1. Introduction
Sponges are the most abundant source of bioactive compounds in marine invertebrates [
1]. About half of the current FDA-approved marine drugs come from sponges or derivatives based on sponge metabolites [
2,
3,
4]. Most of the natural products isolated from sponges belong to bromotyrosine derivatives and exhibit extensive biological activities [
5,
6,
7,
8]. Itampolin A was the first brominated tyrosine alkaloid isolated from the sponge
Iotrochota purpurea [
9]. In our previous study, the total synthesis of itampolin A was achieved for the first time. In an evaluation of the pharmacological properties, the alkaloid showed potent concentration-dependent antitumor activity against non-small cell lung cancer. In addition, its enantiomer (−)-itampolin A exhibited significant inhibitory activity against non-small cell lung A549 cells. The underlying anticancer mechanism was associated with its binding to DFG-out conformation of p38α and then decreasing the phospho-p38 expression in a time-dependent manner [
10].
In recent years, the design of more selective kinase inhibitors, targeting inactive conformations, has been explored. Relative to type I inhibitors that compete directly with ATP, type II inhibitors bind to the inactive DFG-out conformation of kinase induced by the conformational transition of DFG-loop. This opened up a second hydrophobic subcavity formed by the catalytic amino acid triad Asp168, Phe169, and Glu71. A number of studies have demonstrated the advantages of targeting the DFG-out binding mode of kinases in general and p38 MAP kinase (p38 MAPK) in particular such as low toxicity [
11].
Fragment-based drug design (FBDD) is now widely used in academia and industry to obtain small molecule inhibitors for a given target. Moreover, it is established for many fields of research including antimicrobials and oncology [
12,
13,
14]. Many molecules derived from fragment-based approaches are already in clinical trials and two—Vemurafenib and Venetoclax—are on the market [
13]. Unlike other computer aided drug design (CADD) methods, the FBDD theory maintains that the active pockets of the drug target are made up of multiple subcavities, and the fragments are units that combine with these subcavities. Finding these fragments and linking them together often leads to higher active compounds [
15].
