1. Introduction
Cellular senescence, a signal of aging, is stimulated by many different stress conditions, including oxidative, radiative, and inflammatory stress. Both oxidative stress and inflammation contribute to monolithic aging [
1]. Senescent cells secrete inflammatory cytokines, growth factors, and reactive oxygen species (ROS); these molecules contribute to cellular inflammation and oxidative stress, which in turn increase inflammatory cytokines and ROS, resulting in a deleterious cascade [
2]. “Inflamm-aging” is a complex, systemic biological response to harmful stimuli and injury [
3] characterized by the combined activities of leukocytes and macrophages, which help the body resist inflammatory stimuli and repair damage. Lipopolysaccharide (LPS) is a bacterial compound that activates macrophages and stimulates their release of pro-inflammatory cytokines, such as interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α, as well as inflammatory mediators, such as nitric oxide (NO), during oxidative stress. D-galactose (D-gal), a nutrient usually obtained from lactose in milk, has been used in previous studies to induce aging in mice through abnormal metabolism [
4,
5]. Both LPS and D-gal can cause oxidative stress and mitochondrial dysfunction, and damaged mitochondria generate reactive oxygen species (ROS) that have been implicated in inflamm-aging; in addition, increased ROS could lead to additional mitochondrial damage and accelerate the inflamm-aging process.
Evidence indicates that inflammatory signaling stimulates O2 production and oxidative stress via nuclear factor κB (NF-κB), p53, and p21, all of which can induce or facilitate age-related inflammation [
1,
2,
6]. NF-κB activates the transcription of pro-inflammatory cytokines, and oxidative stress and DNA damage have been shown to upregulate the p53/p21 pathway and trigger the secretion of senescence-associated secretory phenotype (SASP) components by increasing NF-κB activity [
7]. Other inflammation-sensitive pathways have also been implicated in aging. While existing anti-inflammatory medications such as glucocorticoids and nonsteroidals (NSAIDs) can significantly suppress the production of pro-inflammatory cytokines [
2,
8], the side effects of these drugs include immune system depression and gastrointestinal ulcerations [
8]. The development of other agents to slow the inflamm-aging process, particularly those from natural sources with fewer adverse effects, remains necessary.
Marine microalgae are a promising sustainable source of bioactive compounds, and several studies have reported on the anti-inflammatory activity of peptides, proteins, and polysaccharides in these microorganisms [
9,
10,
11]. A pigment–protein complex (PPC) thereof is the most abundant source of light-harvesting protein complexes located in algal thylakoid membranes that manage photosynthesis and chlorophyll photoprotection [
12]. Although many experiments have isolated and analyzed the PPC composition and structure from marine microalgae such as
Chlorella vulgaris and
Codium fragile [
12,
13], little is known about the potential bioactivities and pharmacological mechanisms of PPC extracts. In this study, we derived PPC from
Chlorella pyrenoidosa through aqueous and spirituous soaking and gel filtration to investigate the potential anti-aging and anti-inflammatory effects of this extract, and explore their underlying mechanisms. The inflamm-aging-related regulation of IL-β, IL-6, TNF-α, and inducible NO synthase, as well as the impacts on the expression of NF-κB, PPARα, PPARγ, p21, and p53, were thereby evaluated.
