Clerodane Diterpenes from the Marine Sponge Raspailia bouryesnaultae Collected in South Brazil

 ,
,  , ,
, ,
Abstract
:
1. Introduction
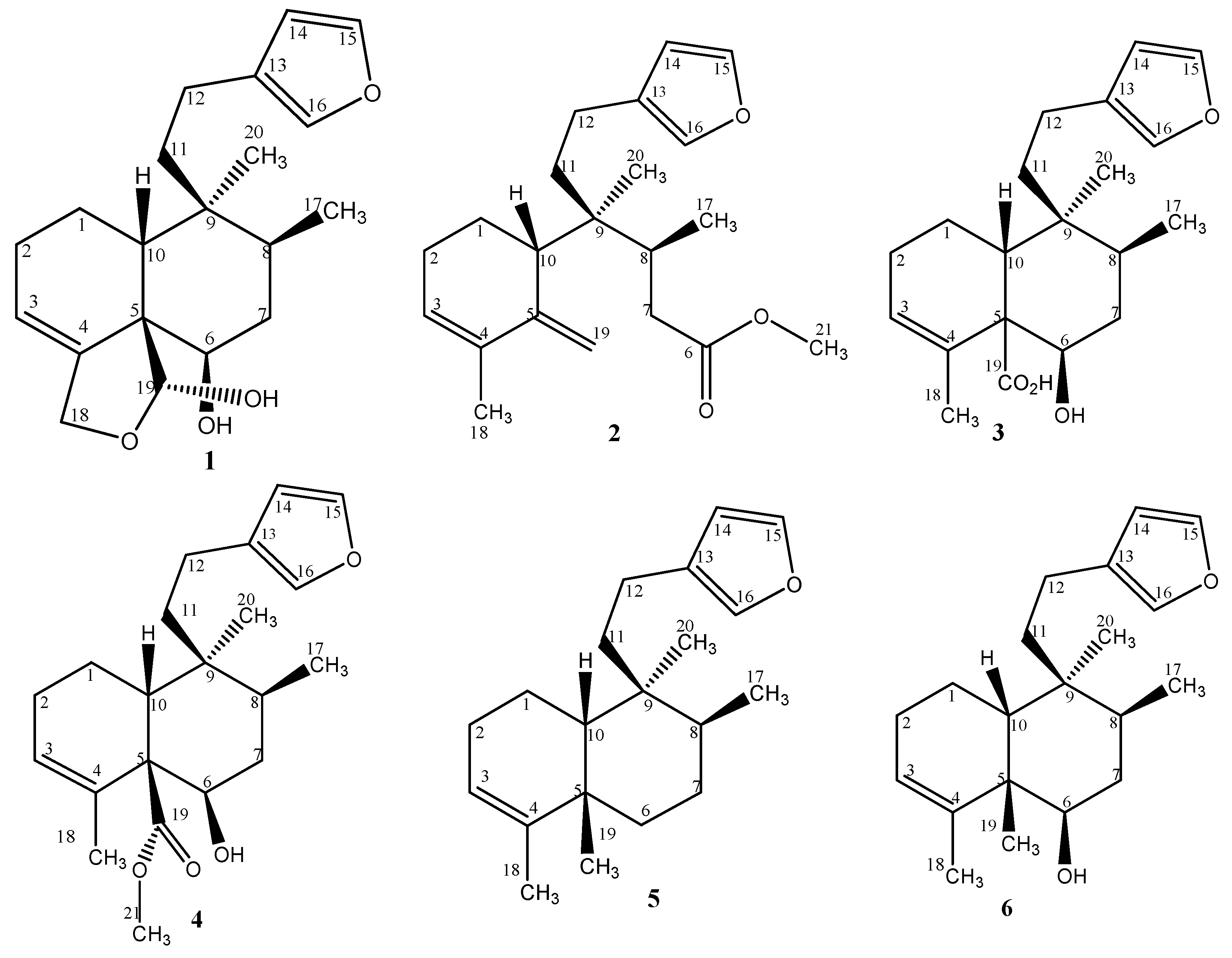
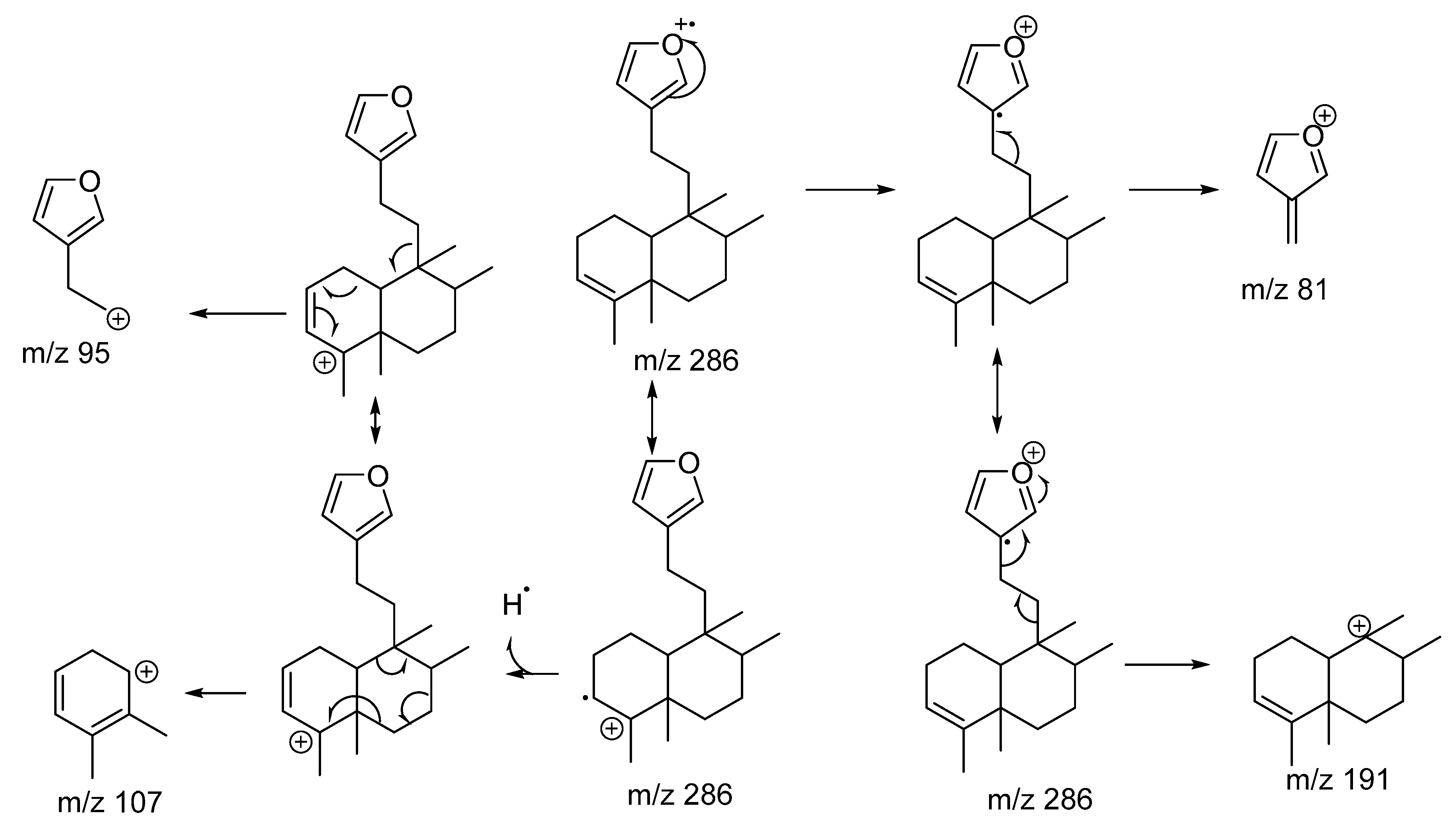
2. Results and Discussion
2.1. Chemistry
2.2. Antiproliferative Effects
2.3. Anti-Herpes Activity
3. Materials and Methods
3.1. General
3.2. Biological Material Collection
3.3. Extraction and Isolation
3.4. Antiproliferative Assays
3.5. Anti-Herpes Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Perdicaris, S.; Vlachogianni, T.; Valavanidis, A. Bioactive natural substances from marine sponges: New developments and prospects for future pharmaceuticals. Nat. Prod. Chem. Res. 2013, 1, 1–8. [Google Scholar] [CrossRef]
- Sagar, S.; Kaur, M.; Minnerman, K.P. Antiviral lead compounds from marine sponges. Mar. Drugs 2010, 8, 2619–2638. [Google Scholar] [CrossRef] [PubMed]
- Vila, F.A.; Gerwick, L. Marine natural product drug discovery: Leads for treatment of inflammation.cancer.infections and neurological disorders. Immunopharmacol. Immunotoxicol. 2010, 32, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Frota, M.J.; Silva, R.B.; Mothes, B.; Henriques, A.T.; Moreira, J.C. Current status on natural products with antitumor activity from Brazilian marine sponges. Curr. Pharm. Biotechnol. 2012, 13, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Hooper, J.N.A.; Van Soest, R.W.M. SystemaPorifera: A Guide to the Classification of Sponges; Kluwer Academic/Plenum: New York, NY, USA, 2002. [Google Scholar]
- Northover, B.J. Natural Products Studies of Marine Organisms of the South Pacific. Master’s Thesis, Victoria University of Wellington, Wellington, New Zealand, 2012. [Google Scholar]
- Li, R.; Morris-Natschke, S.L.; Kuo-Hsiung, L. Clerodanediterpenes: Sources, structures, and biological activities. Nat. Prod. Rep. 2016, 33, 1166–1226. [Google Scholar] [CrossRef] [PubMed]