For the purpose of improving the activity of itampolin A, and increasing the structural diversity of type II inhibitors, we here reported the optimization of (−)-itampolin A as novel p38α inhibitors by using the FBDD method. This strategy involved interrogation of structural information that was available for different in-house chemotypes [
16]. The challenge included three aspects. The first one was deconstruction of known p38α inhibitors to identify highly efficient interactions in the binding site. The second one was screening out suitable units that fit the second hydrophobic subcavity. The last one was exploring the effects on the activity of some atom or fragment substitutions of the brominated tyrosine skeleton.
3. Discussion
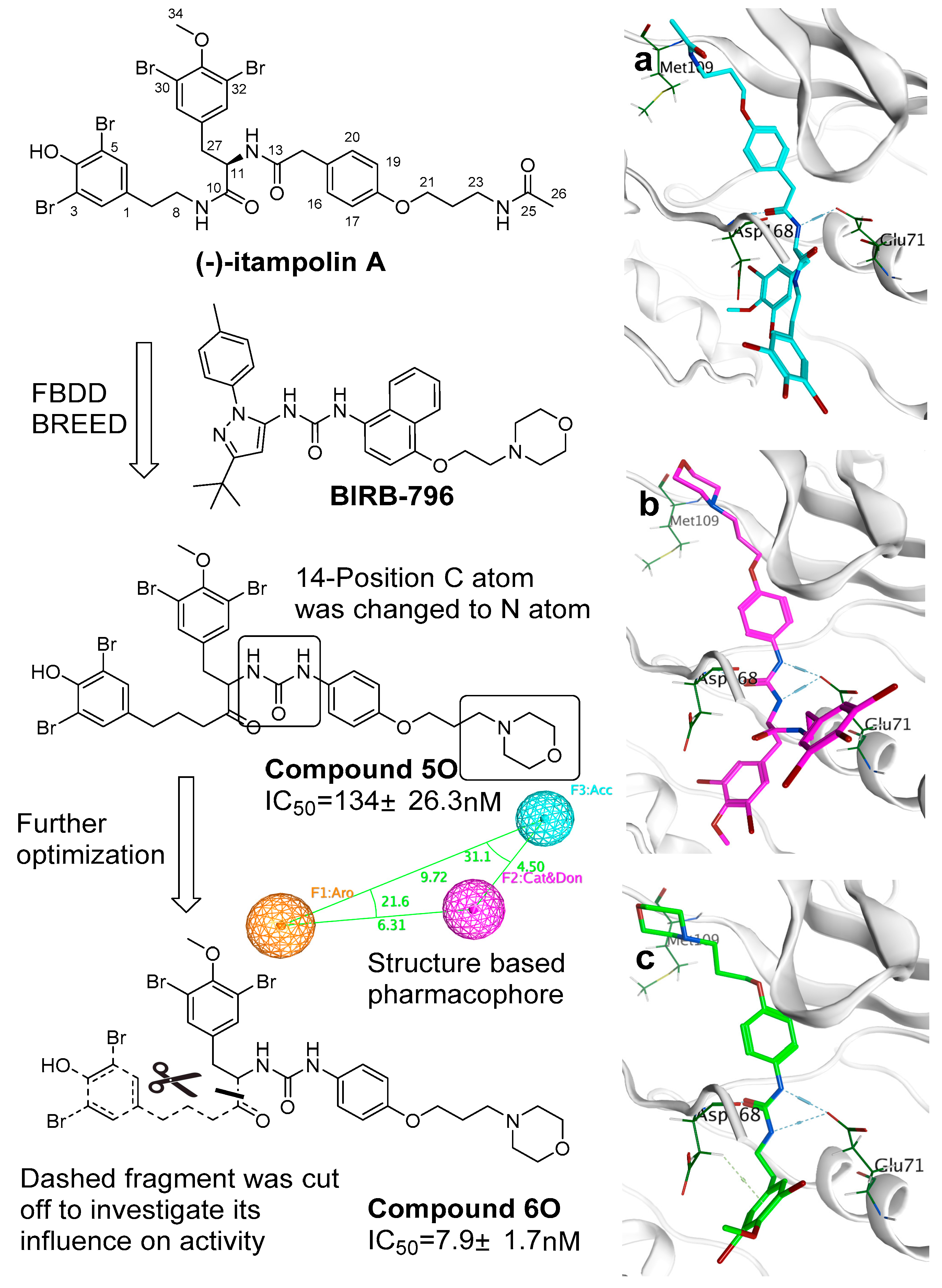
Based on the aligned 3D ligand structures binding to p38α, a novel skeleton was generated based on the structure of (−)-itampolin A (
Figure 5). The carbon atom of 14-position was changed to a nitrogen atom, making the amide unit (12-position, 13-position) convert to a disubstituted urea group. As a result, the new skeleton was capable of binding Glu71 residue. It stabilized the conformation of the molecule in the active site. This binding was crucial, because the interaction between the urea unit and Glu71 ensured that bromoaromatic groups enter the second hydrophobic pocket unique to DFG-out conformation. The atoms between 23-position and 26-position were replaced with a morpholine unit (there were also other units such as fluorine-substituted aromatic ring that are listed in
Table S1). The urea group probably came from the inhibitors which contained the urea unit. The morpholine unit came from BIRB-796. The fluorine-substituted aromatic came from the inhibitor of 3IW5 or 4FA2. Most inhibitors supported the change to the urea unit. However, each inhibitor supported different changes in the atoms between 23-position and 26-position (
Figure 5 shows a more active one with the morpholine unit). Therefore, it was suggested that it is necessary to explore more units to replace the atoms between 23-position and 26-position. The results of molecular docking showed that most of the brominate tyramine fragment was exposed outside the active pocket. Moreover, excessive hydrophobic fragments might hinder the binding of the molecule to Glu71 (
Figure 5a). After the discovery of defects in the skeleton, a filter was added to iterate the process of FBDD. This filter included the Lipinski’s rule of five and a structure-based pharmacophore, worked out by MOE 2015. The screening results showed that cutting off the fragment can further improve the activity of the compounds with a novel skeleton.
