H.; Lu, X.; Jiao, B. Inhibitors of Protein Tyrosine Phosphatase 1B from Marine Natural Products. Chem. Biodivers. 2017, 14. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Nakayama, W.; Takahashi, O.; Kirikoshi, R.; Izumikawa, Y.; Iwasaki, K.; Toraiwa, K.; Ukai, K.; Rotinsulu, H.; Wewengkang, D.S.; et al. Verruculides A and B, two new protein tyrosine phosphatase 1B inhibitors from an Indonesian ascidian-derived Penicillium verruculosum. Bioorg. Med. Chem. Lett. 2015, 25, 3087–3090. [Google Scholar] [CrossRef] [PubMed]
- Sumilat, D.A.; Yamazaki, H.; Endo, K.; Rotinsulu, H.; Wewengkang, D.S.; Ukai, K.; Namikoshi, M. A new biphenyl ether derivative produced by Indonesian ascidian-derived Penicillium albobiverticillium. J. Nat. Med. 2017, 71, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Imperatore, C.; Luciano, P.; Aiello, A.; Vitalone, R.; Irace, C.; Santamaria, R.; Li, J.; Guo, Y.W.; Menna, M. Structure and Configuration of Phosphoeleganin, a Protein Tyrosine Phosphatase 1B Inhibitor from the Mediterranean Ascidian Sidnyum elegans. J. Nat. Prod. 2016, 79, 1144–1148. [Google Scholar] [CrossRef] [PubMed]
- Luciano, P.; Imperatore, C.; Senese, M.; Aiello, A.; Casertano, M.; Guo, Y.W.; Menna, M. Assignment of the Absolute Configuration of Phosphoeleganin via Synthesis of Model Compounds. J. Nat. Prod. 2017, 80, 2118–2123. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.H.; Field, J.J.; Kanakkanthara, A.; Owen, J.G.; Singh, A.J.; Northcote, P.T. Marine Invertebrate Natural Products that Target Microtubules. J. Nat. Prod. 2018, 81, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Monserrate, Z.; Vervoort, H.C.; Bai, R.; Newman, D.J.; Howell, S.B.; Los, G.; Mullaney, J.T.; Williams, M.D.; Pettit, G.R.; Fenical, W.; et al. Diazonamide A and a synthetic structural analog: Disruptive effects on mitosis and cellular microtubules and analysis of their interactions with tubulin. Mol. Pharm. 2003, 63, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, M.; Tcherkezian, J.; Bernier, C.; Prota, A.E.; Chaaban, S.; Rolland, Y.; Godbout, C.; Hancock, M.A.; Arezzo, J.C.; Ocal, O.; et al. The synthetic diazonamide DZ-2384 has distinct effects on microtubule curvature and dynamics without neurotoxicity. Sci. Transl. Med. 2016, 8, 365ra159. [Google Scholar] [CrossRef] [PubMed]
- David, N.; Pasceri, R.; Kitson, R.R.; Pradal, A.; Moody, C.J. Formal Total Synthesis of Diazonamide A by Indole Oxidative Rearrangement. Chem. Eur. J. 2016, 22, 10867–10876. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, G.; Garrone, B.; Deacon, E.; Owen, P.; Pongracz, J.; Mead, G.; Bradwell, A.; Watters, D.; Lord, J. The Polyether Bistratene A Activates Protein Kinase C–δ and Induces Growth Arrest in HL60 Cells. Biochem. Biophys. Res. Commun. 