- West, L.M.; Northcote, P.T.; Battershill, C.N. Two New ClerodaneDiterpenes from the New Zealand Marine Sponge Raspailia sp. Aust. J. Chem. 1998, 51, 1097–1101. [Google Scholar] [CrossRef]
- Ryan., J.M. Novel Secondary Metabolites from New Zealand Marine Sponges. Ph.D. Thesis, Victoria University of Wellington, Wellington, New Zealand, 2007. [Google Scholar]
- Lerner, C.; Carraro, J.L.; Soest, R.V. Raspailia (Raspaxilla) bouryesnaultae, a new name for Brazilian Raspaxillaelegans Boury-Esnault, 1973 (Demospongiae, Poecilosclerida, Raspaileedae) with a redescription and a new record. Zootaxa 2006, 1129, 37–45. [Google Scholar]
- Rangel, M.; Sanctis, B.; Freitas, J.C.; Polatto, J.M.; Granato, A.C.; Berlinck, R.G.S.; Hajdu, E. Cytotocic and neurotoxic activities in extracts of marine sponges (Porifera) from southeastern Brazilian coast. J. Exp. Mar. Bio. Ecol. 2001, 262, 31–40. [Google Scholar] [CrossRef]
- Rodríguez-Hahn, L.; García, A.; Esquivel, B.; Cárdenas, J. Structure of kerlinic acid from Salvia keerlii. Chemical correlation with melisodoric acid. Can. J. Chem. 1987, 65, 2687–2690. [Google Scholar] [CrossRef]
- Zdero, C.; Bohlmann, F.; King, R.M. Diterpenes and Norditerpenes from the Aristeguetia Group. Phytochemistry 1991, 30, 2991–3000. [Google Scholar] [CrossRef]
- Ferrari, M.; Pelizzoni, F. New diterpenoids with clerodane skeleton. Pergamon 1971, 10, 3267–3269. [Google Scholar] [CrossRef]
- Silveira, E.R.; Mcchesney, J.D. 6,7-Oxygenated neo-clerodane furan diterpenes from Croton sonderianus. Phytochemistry 1994, 36, 1457–1463. [Google Scholar] [CrossRef]
- Itokawa, H.; Totsuka, N.; Morita, H.; Takeya, K.; Iitaka, Y.; Schenkel, E.P.; Motidome, M. New Antitumor Principles, Casearins A-F, for Casearia sylvestris Sw. (Flacourtiaceae). Chem. Pharm. Bull. 1990, 38, 3384–3388. [Google Scholar] [CrossRef] [PubMed]