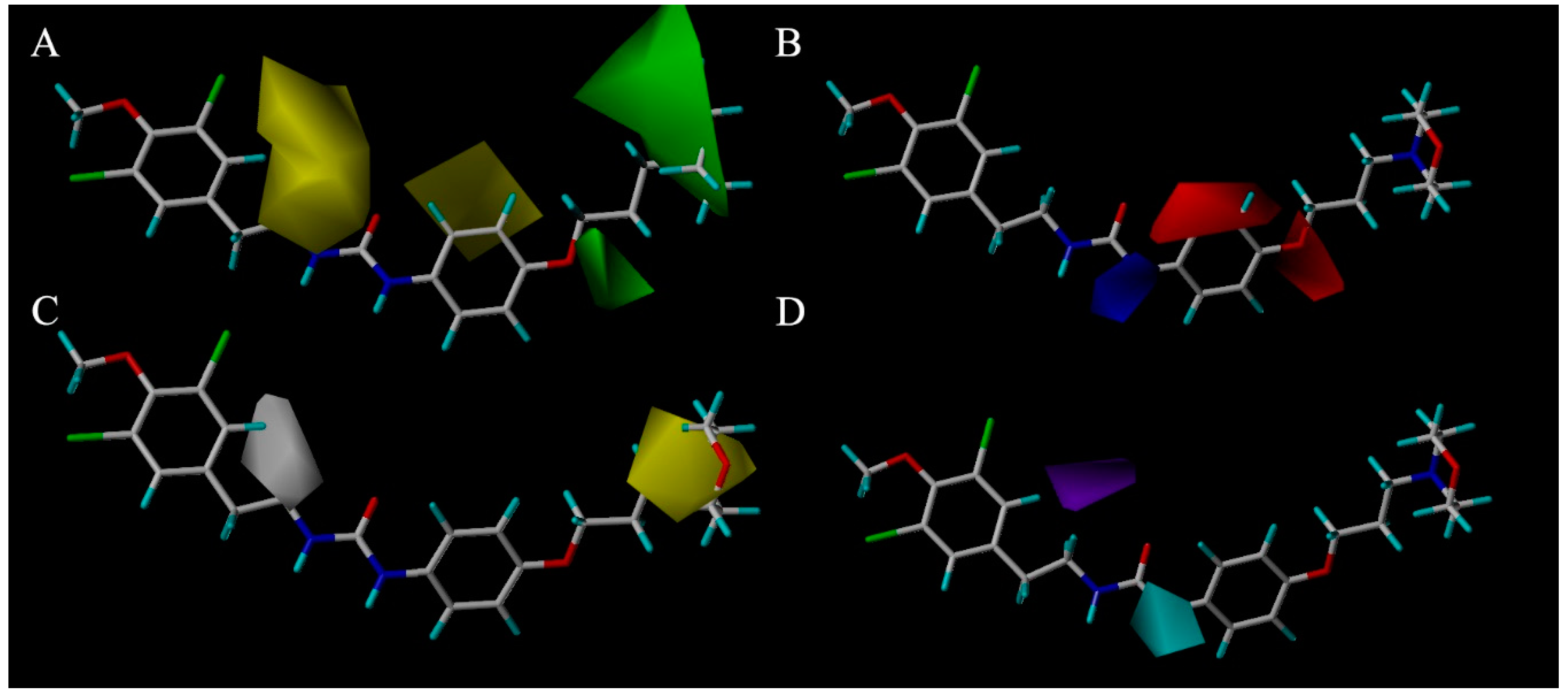
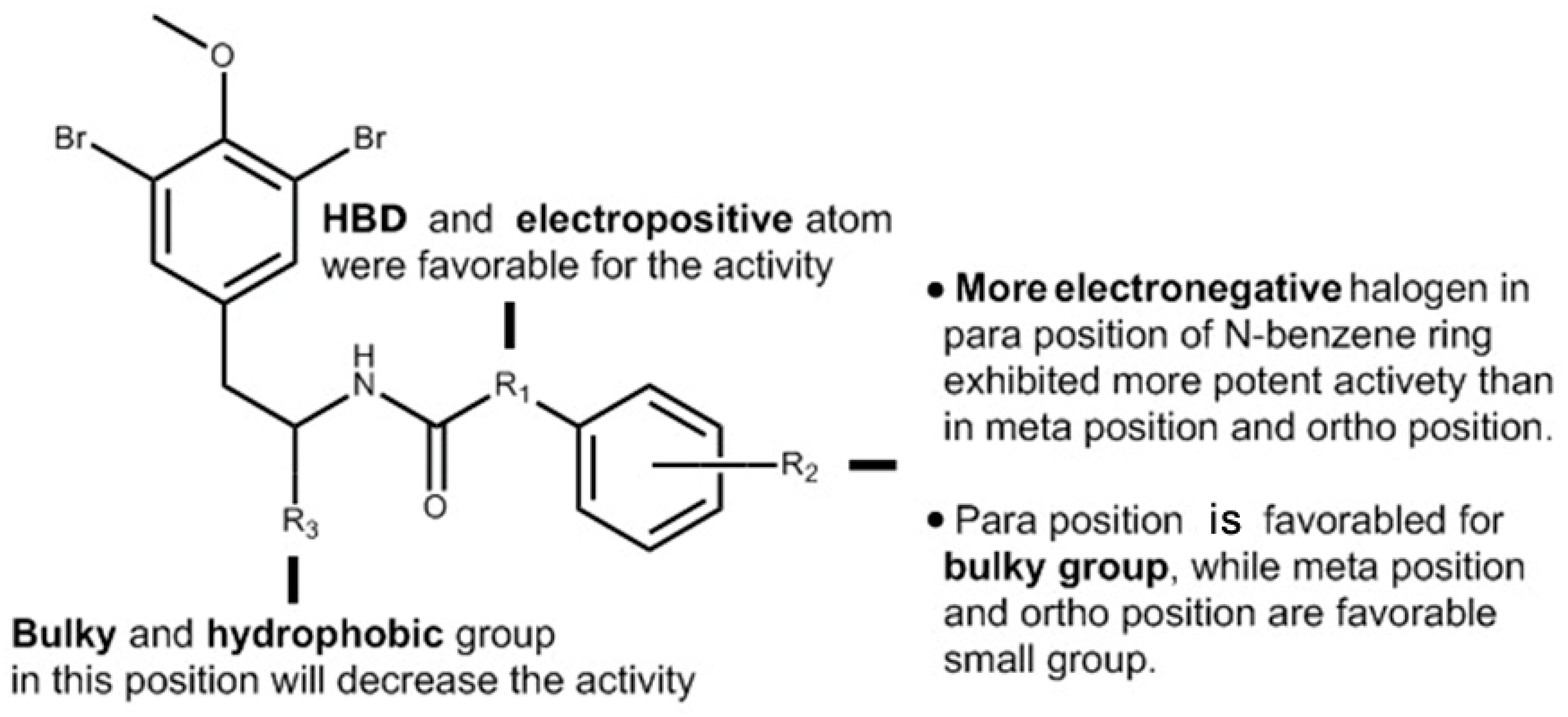
In the case of CoMFA analysis, the contour maps around
6o (
Figure 4a,b) were generated from the CoMFA model. The steric contour map of
6o is shown in
Figure 4a. Two green regions near the
N-substituents of the morpholine proved that this length of hydrocarbon chain preferred sterically favorable functional groups. In contrast, a yellow region over the 11-C showed that a large group such as segment II leads to a decrease in activity. Consistent with this result, after cutting off the brominate tyramine fragment, the activities of compounds 3a–3o were found to be improved. This may be attributable to the excessive volume of the fragment. It was exposed to the outside of the active site pocket, when (−)-itampolin A was docked into p38α, as shown in
Figure 5a. The electrostatic contour map of
6o is shown in
Figure 4b. The red region over the benzene ring revealed that substituents on the benzene ring preferred electronegative groups, such as halogen atoms. The blue region around the 14-N showed that electropositive groups possessed good anticancer activity. Consistent with the QSAR analysis result, the molecular docking study showed that the urea group forms one more hydrogen bond to Glu71 than the amide group (
Figure 5b). These results explained the fact that almost all of the compounds in the second series exhibited more potent inhibitory effects than those in the first series after the 14-position carbon atom was changed to a nitrogen atom.