3. Discussion
In the process of senescence, the functionality of cells and tissues becomes markedly impaired, and oxidative stress and a weakened immune system are characteristic signs of senescence. In the different aging models, increased amounts of other cellular senescence markers such as ROS level, disordered immune system, and poor cell morphology have been observed [
18,
19]. Previous studies have implicated reactive oxygen species (ROS) in inflammatory and aging processes [
18,
19]. In this study, the anti-aging effects of PPC were evaluated in a reliable aging model induced by D-gal. The biochemical and physiological changed in the D-gal-induced aging processes include increases in senescence, oxidative stress, mitochondrial dysfunction, and apoptosis; therefore, the model very much represents the natural aging cell and animal models [
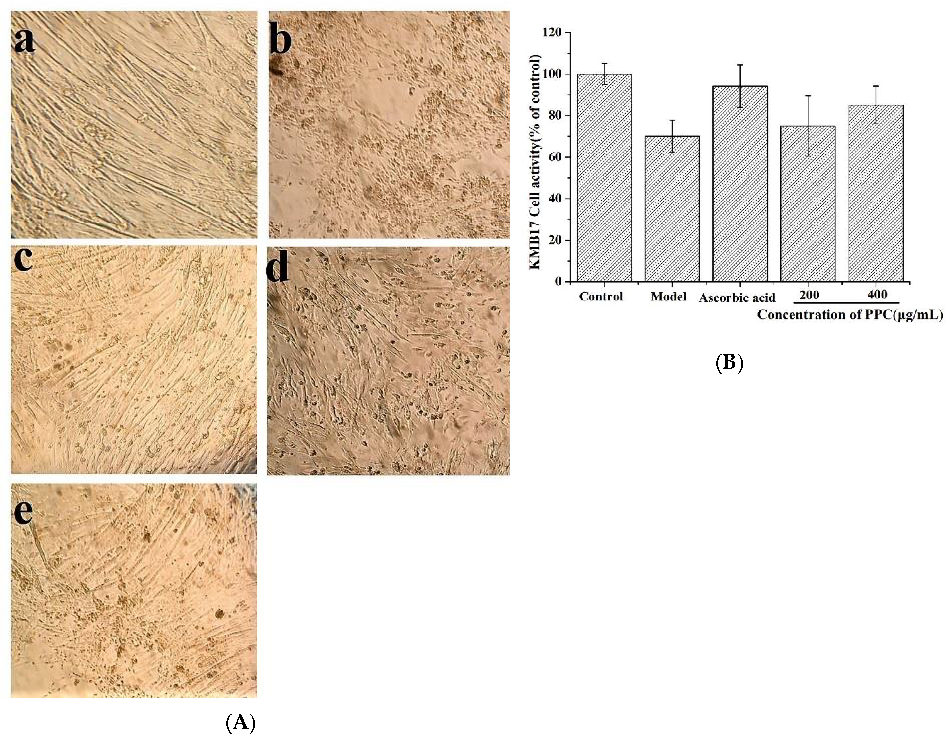
18]. Our results suggested that PPC increased cell proliferation and ameliorated cell morphology damage caused by D-gal in KMB17; PPC could improve immune cells function by increasing their anti-inflammatory capacity and increasing the antioxidant enzymes activities to ameliorate the inflammation associated with the aging process.
PPC administration diminished oxidative damage, decreased MAD activity, and increased SOD activity in a rat model. It has been shown that D-gal increases peroxidation and SOD activity while decreasing catalase and glutathione peroxidase directly or via activation of the NF-κB pathway [
