Abstract
Chemical investigation of the secondary metabolites of a rare New Zealand deep-sea sponge, Lamellomorpha strongylata, resulted in the isolation of twenty-one indole alkaloids, including two new bisindoles—(Z)-coscinamide D (1), (E)-coscinamide D (2)—and four compounds isolated for the first time as natural products—lamellomorphamides A (3), B (4), C (5) and D (6). In addition, fifteen previously reported natural products were isolated, seven of which are seco analogs of hamacanthin alkaloids. The one sponge produces enantiomerically pure but opposite configurations of compounds that only differ in the number of bromines, suggesting enantiodivergent biosynthesis. In addition, four compounds were isolated as partial racemates, suggesting these compounds are biosynthesized via two independent routes.
1. Introduction
In our continuing exploration of marine sponges as sources of novel brominated alkaloids [1,2,3], we investigated a New Zealand deep-sea sponge collected from the western continental slope. Lamellomorpha strongylata belongs to the rare family Vulcanellidae within the order Tetractinellida [4]. There are only two reports of natural products from the genus Lamellomorpha, both from New Zealand from the same 1995 collection from the Chatham Rise, off the east coast of the South Island. Bioassay-guided separation of the extracts from that collection resulted in the isolation of three distinct classes of cytotoxic polyketides: calyculins (calyculins A, B, E, F, and calyculinamides A and B), swinholide H, and theonellapeptolide IIIe [5,6]. Surprisingly, our collection of the same species yielded no polyketides, rather indole alkaloids. In this paper, we report the isolation of four new bisindole alkaloids, as well as seventeen other indole alkaloids, from L. strongylata.
2. Results and Discussion
The freeze-dried sponge was sequentially extracted with hexane, ethyl acetate, and n-butanol, and twenty-one secondary metabolites (Chart 1) were isolated from the ethyl acetate and n-butanol extracts, including two new compounds, (Z)-coscinamide D (1) and (E)-coscinamide D (2). Lamellomorphamides A–D (3–4) were isolated for the first time as natural products, although they were previously reported as intermediates in the synthesis of 6′,6′′-didebromo-cis-3,4-dihydrohamacanthin B, 6′-debromo-cis-3,4-dihydrohamacanthin B, and hamacanthin analogues [7,8]. Eight previously reported compounds, (E)-coscinamide B (7) [9], (Z)-coscinamide B (8) [9], deoxytopsentin (9) [10], isobromodeoxytopsentin (10) [11], bromodeoxytopsentin (11) [11], dibromodeoxytopsentin (12) [11], 6-bromoindole-3-carboxylic acid (13) [12], and (6-bromo-1H-indol-3-yl) oxoacetamide (14) [13], and seven trifluoroacetic acid (TFA)-catalyzed artifacts of the hamacanthin A and B class of compounds, 3,4-seco-(8R)-6′′-debromohamacanthin A (15), 3,4-seco-(8R)-6′,6′′-didebromohamacanthin A (16), 3,4-seco-(8S)-hamacanthin A (17), 3,4-seco-(8S)-hamacanthin B (18), 3,4-seco-(8S)-6′′-debromohamacanthin B (19), 3,4-seco-(8R)-6′-debromohamacanthin B (20), and 3,4-seco-(8R)-6′,6′′-didebromohamacanthin B (21), were also isolated. The structures of 1–21 were determined by analysis of their spectroscopic data, including NMR in DMSO-d6, UV, IR, ECD, and MS in combination with high-level TDDFT calculations. The structures of the eight previously reported compounds 7–14 were also confirmed by comparison to literature data [9,10,11,12,13].
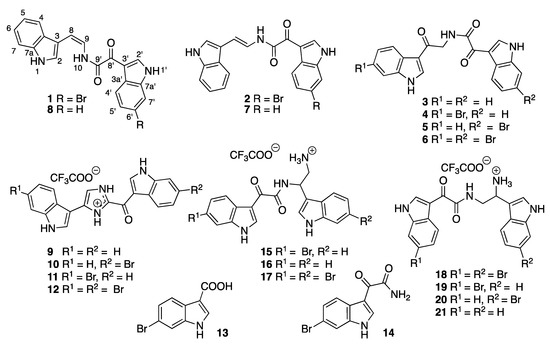
Chart 1.
Structures of compounds 1–21.