1996, 222, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Statsuk, A.V.; Bai, R.; Baryza, J.L.; Verma, V.A.; Hamel, E.; Wender, P.A.; Kozmin, S.A. Actin is the primary cellular receptor of bistramide A. Nat. Chem. Biol. 2005, 1, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.A.; Tereshko, V.; Kossiakoff, A.A.; Kozmin, S.A. Structure of Bistramide A—Actin Complex at a 1.35 Å Resolution. J. Amer. Chem. Soc. 2006, 128, 3882–3883. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.A.; Courson, D.S.; Keller, V.A.; Rock, R.S.; Kozmin, S.A. The dual mode of action of bistramide A entails severing of filamentous actin and covalent protein modification. Proc. Natl. Acad. Sci. USA 2008, 105, 4088–4092. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.A.; Liu, S.; Chen, Z.; Skau, C.; Pytynia, M.; Kovar, D.R.; Chmura, S.J.; Kozmin, S.A. Rationally simplified bistramide analog reversibly targets actin polymerization and inhibits cancer progression in vitro and in vivo. J. Am. Chem. Soc. 2010, 132, 7288–7290. [Google Scholar] [CrossRef] [PubMed]
- Sauviat, M.P.; Gouiffes-Barbin, D.; Ecault, E.; Verbist, J.F. Blockade of sodium channels by bistramide A in voltage-clamped frog skeletal muscle fibres. Biochim. Biophys. Acta (BBA)-Biomembranes 1992, 1103, 109–114. [Google Scholar] [CrossRef]
- Wrona, I.E.; Lowe, J.T.; Turbyville, T.J.; Johnson, T.R.; Beignet, J.; Beutler, J.A.; Panek, J.S. Synthesis of a 35-member stereoisomer library of bistramide A: Evaluation of effects on actin state, cell cycle and tumor cell growth. J. Org. Chem. 2009, 74, 1897–1916. [Google Scholar] [CrossRef] [PubMed]
- Herkommer, D.; Dreisigacker, S.; Sergeev, G.; Sasse, F.; Gohlke, H.; Menche, D. Design, synthesis, and biological evaluation of simplified side chain hybrids of the potent actin binding polyketides rhizopodin and bistramide. ChemMedChem 2015, 10, 470–489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.M.; Zhang, S.Y.; Tu, Y.Q. Recent progress in the isolation, bioactivity, biosynthesis, and total synthesis of natural spiroketals. Nat. Prod. Rep. 2018, 35, 75–104. [Google Scholar] [CrossRef] [PubMed]
- Jaffarali, H.A.; Akram, S.; Arshan, K.M. DNA barcoding of a colonial ascidian, Lissoclinum fragile (Van Name, 1902). Mitochondrial DNA Part A 2017, 28, 810–813. [Google Scholar] [CrossRef]
- Jaffarali, H.A.; Akram, S.; Arshan, K.M. Identification of four Indian ascidians based on COI gene sequences. Mitochondrial DNA Part A 2018, 29, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Eastman, A. Improving anticancer drug development begins with cell culture: Misinformation perpetrated by the misuse of cytotoxicity assays. Oncotarget 2017, 8, 8854. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J. Developing natural product drugs: Supply problems and how they have been overcome. Pharmacol. Ther. 2016, 162, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Schofield, M.M.; Jain, S.; Porat, D.; Dick, G.J.; Sherman, D.H. Identification and analysis of the bacterial endosymbiont specialized for production of the chemotherapeutic natural product ET-743. Environ. Microbiol. 2015, 17, 3964–3975. [Google Scholar] [CrossRef] [PubMed]