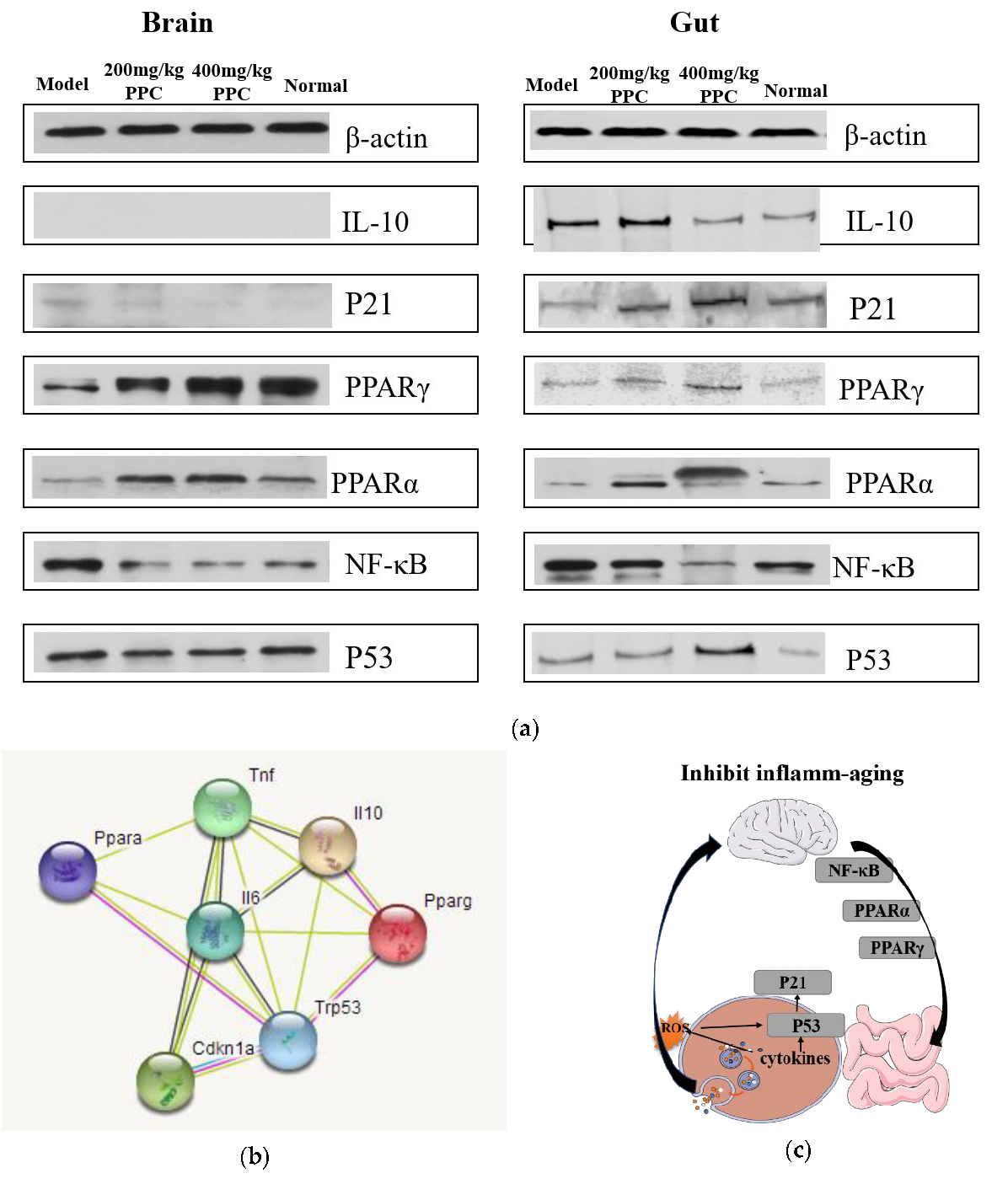
20]. In an animal study, the inflammation- and senescence-associated proteins NF-κB, PPARα, PPARγ, p53, and p21 were measured by Western blot to elucidate potential mechanisms of PPC. Previous studies have indicated that the master regulator nuclear factor NF-κB is crucial in inflammatory responses and cellular senescence [
21], and many effective drugs could alleviate inflammation-aging by upregulated NF-κB expression. Our results show that compared with controls, the levels of NF-κB were prominently increased in the model mice, but effectively reverted in mice treated with PPC; therefore, it can be interpreted that PPC could potentially regulate and maintain the cellular homeostasis with respect to D-gal induced inflammation and oxidative stress. PPARs are another family of regulators implicated in senescence-associated pathophysiological processes related to energy metabolism and oxidative stress [
20,
21]. The evidence supports an interrelation among ROS, PPARs, and NF-κB. According to early research, ROS facilitate the activation of the NF-κB pathway by inhibiting the phosphorylation of IκBα [
22]. Then, activated NF-κB negatively regulates the PPARs response to an oxidative stress environment [
23]. The data shows that the strongest NF-κB signal was accompanied with the weakest signals of PPARα and PPARγ in the model group; the opposite was in the high-dose PPC group, which is consistent with the research.