(Z)-coscinamide D (1) was isolated as a yellow amorphous solid. ESIMS analysis revealed a protonated molecular isotopic doublet at m/z 408/410 in a ratio of 1:1, characteristic of a monobrominated compound. HRESIMS analysis of 1 established a molecular formula of C20H14BrN3O2, based on the exact mass of the [M + Na]+ adduct ion (Δmmu −0.6). The presence of a 6-bromoindole moiety was apparent from a comparison of the 1H and 13C NMR (Table 1) and UV–vis spectroscopic data with those of the previously reported compound coscinamide B (7) [9]. Proton signals at δH 12.43 (1′-NH) and 8.92 (H-2′) and the ABX spin system comprised of signals at δH 7.41 (H-5′, dd, J = 8.4, 1.8 Hz), 7.76 (H-7′, d, J = 1.8 Hz), and 8.16 (H-4′, d, J = 8.4 Hz) indicated the presence of a 3,6-disubstituted indole moiety. The 1′-NH signal showed a COSY correlation with the H-2′ signal at δH 8.92, which in turn showed HMBC correlations to C-3′ (δC 111.8), C-3a′ (δC 125.3), and C-7a′ (δC 137.2) (Figure 1, Table S1). A COSY correlation was also observed between H-4′ and H-5′. Furthermore, 3-bond HMBC correlations from H-4′ to C-3′, and to the non-protonated carbons C-6′ (δC 116.1) and C-7a′ (δC 137.2), confirmed the attachment of bromine at C-6′ and established the 3,6-disubstituted bromoindole moiety. Another set of proton signals at δH 11.45 (1-NH), 7.64 (H-2), 7.43 (H-4), 7.16 (H-5), 7.07 (H-6), and 7.63 (H-7) was assigned to the 3-substituted indole residue. The 1-NH signal showed a COSY correlation with H-2, which in turn showed HMBC correlations to C-3 (δC 109.6), C-3a (δC 126.4), and C-7a (δC 135.8). COSY correlations defined the spin system from H-4 to H-7, confirming that 1 contained a 3-substituted indole. This interpretation was supported by 3-bond HMBC correlations from H-2 and H-4 to C-7a. IR absorption bands at 1670 and 1623 cm−1, along with non-protonated 13C NMR resonances at δC 159.7 and 180.0, suggest the presence of two carbonyl groups, one being an amide carbonyl. Another set of resonances was assigned to a vinylamide based on COSY correlations from H-9 to H-8 and 10-NH, and an HMBC correlation from 10-NH to the amide carbonyl carbon at C-9′ (δC 159.7). The observed 3JH-8,H-9 coupling constant (9.2 Hz) established the geometry of the olefinic bond as Z, which was supported by a strong ROESY correlation between H-8 and H-9. HMBC correlations from H-8 to C-2 (δC 123.8) and C-3a (δC 126.4), and from H-9 to C-3, indicated direct attachment of this fragment to the unsubstituted indole at C-3 (Figure 1). The remaining carbonyl substituent (δC 180.0) could be placed on C-3′ of the bromoindole moiety. Finally, the two indole-containing fragments were linked through the α-ketovinylamide to give the structure of 1. While no HMBC correlations were observed to C-8′ (δC 180.0), the chemical shifts of 1 closely resembled those reported for coscinamides A and B (7) [9]. It should also be noted that there are no literature reports of HMBC correlations to C-8′ in analogous known compounds, even for experiments optimized for small range coupling constants [9].

Table 1.
NMR Data (DMSO-d6; 1H 600 MHz, 13C 150 MHz) for (Z)-coscinamide D (1) and (E)–coscinamide D (2).
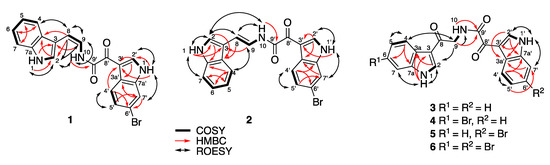
Figure 1.
Key HMBC, COSY, and ROESY NMR correlations for 1–6.
(E)-Coscinamide D (2) was isolated as a yellow amorphous solid. HRESIMS analysis of 2 established the same molecular formula as 1 (C20H14BrN3O2) based on the deprotonated molecule [M − H]− (Δmmu +0.2). The NMR data for 2 were very similar to those for 1 (Table 1), with the only significant differences being in the chemical shifts and coupling constants of the vinyl protons H-8 and H-9. The observed 3JH-8,H-9 coupling constant (14.7 Hz) suggested an E geometry, which was supported by the absence of a ROESY correlation between H-8 and H-9 (Figure 1, Table S2), confirming that 2 is the E isomer of 1. All other spectroscopic data were consistent with this structure.