The CoMSIA contour hydrophobic map is shown in
Figure 4c. The yellow contour map in the morpholine indicated the region where addition of the hydrophobic groups would increase the inhibitory activity while the white contour map over the 11-C proved that segment II in this position will decrease the inhibitory activity. The contour map of the CoMSIA hydrogen bond donor (HBD) is shown in
Figure 4d; the purple region above 11-C represents the HBD atom substitution which was adverse to the activity. This might be attributed to the hydroxyl of segment II. The cyan region around the 14-N atom indicates that the addition of the HBD atom was conducive to the activities. The CoMSIA HBD contour map can be validated by the fact that almost all of the compounds in the second series exhibited more potent inhibitory effects than those in the first series after the 14-position carbon atom was changed to a nitrogen atom.
Finally, we combined the ligand-based 3D-QSAR analysis with the structure-based molecular docking study, to identify the necessary moiety of the brominated tyrosine derivatives (
Figure 6).
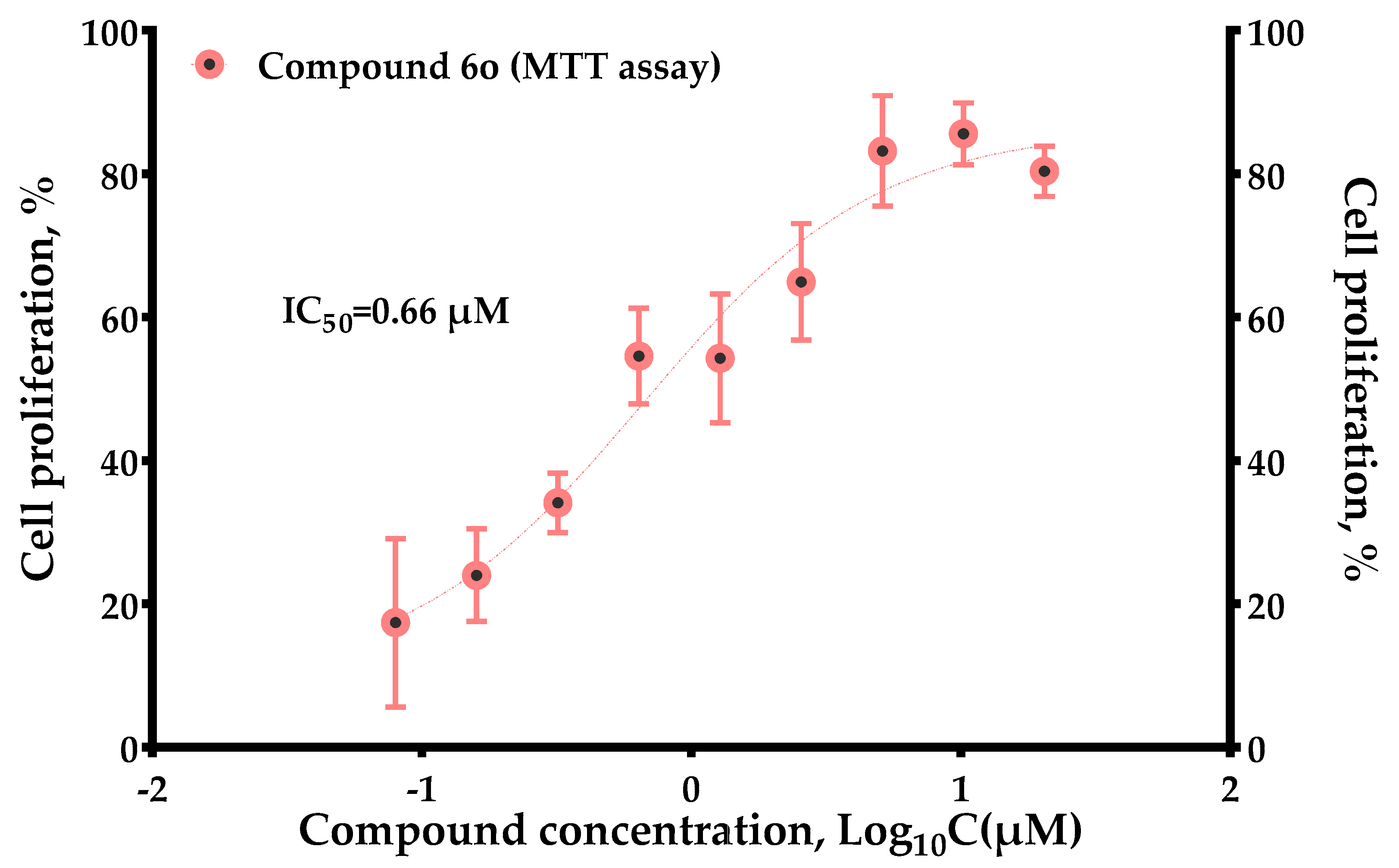
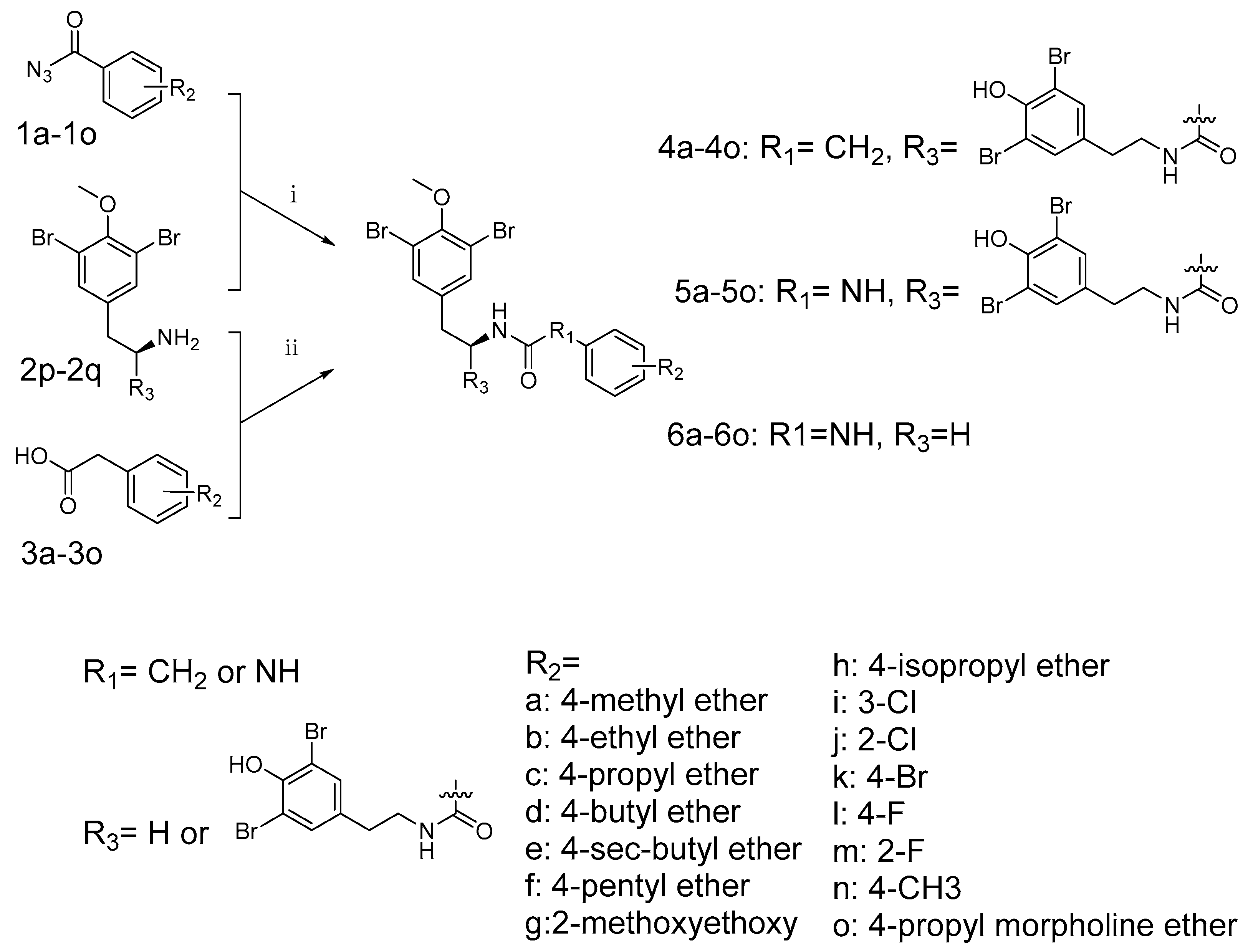