The p53/p21 pathway is not only the primary mediator of cellular senescence, but also essential for the cellular damage response pathways [
19]. The pathway potentiates inflammatory responses and inhibits both apoptosis and proliferation, leading to cellular senescence [
20]. The literature shows that the regulation of anti-aging occurs by activating the pathway p53/p21. However, in our results, levels of p21 and p53 in the brain and gut were somewhat inconclusive, with the strongest intensities of p21 and p53 in high concentration in the PPC gut, and little change in the PPC brain. We surmise that PPC may exert its protective effect against age-associated inflammatory factors by downregulating the p53/p21 pathway in the gut, if not in the brain.
STRING-based network analysis revealed that the cytokines TNF-α, IL-6, and IL-10, and proteins PPARa, PPARg, p53, and p21 were mapped to a network (
Figure 5b) that directly relates to inflammatory and aging processes, including inflammatory bowel disease (IBD) and cellular senescence. Cancer, viral infection, non-alcoholic fatty liver disease (NAFLD), and insulin resistance pathways were also involved, suggesting that PPC efficacy may extend to other conditions. Overall, the present data suggest that PPC can slow down the inflamm-aging process by lowering NF-κB and PPARs signaling in the brain and gut, as well as activating the p53/p21 pathway in the intestine (
Figure 5c); however, further studies are needed to confirm this.
4. Materials and Methods
4.1. Materials and Chemicals
C. pyrenoidosa powder (62.4% ± 1% total protein content) was obtained from Dr. Zhang Daojing at the East China University of Technology, Shanghai, China. Bicinchoninic acid (BCA) protein, superoxide dismutase (SOD), and malondialdehyde (MDA) kits were obtained from the Nanjing Jiancheng Bioengineering Institute (China). LPS and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Primary and secondary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Methanol and acetonitrile used in liquid chromatography were of HPLC grade. All other chemicals and reagents were also of analytical grade and commercially available.
4.2. Pigment–Protein Complex (PPC)
Crude pigment–protein mixture was derived as described in previous studies [
14], and the extracted pigment–protein complement was then lyophilized (FDU-1200, Tokyo Rikakikai Co., Ltd., Tokyo, Japan). The protein content therein was evaluated with a BCA kit (Nanjing Jiancheng Bioengineering Institute, China), and the extraction ratio was calculated.
4.3. Gel Filtration Chromatography
The fraction with the highest radical-scavenging activity was separated by gel filtration chromatography. Two milliliters of bioactive fraction dissolved in distilled water at a concentration of 20 mg/mL were loaded onto a Sephadex G-25 (1.6, 3 × 30 cm). The column was eluted with distilled water at a flow rate of 0.5 mL/min. The absorbance of the collected solution was evaluated at 280 nm. All eluates with the same peak were combined and freeze-dried.
4.4. Characterization of Pigment–Protein Complex
4.4.1. Spectrophotometric Measurement
Absorption spectra of the pigment–protein fractions were recorded over a wavelength range of 200 to 800 nm on a Shimadzu UV-2550 spectrophotometer at room temperature.
4.4.2. Pigment Determination
Five volumes of methanol were added to the protein solution, and the precipitate was subsequently removed. For high-performance liquid chromatography (HPLC) analysis, the PPC was eluted with a Shimadzu LC-20A system equipped with a Unimicro SP-120-5-C18 (4.6 × 180 mm, 3 μm) column. The mobile phase contained variational proportions of acetonitrile/ultrapure water (from 0:100% to 90%:10%), and absorbances were monitored at a wavelength of 450 nm.
4.4.3. Protein Determination
The protein content of the fractions was quantified with the BCA kit in accordance with the manufacturer’s instructions.
4.5. Cell Culture
RAW 264.7 macrophages were maintained at 37 °C in a humidified atmosphere of 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin solution. The KMB17 cell line (purchased from the Institute of Laboratory Animal Science, Jinan University), which originated from a human embryonic lung, is now widely used for cell senescence evaluations in vitro. KMB17 cells were grown in DMEM supplemented with 10% fetal bovine serum.
4.5.1. PPC Toxicity Tests
RAW 264.7 cells were seeded in 96-well plates at a density of 2 × 105 cells per well. After 24 h, the medium was exchanged for that with stepped concentrations of PPC, and cells were incubated for another 24 h. Then, 20 μL of MTT (1 mg/mL) was added to each well, and cells were incubated for another 4 h at 37 °C. The supernatant was subsequently decanted, and 180 μL of dimethyl sulfoxide (DMSO) was added to the cells. Then, absorbances were measured with a microplate reader at a wavelength of 570 nm.