Lamellomorphamide A (3) was isolated as a yellow amorphous solid. HRESIMS analysis revealed a protonated molecule [M + H]+ consistent with a molecular formula C20H15N3O3 (Δmmu −0.3). The NMR data for 3 (Table 2) also showed the presence of two 3-substituted indole residues, which was reminiscent of those of coscinamide B (7). The main difference from the coscinamide derivatives was the presence of an extra carbonyl carbon. IR absorption bands at 1670 and 1623 cm−1 along with non-protonated 13C NMR resonances at δC 163.8, 181.9, and 189.2, suggest the presence of one amide and two conjugated ketones. HMBC correlations from H-2 (δH 8.50, d, J = 3.2 Hz) to C-3a (δC 125.4) and C-7a (δC 136.3), and from 1-NH (δH 12.07) to C-3 (δC 113.9) and C-3a (δC 125.4), indicated that the pyrrole 1-NH and H-2 were connected to an ABMX aryl spin system comprised of the signals at δH 7.20 (H-5), 7.23 (H-6), 7.49 (H-7), and 8.16 (H-4) (Figure 1, Table S3). HMBC correlations from H-2′ (δH 8.82, d, J = 3.2 Hz) and 1′-NH (δH 12.26) to C-3a′ (δC 126.2) and C-7a′ (δC 136.4) indicated that the other pyrrole protons 1′-NH and H-2′ were connected to another ABMX aryl spin system comprised of the signals at δH 7.27 (H-5′), 7.28 (H-6′), 7.54 (H-7′) and 8.26 (H-4′). This confirmed the presence of the two 3-substituted indole moieties. An HMBC correlation from H-2 to C-8 (δC 189.2) indicated attachment of C-8 at C-3. The amide proton showed an HMBC correlation to its carbonyl C-9′ (δC 163.8) and a COSY correlation to methylene H2-9 (δH 4.63, d, J = 5.9 Hz), which in turn was coupled by HMBC to the carbonyl C-8. ROESY correlations between H-2 and H2-9, and between H2-9 and 10-NH (δH 8.91) verified the attachment of the fragment RCO–NH–CH2–CO on C-3 of the indole moiety (Figure 1). The remaining carbonyl substituent C-8′ (δC 181.9) was placed on C-3′ of the additional indole moiety, and the two indole-containing fragments were linked through the ketone (δC 181.9) and the amide carbonyl (δC 163.8) to give the structure of 3. Analogous to 1, and the literature [11,14], no HMBC correlations were observed to C-8′.
Table 2.
NMR Data (DMSO-d6; 1H 600 MHz, 13C 150 MHz) for lamellomorphamides A (3) and B (4).
Lamellomorphamide B (4) was also isolated as a yellow, amorphous solid. ESIMS analysis revealed a protonated molecule isotopic cluster [M + H]+ at m/z 424/426 in the ratio 1:1, suggesting the presence of one bromine atom. HRESIMS of 4 established a molecular formula of C20H14BrN3O3, based on the [M + Na]+ adduct ion (Δmmu −0.3). The 1H NMR data for 4 were similar to 3, indicating the same basic scaffold. The NMR data for 4 (Table 2) also showed the presence of one 3-substituted indole and one 3,6-disubstituted bromoindole. The main difference from 3 was the presence of a bromine atom, and its attachment on the indole ring of the “left hand” of the compound was confirmed by HMBC and ROESY NMR experiments (Figure 1, Table S4). The 1-NH signal (δH 12.17), the methine H-2 (δH 8.53), and the AMX spin system comprised of the signals at δH 7.35 (H-5, dd, J = 8.5, 1.8 Hz), 7.69 (H-7, d, J =1.8 Hz), 8.09 (H-4, d, J = 8.5 Hz), indicated the presence of a 3,6-disubstituted indole moiety with the bromine atom located on C-6 (δC 115.0). HMBC correlations were observed from 1-NH, H-2, H-5, and H-7 to C-3a (δC 124.5) and from H-2 and H-4 to C-7a (δC 137.4). The connectivity between 3,6-disubstituted bromoindole to the carbonyl C-8 (δC 189.4) was established by a ROESY correlation between H-2 and H2-9 (δH 4.62). The proton signals at δH 12.25 (1′-NH), 8.80 (H-2′), 8.25 (H-4′), 7.27 (H-5′), 7.28 (H-6′), and 7.53 (H-7′) suggested the presence of a 3-substituted indole moiety. Attaching this second indole to the ketone C-8′ (δC 181.8) gave 4 as the only possible structure for lamellomorphamide B.