- Morita, H.; Nakayama, M.; Kojima, H.; Takeya, K.; Itokawa, H.; Schenkel, E.P.; Motidome, M. Structures and Cytotoxic Activity Relationship of Casearins, New Clerodane Diterpenes from Casearia sylvestris SW. Chem. Pharm. Bull. 1991, 39, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Burleson, F.G.; Chamberts, T.M.; Wiedbrauk, D.L. Virology: A Laboratory Manual; Academic, University of San Diego: San Diego, CA, USA, 1992. [Google Scholar]
- Boff, L.; Silva, I.T.; Argenta, D.F.; Farias, L.M.; Alvarenga, L.F.; Padua, R.M.; Braga, F.C.; Leite, J.P.; Kratz, J.M.; Simões, C.M.O. Strychnos pseudoquina A. St. Hil.: A Brazilian medicinal plant with promising in vitro anti-herpes activity. J. Appl. Microbiol. 2016, 121, 1519–1529. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1 | 19.7 | 24.7 | 38.2 | 19.1 | 19.6 | 21.6 |
| 2 | 25.8 | 24.5 | 18.7 | 26.5 | 25.8 | 27.1 |
| 3 | 120.7 | 127.5 | 129.1 | 124.8 | 122.4 | 123.9 |
| 4 | 139.1 | 133.8 | 136.2 | 135.5 | 148.0 | 142.1 |
| 5 | 53.4 | 145.9 | 59.4 | 54.6 | 38.2 | 47.1 |
| 6 | 74.6 | 174.6 | 75.3 | 75.5 | 33.0 | 75.7 |
| 7 | 38.3 | 37.6 | 38.5 | 38.8 | 25.8 | 38.6 |
| 8 | 36.8 | 36.4 | 36.1 | 36.8 | 37.4 | 37.0 |
| 9 | 38.0 | 41.0 | 38.9 | 38.7 | 38.4 | 39.1 |
| 10 | 40.4 | 45.4 | 43.7 | 44.4 | 44.6 | 43.5 |
| 11 | 32.8 | 35.1 | 26.1 | 32.1 | 32.3 | 46.0 |
| 12 | 18.9 | 20.5 | 18.7 | 18.9 | 18.9 | 19.4 |
| 13 | 130.0 | 125.8 | 124.9 | 125.7 | 123.2 | 124.4 |
| 14 | 111.1 | 110.9 | 110.6 | 110.9 | 111.0 | 111.0 |
| 15 | 142.6 | 142.7 | 142.8 | 142.7 | 142.6 | 142.7 |
| 16 | 138.3 | 138.4 | 138.8 | 138.3 | 138.5 | 138.5 |
| 17 | 15.7 | 15.8 | 15.6 | 15.7 | 15.3 | 15.6 |
| 18 | 68.7 | 20.4 | 23.4 | 22.8 | 20.1 | 22.7 |
| 19 | 99.6 | 112.3 | 186.8 | 178.5 | 37.8 | 18.1 |
| 20 | 25.3 | 22.1 | 21.3 | 26.4 | 26.0 | 28.6 |
| 21 | - | 51.4 | - | 52.0 | - | - |
| Position | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| 1β | 1.73 (m) | 2.35 (d, 1.83) | 1.76 (m) | 1.73 (m) | 1.70 (m) | 1.71 (m) |
| 2α | 2.20 (m) | 2.11 (d, 1.83) | 1.48 (m) | 2.16 (m) | 1.95 (m) | 2.04 (m) |
| 2β | 2.20 (m) | 2.35 (d, 1.83) | 1.75 (m) | 2.16 (m) | 2.00 (m) | 2.04 (m) |
| 3 | 5.76 (m) | 5.63 (d, 2.8) | 5.88 (s) | 5.64 (s) | 5.35 (s) | 5.50 (s) |