- Sardar, D.; Tianero, M.D.; Schmidt, E.W. Directing biosynthesis: Practical supply of natural and unnatural cyanobactins. In Methods in Enzymology; Academic Press: Cambridge, Massachusetts, USA, 2016; Volume 575, pp. 1–20. [Google Scholar] [CrossRef]
- Zhang, G.; Li, J.; Zhu, T.; Gu, Q.; Li, D. Advanced tools in marine natural drug discovery. Curr. Opin. Biotechnol. 2016, 42, 13–23. [Google Scholar] [CrossRef] [PubMed]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ascidian Source | Compound Class | Biological Activity | Molecular Target(s) | References |
|---|---|---|---|---|---|
| Ascididemin | Didemnum sp. | Pyridoacridine alkaloid | Cytotoxic | DNA intercalation, stabilizes G4 quadriplexes and inhibits telomerase | [99,103] |
| Bistramides | Lissoclinum bistratum | Spiroketal | Cytotoxic, induces protein phosphorylation | Actin filaments | [183,184,185,186,187,188] |
| Bistratamides | Lissoclinum bistratum | Cyanobactins | Cytotoxic, Metal binding | unknown | [45] |
| Botryllamides | Botryllus sp. | Brominated tyrosine derivatives | MDR reversal | ABCG2 | [127,128] |
| Diazonamide A | Diazona angulata | Cyclic peptide | Cytotoxic | Microtubules | [179,180,181] |
| Didemnin B | Tridemnum solidum | Cyclic depsipeptide | Cytotoxic, Inhibition of protein translation, immunosuppressive, antiviral | eEF1A1PPT1 | [57,58,59] |
| Eudistidines | Eudistoma sp. | Novel alkaloids | Inhibition of protein-protein interaction, anti-malarial | HIF1-p300 | [137,138] |
| Eudistomin C | Eudistoma sp. | β-Carboline alkaloid | Cytotoxic, anti-viral, Inhibition of protein translation | us11 protein on 40S ribosome | [109] |
| Euseynstelamide B | Didemnum candidum | Bis-indole alkaloid | Cytotoxic, causing G2 arrest | Topoisomerase II | [136] |
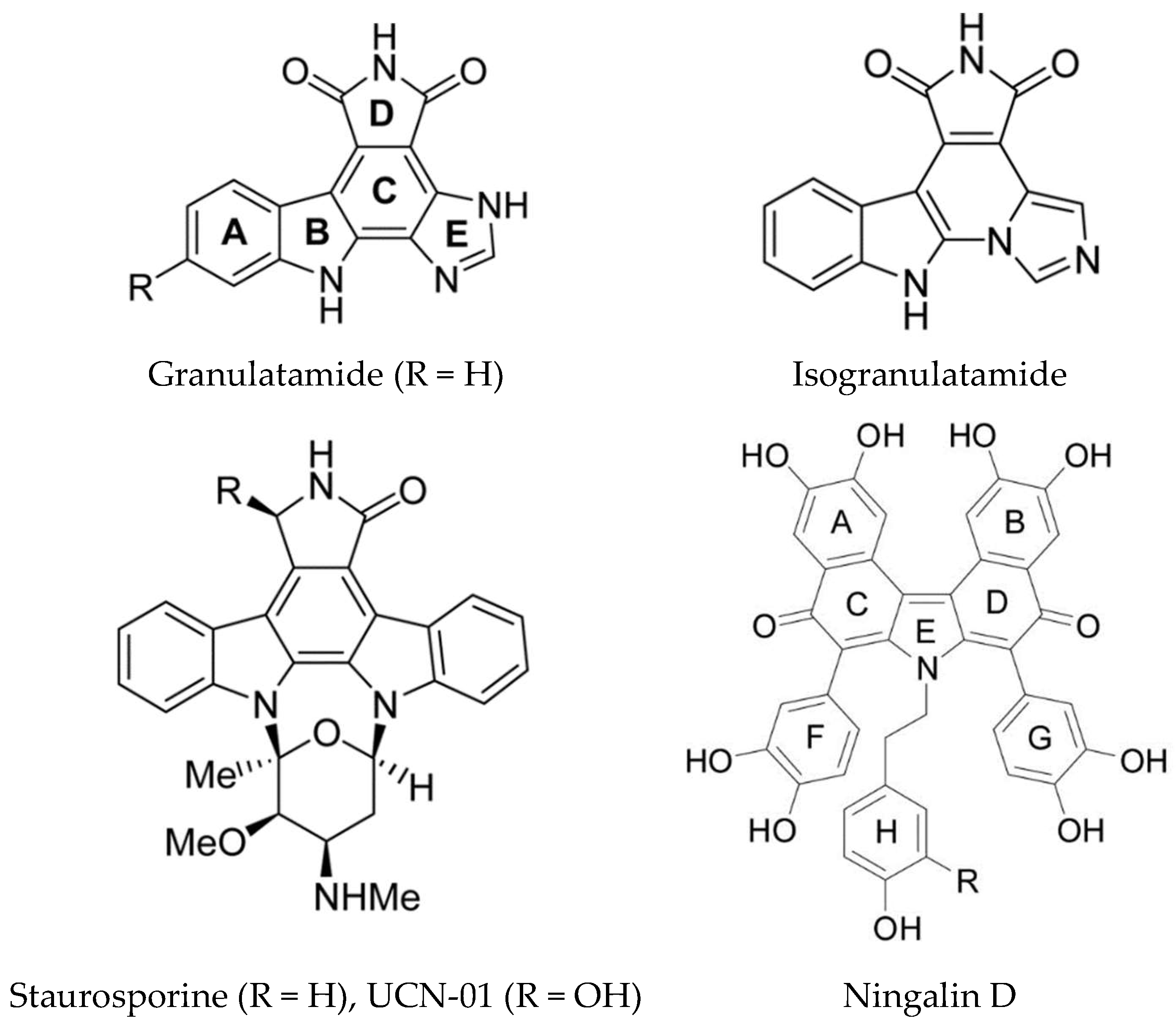
| Granulatimides | Didemnum granulatum | Alkaloids | Kinase Inhibition | Chk1 (kinase) | [160,161,162] |
| Irenecarbolines | Cnemidocarpa irene | β-carbolines | Enhancement of cholinergic neurotransmission | AChE inhibitors | [169] |
| Lamellarins | Didemnum sp. | DOPA/TOPA derived pyrrole alkaloids | Cytotoxic | Multiple targets – Topoisomerase 1, Kinases, Drug efflux pumps e.g. MDR-1, P-glycoprotein | [114,115,116,117,118,119] |
| Lissoclibadins | Lissoclinum cf badium | Polysulfur aromatic alkaloids | Cytotoxic, anti-tumor in mice | unknown | [139] |
| Lissoclimides | Lissoclinum voeltzkowi Michaelsen | Labdane diterpenoids | Cytotoxic, inhibition of elongation step of protein synthesis | LSU Ribosomal E-site | [151] |
| Lissoclinamides | Lissoclinum patella | Cyanobactins | Cytotoxic, Metal Binding | unknown | [51] |
| Mandelalides A & B | Lissoclinum mandelai | Polyketides | Cytotoxic | ATP synthase complex V | [78,79] |