Lamellomorphamide C (5) was isolated as a yellow, amorphous solid. ESIMS analysis revealed a protonated molecule isotopic cluster [M + H]+ at m/z 424/426 in the ratio 1:1, indicating the presence of one bromine atom. HRESIMS of 5 established the same molecular formula as 4 (C20H14BrN3O3) based on the [M + Na]+ adduct ion (Δmmu −0.3). This immediately suggested that 5 is a regioisomer of 4, with the bromine on the other (“right hand”) indole ring (Table 3). This interpretation was supported by ROESY correlations between 1′-NH (δH 12.32) and H-7′ (δH 7.75, d, J = 1.8 Hz) (Figure 1, Table S5). The presence of a small meta coupling between H-7′ and H-5′ (δH 7.42), and the absence of an ortho coupling revealed the attachment of the bromine atom at C-6′ (δC 116.0). The observed proton signals and coupling constants at δH 8.18 (H-4′, d, J = 8.4 Hz), 7.42 (H-5′, dd, J = 1.8, J = 8.4 Hz), and 7.75 (H-7′, d, J = 1.8 Hz), together with HMBC correlations from 1′-NH and H-2′ to C-3a′ (δC 125.3, is characteristic of a 3,6-disubstituted indole moiety with the bromine atom located on the indole ring of the “right hand” of 5. The proton signals at δH 12.07 (1-NH), 8.50 (H-2), 8.16 (H-4), 7.20 (H-5), 7.23 (H-6), and 7.49 (H-7) suggest the presence of the 3-substituted indole moiety, which was confirmed by HMBC correlations from H-2 to C-3, C-3a, and C-7a, and from H-4 to C-3, C-6, and C-7a. The connection of 3-indole to C-8 was confirmed by a strong ROESY correlation between H-2 and H-9. The amide proton showed an HMBC correlation to its carbonyl C-9′ (δC 163.4) and a COSY correlation to methylene H2-9 (δH 4.63, J = 5.9 Hz), which in turn was coupled by HMBC to the carbonyl C-8. Comparison of the chemical shifts of the indole protons with those of 4, together with HMBC correlations from H-2′ to C-3′, C-3a′, and C-7a′, and from H-4′ to C-3′, C-6′, and C-7a′, confirmed the connection of the 3′-bromoindolyl residue to C-8′, even though this carbon, like all others in this series, showed no correlations (Table S5). This left 5 as the only possible structure of lamellomorphamide C that matched the observed spectroscopic data.
Table 3.
NMR Data (DMSO-d6; 1H 600 MHz, 13C 150 MHz) for lamellomorphamides C (5) and D (6).
Lamellomorphamide D (6) was isolated as a yellow, amorphous solid. ESIMS analysis revealed a protonated molecule isotopic cluster [M + H]+ at m/z 502/504/506 in the ratio 1:2:1, indicative of two bromine atoms. HRESIMS of 6 established a molecular formula of C20H13Br2N3O3, based on the [M + H]+ protonated molecule (Δmmu 0.0). The similarity in 1H NMR spectra between 6 and 3–5 suggested 6 had both indoles substituted with bromine. This was confirmed by the presence of two aromatic ABX systems; δH 7.35 (H-5, dd, J = 8.5, 1.8 Hz), 7.69 (H-7, d, J = 1.8 Hz), and 8.09 (H-4, d, J = 8.5 Hz); and δH 7.42 (H-5′, dd, J = 8.4, 1.8 Hz), 7.75 (H-7′, d, J = 1.8 Hz), and 8.18 (H-4′, d, J = 8.4 Hz). ROESY correlations between 1-NH (δH 12.17) and H-7 (δH 7.69, d, J = 1.8 Hz) and from 1′-NH (δH 12.32) and H-7′ (δH 7.75, d, J = 1.8 Hz) were observed (Figure 1, Table S6). The small meta coupling constants of H-7 and H-7′ to H-5 and H-5′, respectively, revealed the attachment of the bromine atoms at C-6 (δC 115.5) and C-6′ (δC 116.0), consistent with the previously identified structures. The remaining spectroscopic data (Table 3) were consistent with 6 being 6,6′-dibromolamellomorphamide A.