| 6α | 3.84 (m) | - | 3.86 (m) | 3.87 (td, 11.8, 4.7) | 1.21 (m) | 3.98 (m) |
| 6β | - | - | - | - | 1.24 (s) | - |
| 7α | 1.48 (m) | 2.08 (m) | 1.62 (m) | 1.62 (m) | 1.95 (m) | 1.56 (m) |
| 7β | 1.65 (m) | 2.57 (m) | 1.76 (m) | 1.73 (m) | 2.00 (m) | 1.75 (m) |
| 8 | 1.81 (m) | 2.30 (m) | 1.76 (m) | 1.83 (m) | 1.51 (m) | 2.16 (m) |
| 10 | 2.00 (dd, 13.2, 3.1) | 2.47 (m) | 2.16 (m) | 2.36 (m) | 1.43 (m) | 2.20 (d, 7.8) |
| 11α | 1.40 (m) | 1.60 (m) | 2.07 (m) | 1.26 (m) | 1.49 (m) | 1.83 (m) |
| 11β | 1.56 (m) | 1.65 (m) | 2.17 (m) | 1.52 (m) | 1.61 (m) | 2.09 (m) |
| 12α | 2.18 (m) | 1.83 (d, 1.51) | 2.19 (m) | 2.36 (m) | 2.35 (m) | 2.52 (m) |
| 12β | 2.61 (td, 12.9, 3.3) | 2.35 (d, 1.51) | 2.39 (m) | 2.45 (m) | 2.45 (m) | 2.52 (m) |
| 14 | 6.27 (s) | 6.24 (s) | 6.25 (s) | 6.27 (s) | 6.28 (s) | 6.27 (s) |
| 15 | 7.33 (t, 1.6) | 7.34 (t, 1.6) | 7.36 (t, 1.5) | 7.35 (t, 1.6) | 7.35 (s) | 7.35 (t, 1.6) |
| 16 | 7.18 (s) | 7.18 (s) | 7.22 (s) | 7.19 (s) | 7.21 (s) | 7.21 (s) |
| 17 | 0.91 (d, 6.8) | 0.91 (s) | 0.91 (d, 6.8) | 0.88 (d, 6.5) | 0.81 (d, 6.8) | 0.85 (d, 6.9) |
| 18α | 4.34 (dq, 11.7, 1.9) | 1.83 (s) | 1.80 (s) | 1.76 (s) | 1.98 (s) | 1.88 (m) |
| 18β | 4.48 (m) | - | - | - | - | - |
| 19α | 5.64 (d, 2.6) | 4.82 (s) | 9.5 (d, 2.9) | - | 1.55 (s) | 1.28 (s) |
| 19β | - | 5.14 (s) | - | - | - | - |
| 20 | 0.95 (s) | 0.96 (s) | 0.87 (s) | 0.97 (s) | 1.07 (s) | 1.03 (s) |
| 21 | - | 3.87 (s) | - | 3.50 (s) | - | - |
| 6OH | 2.92 (ls) | - | - | 2.83 (ls) | - | - |
| Compounds | IC50 µM | CC50 µM | SI |
|---|---|---|---|
| A549 Cell Line (Confidence Interval 95%) | VERO Cell Line (Confidence Interval 95%) | ||
| 1 | 24.12 (15.68 a 37.09) | 148.3 (93.30 a 235.7) | 6.14 |
| 2 | 100.3 (43.54 a 231.2) | >250 | 2.49 |
| 3 | 66.22 (49.16 a 89.21) | 181.5 (140.7 a 234.2) | 2.74 |
| 4 | 20.63 (11.64 a 36.55) | 98.41 (74.08 a 130.7) | 4.77 |
| 5 | 143.7 (107.7 a 191.9) | >250 | >1.74 |
| 6 | 24.51 (17.72 a 33.89) | 40.77 (27.74 a 59.91) | 1.66 |