In conclusion, (−)-itampolin A, as a lead compound, was optimized by the FBDD method based on the interaction between type II inhibitors and p38α. The 14-position carbon atom was changed to a nitrogen atom, making the amide group change to a diaryl urea unit. The brominate tyramine fragment and the substituent groups on the benzene ring were also modified. Thus, 45 brominated tyrosine derivatives of three series were designed and synthesized. Not all the compounds were reported in previous studies, and their structures were confirmed by 1H-NMR and 13C-NMR. Through the analysis of 3D-QSAR, it was found that the conversion of the 14-position carbon atom to a nitrogen atom contributed to improving the activity of the derivatives. Cleavage of the brominate tyramine fragment helped to improve the activities of 6a–6o. The groups on the benzene ring preferred electron-withdrawing groups, followed by meta-substituted halogen atoms. The alkane chains on the aromatic ring were not beneficial to increasing inhibitory activity. Moreover, we discovered that a novel derivative, 6o (IC50 = 0.66 μM), exhibited significant antitumor activity against A549.
4. Experimentation
4.1. Fragment-Based Drug Design
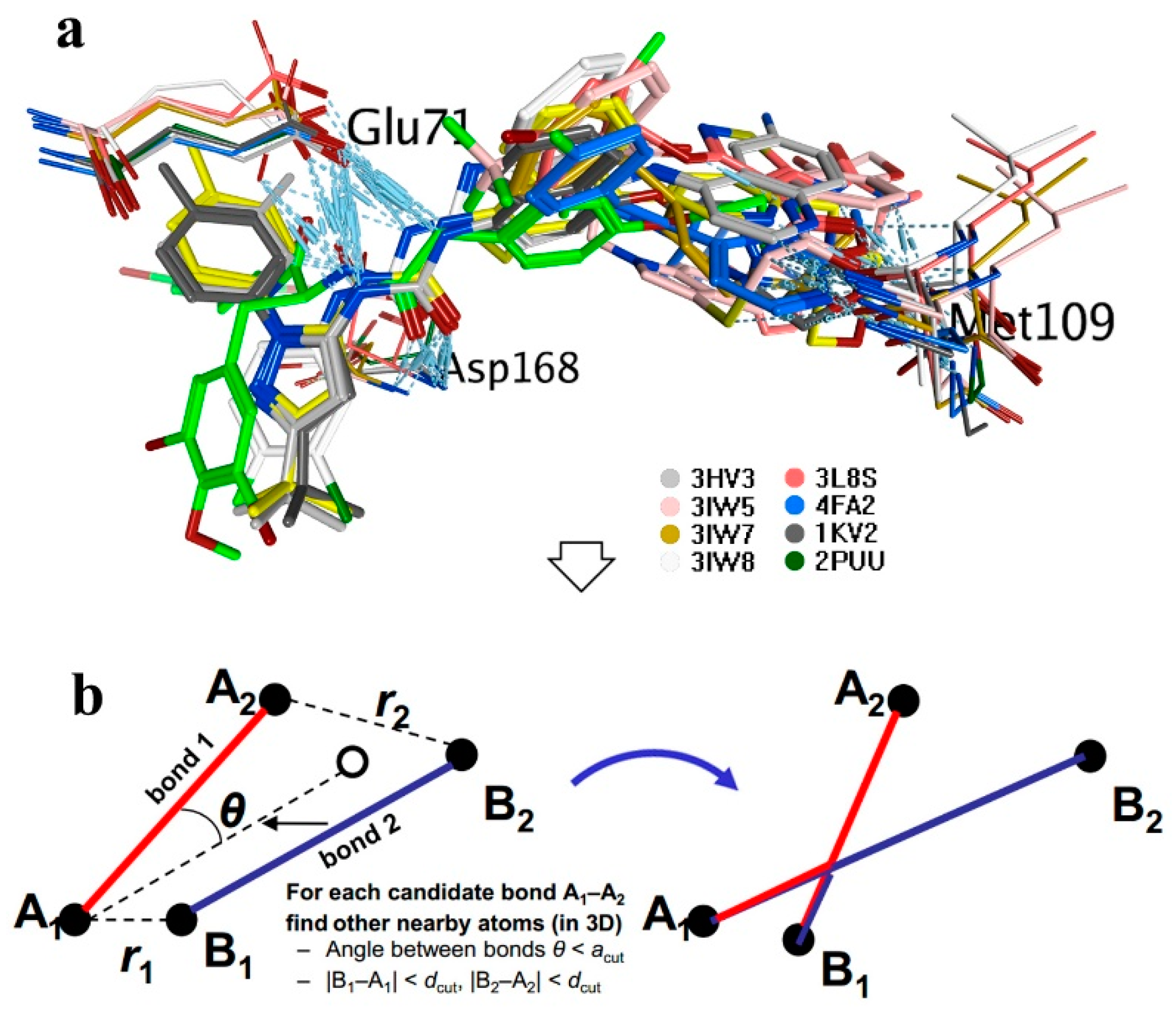
Fragment-based drug design was performed using the BREED module in Molecular Operating Environment package (MOE 2015.1001.). Eight molecules (ligands in 3HV3, 3IW5, 3L8S, 3IW7, 3IW8, 4FA2, 2KV2, 2PUU) after alignment were set as novel bond producers. (−)-Itampolin A was set as the parent compound. During the generation, a filter included the Lipinski’s rule of five and a structure-based pharmacophore was toggled ON. The novel compounds were generated based on candidate bonds.