Compounds 3 and 4 have previously been reported as intermediates in the synthesis of cis-3,4-dihydrohamacanthin B [7], and compounds 5 and 6 have previously been reported as intermediates in the synthesis of hamacanthin B anologues [8] but to date have not been reported as natural products. The reported 1H and 13C NMR data for 4 matched our data exactly. Although no HMBC correlations to C-8′ were detected for any of the compounds 1–6, the connectivity between the keto enamide and the indole moieties was determined by comparison of our NMR data with those of coscinamide B. The absence of any correlations to C-8′ is also consistent with the topsentins [11] and spongotines [14]. Coscinamides are the only known natural products that contain the unusual α-ketovinylamide functionality, and this is the first example of naturally occurring E isomers, although both coscinamide B and its Z isomer have been synthesized as intermediates in the synthesis of dihydrohamacanthins [15].
Deoxytopsentin (9) [10] was the major metabolite of L. strongylata and was obtained pure after Sephadex LH-20 gel permeation chromatography. As previously reported for topsentins [16], the doubling of 1H NMR signals was observed in DMSO-d6 due to slow tautomerization of the imidazole. The addition of a trace amount of TFA to the NMR sample resulted in one set of signals (Figure S47), but with broadening of the imidazolium methine. In methanol-d4 (Figure S48), all peaks were sharp and could be easily assigned.
Hamacanthins A (24) and B (25) [14,17] are part of a series of marine alkaloids that contain a six-membered nitrogen heterocycle (5,6-dihydro-2-(1H)-pyrazinone) as a linker unit between two variously substituted indole groups. Seven putative acid-catalyzed artifacts of the hamacanthin A and B class of compounds were isolated from the ethyl acetate and n-butanol extracts: 3,4-seco-6′′-debromohamacanthin A (15), 3,4-seco-6′,6′′-didebromohamacanthin A (16), 3,4-seco-hamacanthin A (17), 3,4-seco-hamacanthin B (18), 3,4-seco-6′′-debromohamacanthin B (19), 3,4-seco-6′-debromohamacanthin B (20), and seco-6′,6′′-didebromohamacanthin B (21). NMR spectroscopy of these HPLC purified compounds (containing traces of TFA) showed only one set of peaks corresponding to the acyclic form (Figures S56–S69, Tables S7–S9), while LC-MS analysis (with or without formic acid) showed two peaks (Figure S1). The first peak (m/z 425/427; λmax 214, 262, 278, 329 nm) corresponded to the acyclic form and the second peak (m/z 407/409, Figure S75; λmax 216, 280, 385 nm;) corresponded to the cyclic form (Figure S2). Compounds 15–21 were isolated by HPLC purification with 0.05% TFA in the mobile phase. An LC-MS analysis of crude n-butanol and ethyl acetate fractions did not reveal masses corresponding to the acyclic compounds (Figures S3 and S4), but rather the cyclic hamacanthin A and B class of compounds. We therefore propose that the TFA-catalyzed ring-opening occurred during HPLC purification of 15–21. A similar mechanism has been proposed by Kuoko et al. [18] in the total synthesis of optically active hamacanthins A and B from the acyclic intermediates. Alternatively, the previously isolated hamacanthin A and B type alkaloids could be artifacts of isolation as previous reports all used silica gel chromatography, which we found efficiently cyclizes compounds 15–21.
Given there are no specific rotation data reported for any compounds related to 15–21, our ORD data could not be compared to any literature values. Therefore, 15–21 were passed through a silica SPE column, eluting with dry 20% ethyl acetate in hexane, to facilitate dehydration and ring closure (Scheme 1). The specific rotations of the resulting cyclized compounds 22–28 were measured for comparison to the hamacantins (Table 4). The specific rotation of 22 indicated it was (R)-6′′-debromohamacanthin A and that 15 must therefore also have an R configuration at C-8. Compound 23 had a specific rotation suggesting it was (R)-didebromohamacanthin A, the enantiomer of natural (S)-bisdebromohamacanthin A. Compound 24 had a specific rotation similar to synthetic and natural (S)-hamacanthin A (Table 4) and opposite to synthetic (R)-hamacanthin A (−79 deg cm2 g–1 [19]). The specific rotations of natural and synthetic hamacanthin B (25) matched the sign but not the amplitude of the specific rotation for cyclized 18. Similarly, 28 had a negative specific rotation but much smaller than expected from the literature. In contrast, the specific rotation of 26 was a reasonable match for the published specific rotation of (S)-6′′-debromohamacanthin B. Compound 27, did not match the amplitude and was opposite in sign, suggesting 27 (and 20) had the opposite configurations to the previously isolated compounds. Kuoko et al. [18] in the total synthesis (+)-hamacanthins A and B, reported the specific rotations of the Boc-protected acyclic amide intermediates as [α]D −2.6, +9.8, and −5.0 deg.cm2.g–1, which are very small compared to the specific rotations of the cyclic (S)-hamacanthins A and B (+83.7 and +170.1 deg.cm2.g–1, respectively) and generally consistent with the fact that specific rotation is not a useful indicator of absolute configuration in this series of closely related compounds.