| Compounds | Tested Concentration (µg/mL) | % HSV-1 Inhibition ± SD | |
|---|---|---|---|
| KOS Strain | 29R Strain | ||
| 1 | 100 | 34.56 ± 7.36 | 0 |
| 2 | 100 | 83.41 ± 16.12 | 74.21 ± 6.14 |
| 3 | 100 | 24.42 ± 4.04 | 32.03 ± 6.12 |
| 4 | 50 | 50.23 ± 8.24 | 14.45 ± 5.46 |
| 5 | 100 | 15.20 ± 4.65 | 30.85 ± 9.07 |
| 6 | 25 | 0 | 0 |
| Compounds | CC50 | IC50 | SI | ||
|---|---|---|---|---|---|
| Vero Cells | (KOS Strain) | (29R Strain) | (KOS Strain) | (29R Strain) | |
| 2 | >250 | 81.39 ± 9.82 | 74.93 ± 7.30 | >3.07 | >3.33 |
| 4 | 98.41 (74.08–130.7) | 52.38 ± 4.78 | ND | 1.87 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lhullier, C.; de Oliveira Tabalipa, E.; Nienkötter Sardá, F.; Sandjo, L.P.; Zanchett Schneider, N.F.; Carraro, J.L.; Oliveira Simões, C.M.; Schenkel, E.P. Clerodane Diterpenes from the Marine Sponge Raspailia bouryesnaultae Collected in South Brazil. Mar. Drugs 2019, 17, 57. https://doi.org/10.3390/md17010057
Lhullier C, de Oliveira Tabalipa E, Nienkötter Sardá F, Sandjo LP, Zanchett Schneider NF, Carraro JL, Oliveira Simões CM, Schenkel EP. Clerodane Diterpenes from the Marine Sponge Raspailia bouryesnaultae Collected in South Brazil. Marine Drugs. 2019; 17(1):57. https://doi.org/10.3390/md17010057
Chicago/Turabian StyleLhullier, Cintia, Eliane de Oliveira Tabalipa, Fernanda Nienkötter Sardá, Louis Pergaud Sandjo, Naira Fernanda Zanchett Schneider, João Luis Carraro, Cláudia Maria Oliveira Simões, and Eloir Paulo Schenkel. 2019. "Clerodane Diterpenes from the Marine Sponge Raspailia bouryesnaultae Collected in South Brazil" Marine Drugs 17, no. 1: 57. https://doi.org/10.3390/md17010057
APA StyleLhullier, C., de Oliveira Tabalipa, E., Nienkötter Sardá, F., Sandjo, L. P., Zanchett Schneider, N. F., Carraro, J. L., Oliveira Simões, C. M., & Schenkel, E. P. (2019). Clerodane Diterpenes from the Marine Sponge Raspailia bouryesnaultae Collected in South Brazil. Marine Drugs, 17(1), 57. https://doi.org/10.3390/md17010057