4.2. Molecular Docking
Molecular docking was performed using MOE. The ligands were docked into p38α after energy minimization. Their conformations were generated with the bond rotation method. In order to validate the docking protocol, a self-docking of BIRB-796 into the binding pocket was firstly performed. The triangle matcher was selected as the ligand placement method, the London dG was selected as the scoring function, the rigid receptor was used as the type of post-placement refinement, the GBVI/WSA dG was used as the refinement scoring function. The one with the best docking conformation was selected from 30 poses according to E-strain. The RMSD value between the docking conformation and actual conformation was 0.9695, as shown in
Figure S1.
4.3. The p38α MAP Kinase Activity Based on the Rate of Phosphorylation of ATF-2
Kinase reaction buffer composition: 50 mM HEPES (pH 7.5), 10 mM MgCl2, 0.01% BRIJ-35 and 1 mM EGTA. Titration MAPK14/p38α at 90 μM ATP: Prepare MAPK14/p38α in kinase buffer at a concentration of 500 ng/mL. Perform two-fold serial dilution using kinase buffer from 500 ng/mL, 16 dose points. Add 5 μL of the serially diluted MAPK14/p38α into the 384-well plate in triplicate. Prepare 1 mL of 0.8 μM substrate GFP-ATF2 (19–96) and 180 μMATP in kinase reaction buffer. The reaction was carried out by adding 5 μL of the