| Meridianins | Aplidium meridianum | Indole alkaloids | Kinase inhibition | GSK-3β, CK1, CDKs | [129,130,131] |
| Meridine | Amphicarpa meridiana | Pyridoacridine alkaloid | Cytotoxic | DNA, stabilizes G4 quadriplexes and inhibits telomerase | [104] |
| Ningalins | Didemnum sp. | DOPA/TOPA derived pyrrole alkaloids | MDR reversal, kinase inhibition | MDR-1, P-glycoprotein | [124,125,167] |
| Patellamides | Lissoclinum patella | Cyanobactins | Cytotoxic, metal binding | MDR-1, others unknown | [46,47,49] |
| Patellazoles A–C | Lissoclinum patella | Polyketides | Cytotoxic, chemical defense | unknown | [36,72,73] |
| Phosphoeleganin | Sidnyum elegans | Polyketide | Phosphatase inhibition | PTP1B | [177,178] |
| Pibocin, Varacin, Pictamine, Lepadin | Eudistoma sp. Lissoclinum sp. Clavelina picta Clavelina lepadiformis | Ergoline alkaloid Benzopentathiepin Quinolizidine alkaloid Decahydroquinoline alkaloid | Inhibition of cholinergic neurotransmission Inhibition of cholinergic neurotransmission | nAChR antagonistsn AChR antagonists | [171] |
| Plitidepsin (dehydrodidemnin B) Aplidin® | Aplidia albicans | Cyclic depsipeptide | Anticancer drug | eEF1A2 | [63] |
| Polyandrocarpamines A & B | Polyandrocarpa sp. | 2-aminoimidazolone alkaloid | Kinase inhibition | CLK1, CLK2, DYRK | [164] |
| Pulmonarins A & B | Synoicum pulmonaria | Dibrominated tyrosine derivatives | Enhancement of cholinergic neurotransmission | AChE inhibitors | [168] |
| Ritterazines | Riterella tokiada | Dimeric steroidal pyrazine alkaloids | Cytotoxic | Hsp70s, GRP78 | [17,82,143] |
| Tamandarins | Unidentified Brazilian species | Cyclic depsipeptides, closely related to didemnin B | Cytotoxic | Unknown but may be similar to didemnin B | [68] |
| Thiaplidiaquinones | Aplidium conicum | Thiazinoquinones | Cytotoxic, anti-malarial | DNA, stabilizes topoisomerase II, ROS generation. FTase | [154,155,156,157] |
| Trabectidin (ET-473) Yondelis® | Ecteinascidia turbinata | Tetrahydroisoquinoline alkaloid | Anticancer drug, Induces apoptosis in tumor associated macrophages | DNA, minor groove, interference with transcription factors and DNA repair proteins | [49,86,87,88,89] |
| UCN-01 (7-hydroxystaurosporine) | Eudistoma sp. | Alkaloid | Kinase inhibition | Multiple kinases | [159] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watters, D.J. Ascidian Toxins with Potential for Drug Development. Mar. Drugs 2018, 16, 162. https://doi.org/10.3390/md16050162
Watters DJ. Ascidian Toxins with Potential for Drug Development. Marine Drugs. 2018; 16(5):162. https://doi.org/10.3390/md16050162
Chicago/Turabian StyleWatters, Dianne J. 2018. "Ascidian Toxins with Potential for Drug Development" Marine Drugs 16, no. 5: 162. https://doi.org/10.3390/md16050162
APA StyleWatters, D. J. (2018). Ascidian Toxins with Potential for Drug Development. Marine Drugs, 16(5), 162. https://doi.org/10.3390/md16050162