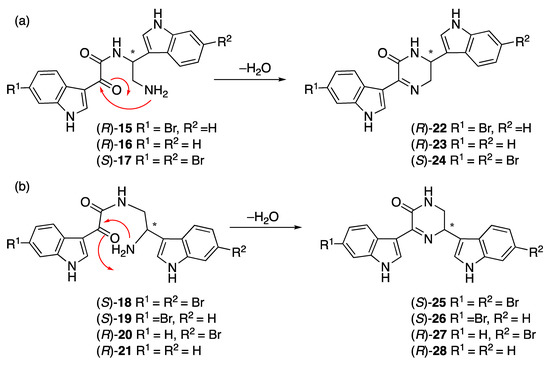
Scheme 1.
Proposed mechanism for ring closing of 15–21 to hamacanthins a and b derivatives.
Table 4.
Specific rotation comparison of cyclized compounds 22–28.
Turning to electronic circular dichroism (ECD; Figure 2 and Figures S5–S11), it is clear that 15, 16, 20, and 21 have the same configurations, while 17–19 have the opposite configurations. The weak ECD for 17, 18 and 20, 21 suggests that they are partial racemates, supporting the conclusions above (Table 4). The ECD spectra of 15–21 showed negative Cotton effects at 280 and 330 nm and positive Cotton effects at 360 nm for all the compounds except 17–19, which were the opposite. This is consistent with the specific rotations found for 27 and 28 (derived from 20 and 21 respectively) supporting the assignment of 15 and 16 as R, and 17–19 as S. Based on the ECD data, 20 and 21 should have R configurations, consistent with a negative specific rotation, but not consistent with the magnitude of the published specific rotations. Taken together, these data suggest that 20 and 21 are partial racemates with the R form predominating and that 18 and 19 are partial racemates with the S form predominating. This result is intriguing for a number of reasons. Firstly, the one sponge produces enantiomerically pure but opposite configurations of compounds that only differ in the number of bromines, suggesting parallel rather than sequential biosynthesis. Secondly, four compounds are partial racemates, which (if the above is true) suggests these compounds are biosynthesized via two routes in non-equivalent amounts. This type of enantiodivergent biosynthesis has previously been observed in related bromotyrosine and bromopyrrole alkaloids [3,23,24,25]. While it is possible that this chiral center could undergo epimerization during handling, this is unlikely as partial racemization was seen in only four out of the seven compounds isolated from the same extract, and all compounds were handled identically. Finally, our results explain the range of optical rotations published for these compounds. For example, synthetic (S)-hamacanthin B (25) has been reported with a specific rotation (methanol) of +170–183 deg.cm2.g–1 [18,22] whereas natural 25 has been reported with +172 deg.cm2.g–1 [17] or +56 deg.cm2.g–1 [20]. The latter is close to the value we obtained (+46 deg.cm2.g–1) for the partial racemate, suggesting that the compound isolated by Bao et al. was also a partial racemate. Enantiodivergent biosynthesis of natural products may be more common than currently recognized.
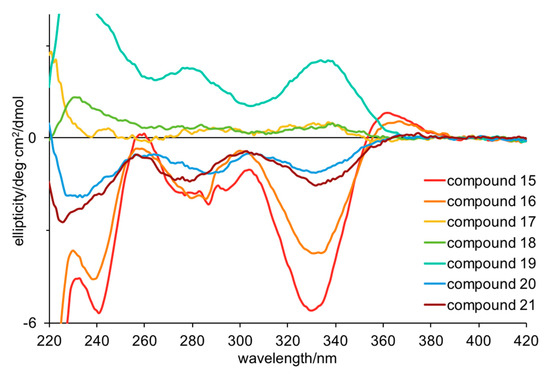
Figure 2.