substrate GFP-ATF2 (19–96) and ATP solution into each well of the assay plate. Seal the assay plate and incubate for 1 h at room temperature (RT). Add 10 μL of the antibody solution prepared in TR-FRET dilution buffer (20 mM EDTA and 4 nM Tb-antipATF2 (pThr71), and mix gently. The final EDTA and Tb-antipATF2 (pThr71) concentrations were 10 mM and 2 nM, respectively. Seal the assay plate and incubate for 30 min at RT. Read the TR-FRET signal on the Envision 2104 plate reader.
Determination of IC50 values of inhibitors: add 2 μL/well of inhibitor in 0.5% DMSO at five-fold the final assay concentration to the 384-well assay plate. For the first cycle inhibitor screening, the final concentrations of inhibitors were 1117, 279.25, 69.81, 17.45, and 4.36 nM (three-fold dilution, five dose points, two replicates for each dose). Adjust the inhibitor concentration based on the results of the first cycle. An amount of 4 μL MAPK14/p38α was added to each well of the 384-well assay plate. Incubate for 15 min at RT. Finally, 4 μL substrate GFP-ATF2 (19–96) and ATP in kinase reaction buffer were added to start the reaction. The final concentrations of MAPK14/p38α, Substrate, and ATP were 1 ng/mL, 0.4 μM, and 90 μM, respectively. Incubate for 1 h at RT. Add 10 μL/well of antibody solution. The final concentrations of EDTA and antibody were 10 mM and 2 nM respectively. Incubate for 30 min at RT. Read the TR-FRET signal on the Envision 2104 plate reader.