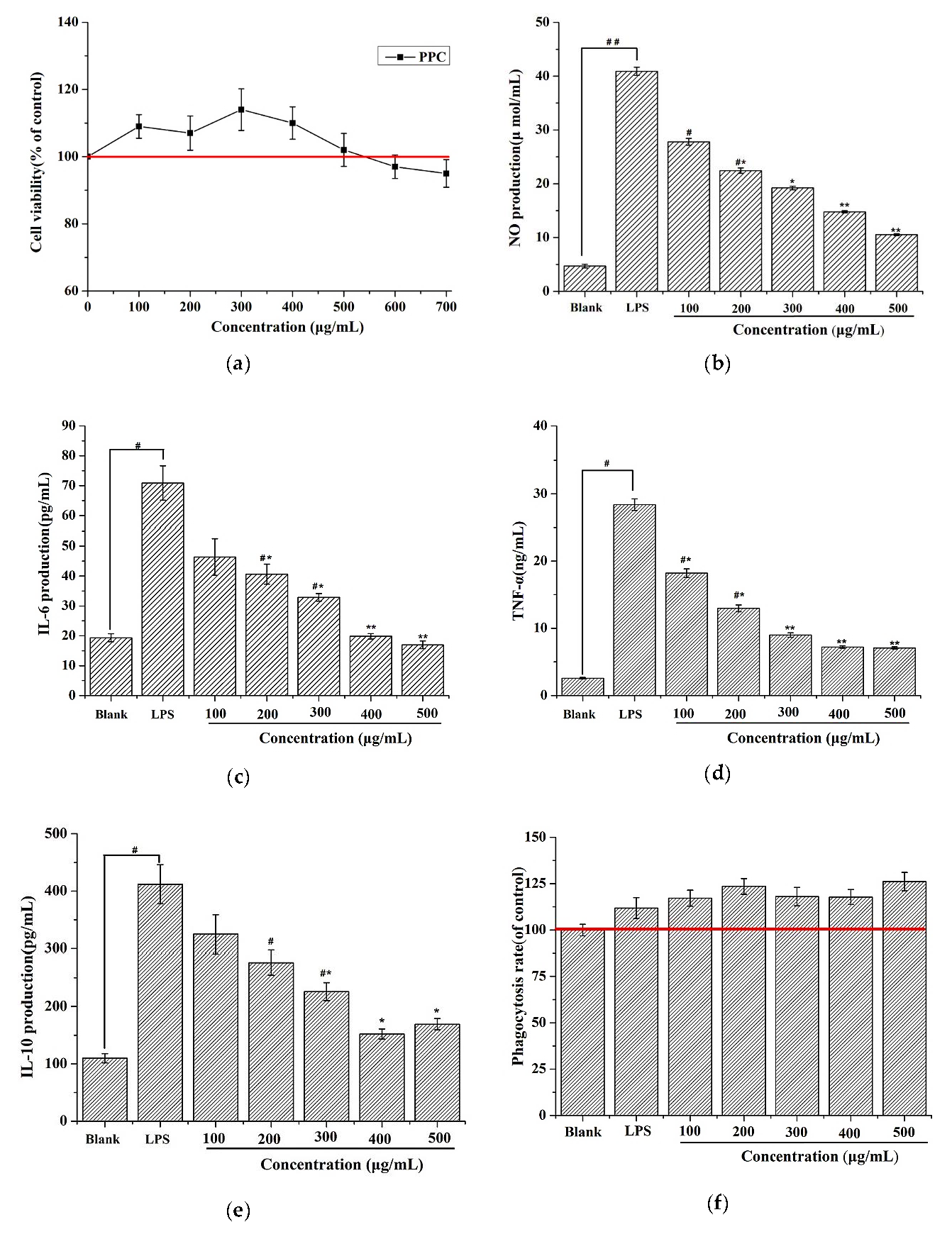
4.5.2. Measurement of Cytokines and NO Production
RAW 264.7 cells were plated into 96-well plates at 2 × 105 cells per well and stimulated with 2 μg/mL LPS for 24 h. Then, cells were treated with stepped concentrations of PPC for 24 h, followed by collection of the culture medium. Levels of TNF-α, IL-6, and IL-10 in the supernatants were assessed with enzyme-linked immunosorbent assay (ELISA) kits according to the manufacturer’s instructions.