Electronic circular dichroism spectra for compounds 15–21 in methanol (50–70 μM).
Finally, turning to time-dependent density functional theory (TDDFT) calculations, we were able to confirm the R configuration for 16 and 21. TDDFT-D3//pbe0/TZVPP-COSMO(methanol) was able to qualitatively reproduce the UV (Figure S12) and ECD (Figure 3) spectra for 16 and 21. Specifically, the positive Cotton effect at 360 nm and negative Cotton effect at 330 nm were reproduced, albeit with differing oscillator strengths. These results add weight to the interpretation that 17–19 have S configurations while the others have R configurations.
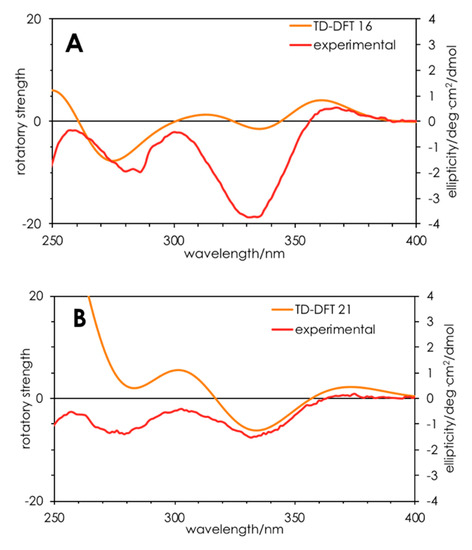
Figure 3.
Calculated (orange) and experimental (red) ECD spectra for compounds (R)-16 (A) and (R)-21 (B). Calculations were performed on ground state-optimized (TDDFT-D3//pbe0/TZVPP) structures in a solvent continuum model (COSMO; methanol).
Bisindole alkaloids are a rapidly growing group of sponge metabolites that exhibit potent and diverse bioactivities, including cytotoxicity, antiviral, antifungal, and anti-inflammatory activities [20]. For example, the bisindole alkaloids vinblastine and vincristine have been developed as effective anticancer drugs [26]. Examples of bisindole alkaloids reported from marine sponges include the topsentins [16], which have a ketone and an imidazole moiety as a linker between two indole rings; the nortopsentins [27], which lack the central ketone; and dragmacidins [17] and the hamacanthins (22–28) [17,18], which have a piperazine–piperazinone linker. Compounds 1–9 and 13–21 were screened against Methicillin-resistant Staphylococcus aureus (MRSA, ATTC 43300), Escherichia coli (ATCC 25922), Klebsiella pneumoniae (MDR, ATCC 700603), Acinetobacter baumannii (ATCC 19606), Pseudomonas aeruginosa (ATCC 27853), Candida albicans (ATCC 90028), and Cryptococcus neoformans var. grubii (ATCC 208821). Compounds 2, 3, 6, 13, and 18 showed some activity against MRSA (Table S10).
3. Materials and Methods
3.1. General Experimental Procedures
Specific optical rotations were measured on a Jasco P-1010 Polarimeter (Jasco, Tokyo, Japan). Infrared spectra were recorded on a Nicolet iS10 ATR FTIR Spectrometer (Thermo Scientific, Scorsby, Australia) as thin films. 1H NMR and 13C NMR spectra were acquired at 25 °C on a Bruker Avance AVII 600 MHz NMR spectrometer in DMSO-d6, processed using Bruker Topspin v3.2 software and referenced to residual monoprotonated solvent (DMSO-d6 δH 2.49; δC 39.52). LC-MS was performed on an Agilent 1260 Infinity UHPLC coupled to an Agilent 6130B single quadrupole mass spectrometer by electrospray ionization in positive and negative polarity modes. UV–vis spectra were acquired on an Agilent 1260 Infinity diode array detector. High-resolution mass spectra were acquired on a Q Exactive Plus hybrid quadrupole-orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) by direct infusion in CH3CN. HPLC separations were achieved on a Gilson 506C HPLC system. Analytical and preparative HPLC were performed on Synergi Max-RP C12 HPLC columns (Phenomenex, Sydney, Australia): Synergi 10 μm Max-RP 250 × 4.6 mm (1.0 mL/min), Synergi 10 μm Max-RP 250 × 10.0 mm (3.0 mL/min), and Synergi 10 μm Max-RP 250 × 21.2 mm (14.0 mL/min). LC-MS was performed on a C18 analytical column (Phenomenex Gemini 3 μm C18, 150 × 2.0 mm) with a gradient from 5%–95% CH3CN in H2O with 0.025% formic acid, flow rate 0.2 mL/min, and UV detection at 254 nm.