4.4. MTT Assay
The A549 cell lines were cultured using DMEM and RPMI, respectively, in standard humidified incubation conditions at 37 °C in 5% CO2. Then, the cancer cells (1000/well) were placed on 96-well plates in triplicate. After 24 h incubation, cells were treated with a series of concentrations of the brominated tyrosine derivatives (1, 10, 20, 80 μmol/L). The positive control contained the same concentrations of BIRB-796, and the negative control received the same volume of DMSO. After 48h incubation, cells were stained with 5% MTT, and the optical density was recorded at an absorbance wavelength of 570 nm.
4.5. 3D-QSAR Study
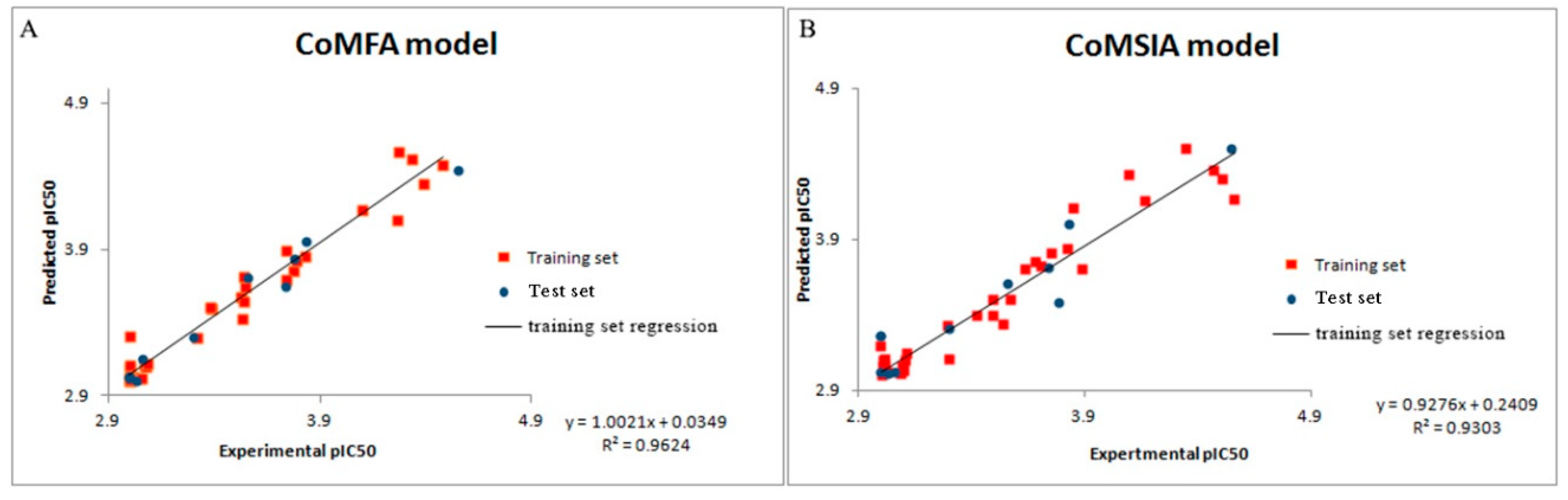
The SKETCH function of Sybyl-X2.0 was utilized for drawing the structure and charges were calculated by the Gasteiger–Huckel method, and tripos force field was utilized for energy minimization of these molecules. These 45 synthetic molecules were divided into the training set and test set in the ratio of 80:20. The training set was used to build the 3D-QSAR model, and the test set was used to test the predictions of the model [
17,
18]. In the case of building the Topomer CoMFA QSAR model, a carbon sp3 probe was applied for calculating steric and electrostatic parameters. The database alignment was used to build the CoMSIA QSAR model [
19].
4.6. Chemsitry
1H-NMR and 13C-NMR spectra were recorded on Varian NMR spectrometers operating at 600 MHz for 1H, and 150 MHz for 13C. All chemical shifts were measured in DMSO-d6 as solvent. All chemicals were purchased from Sinoreagent Chemcal Reagent (Beijing, China) and were used as received, unless stated otherwise. Analytical TLC was performed on Haiyang (Qingdao Haiyang Chemcal Co., Ltd. (Qingdao, China)) silica gel 60 F254 plates and visualized by UV and potassium permanganate staining. Flash column chromatography was performed on Haiyang (Qingdao Haiyang Chemcal Co., Ltd.) gel 60 (40–63 mm). HPLC was performed on Agilent 1260 Infinity II. Chromatographic separation was performed on a C18 column (3.0 × 50 mm, 1.7 µm) and the column temperature was maintained at 25 °C. The mobile phase consisted of methanol (50%) and water (50%).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}