4.5.3. Phagocytosis rate
RAW 264.7 cells (1 × 10
5 cells/well) were plated on 96-well plates and stimulated with 2 μg/mL LPS for 24 h. Then, cells were treated with stepped concentrations of PPC for 24 h followed by supplementation with 100 μL of 0.075% neutral red saline solution and subsequent incubation for 1 h at 37 °C. Then, cells were washed three times with PBS, and 100 μL of cell lysate was added into each well. After 2 h, absorption values were measured at 540 nm, and the phagocytosis rate was calculated with the following formula:
where
ODb refers to the value of the treated wells,
OD0 is the value of a blank well, and
ODa is the value of the control well.
4.5.4. KMB-17 Cell Senescence in Vitro
KMB-17 cells were seeded in 6-well plates at a density of 5 × 103 cells per well and stimulated with 2 μg/mL D-gal. After 24 h, the medium was exchanged for that with stepped concentrations of PPC followed by incubation for another 24 h. Then, cells were observed with light microscopy, and cell viability was measured.
4.6. Murine Model
A total of 30 C57BL/6 male and female mice (50%/50%) were randomly divided into five groups (n = 6). In the D-gal-administration group (model group), the mice were intraperitoneally (IP) injected with 120 mg/kg D-galactose once daily for 30 days. In the D-gal + PPC group, mice were IP injected with D-gal combined with either 200 or 400 mg/kg of PPC once daily for 30 days. In the control group, mice received IP saline at the same dose and time as their experimental counterparts. In the ascorbic acid group, mice were injected with D-galactose and 40 g/kg ascorbic acid once daily for 30 days. On the 31st day, the mice were anesthetized with sodium pentobarbital and euthanized by cervical dislocation. Then, brain and gut samples from the mice were collected for further analysis. The study was completed at the Institute of Laboratory Animal Science. The protocol was approved by the Laboratory Animal Ethics Committee Jian University on 2 April 2018 (the project identification code is 20180402-08).
4.7. Histological Analysis
Gut tissues were fixed in fresh 4% paraformaldehyde (pH 7.4) for 3 days and then embedded in paraffin. The sections were cut and stained with hematoxylin and eosin (H&E) and examined under light microscopy (Axioskop 40, Zeiss, Germany).
4.8. Measurement of Enzyme Levels
Levels of SOD and MDA were analyzed with the appropriate enzymatic kits, and protein content was measured with a BCA assay kit. Levels are expressed as nmol per mg of protein.
4.9. Western Blot Analysis
Brain and gut tissues were washed and lysed in RIPA (Radio Immunoprecipitation Assay) buffer for 30 min, and the lysates were centrifuged at 12,000× g for 25 min at 4 °C. Supernatants were decanted, mixed with 4X SDS sample buffer, and boiled for 10 min. Samples were run through a 4%/15% SDS polyacrylamide gel and then transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was blocked with 5% (w/v) BSA for 30 mins, and then incubated with primary antibody at 4 °C for 12 h. After 3 washings with TBST (Tris-Buffered Saline Tween-20), the membrane was incubated with secondary antibody for 1 h. The membrane was again washed with TBST, and bands were detected with a microplate scanner (Thermo Fisher Scientific, Inc.). Fluorescence intensities were measured using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
4.10. Statistical Analysis
To ensure the reliability and credibility of the results, all experiments were performed in triplicate and independently repeated at least three times. Quantitative data are expressed as mean ± standard deviation. One-way ANOVA or Duncan’s new multiple range tests were conducted to test for significance using SPSS 10.0 software. A p-value of less than 0.05 was considered statistically significant.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}