3.2. Biological Material
The sponge was collected from the Western Continental Slope (Station J954), Northland, New Zealand, (34.6333° S, 172.2250° E) at a depth of ~200 m, by gear rock dredge in June 1981. The sample was frozen upon collection, freeze-dried and stored at room temperature. It was identified as Lamellomorpha strongylata Bergquist, 1968 (class Demospongiae, order Tetractinellida, suborder Astrophorina, family Vulcanellidae), and deposited at the New Zealand National Institute of Water and Atmospheric Research (NIWA) under the accession number NIWA 92907.
3.3. Extraction and Isolation
The freeze-dried sponge (150 g) was blended to a powder and extracted sequentially with hexane (3 × 500 mL), ethyl acetate (3 × 500 mL), and n-butanol (2 × 200 mL) to yield 1.2, 1.2, and 0.6 g of crude extracts, respectively. The ethyl acetate extract was subjected to gel permeation chromatography (Sephadex LH-20; 22 × 400 mm; 1:1 CHCl3:MeOH), to afford seven subfractions (E1–E7), which were subjected to LC-MS analysis. Fractions E3–E5 and E7 were individually passed through an Alltech high capacity C18 solid-phase extraction cartridge, eluting with 90% CH3CN in H2O, and the eluates were purified separately by preparative HPLC columns (Synergi Max-RP C12, 10 μm, 250 × 22 mm) using a gradient from 20%–95% CH3CN in H2O with 0.01% TFA. Fraction E3 (120.8 mg) afforded 1 (tR 33.2 min; 0.5 mg), 3 (tR 22.5 min; 0.5 mg), 4 (tR 25.5 min; 0.9 mg), 5 (tR 26.0 min; 2.2 mg), 6 (tR 31.0 min; 1.7 mg), 7 (tR 28.5 min; 3.2 mg), 15 (tR 13.5 min; 10.5 mg), and 16 (tR 10.2 min; 5.5 mg). Fraction E4 (78.9 mg) afforded 8 (tR 30.0 min; 8.5 mg), 13 (tR 21.0 min; 1.9 mg), 14 (tR 19.8 min; 1.9 mg), 18 (tR 23.5 min; 2.0 mg), 19 (tR 15.5 min; 1.9 mg), and 20 (tR 18.0 min; 5.2 mg). Fraction E5 (111.5 mg) afforded 2 (tR 22.4 min; 0.5 mg). Fraction E6 (282.7 mg) did not require further purification and was identified as 9 by NMR spectroscopy. Fraction E7 (128.6 mg) afforded 10 (tR 19.6 min; 6.2 mg), 11 (tR 17.4 min; 2.4 mg), and 12 (tR 21.2 min; 5.8 mg). The n-butanol extract was also subjected to gel permeation chromatography (Sephadex LH-20; MeOH) to afford six subfractions (B1–B6), which were subjected to LC-MS analysis. Fractions B5 and B6 were individually passed through a C18 Sep-Pak and eluted with 90% CH3CN in H2O. The eluates were purified by HPLC (Synergi Max-RP C12, 10 μm, 250 × 22 mm) using gradients from 20% CH3CN to 95% CH3CN in H2O with 0.01% TFA. Fraction B5 (45.6 mg) afforded 15 (tR 14.0 min; 19.1 mg) and 16 (tR 10.2 min; 14.9 mg) and fraction B6 (38.2 mg) afforded 17 (tR 18.0 min; 1.7 mg) and 21 (tR 10.5 min; 1.7 mg). Please refer to Figures S13–S75 for NMR spectra of all isolated compounds.
(Z)-Coscinamide D (1): yellow amorphous powder (0.5 mg); UV (MeOH) λmax 214, 262, 278, 340 nm; IR (neat film) νmax 3437, 3195, 2987, 2939, 1647, 1594, 1541, 1489, 1236, 1130, 924, 738 cm−1; 1H and 13C NMR data, see Table 1, Table S1; ESIMS m/z 408/410 [M + H]+ isotopic cluster in the ratio 1:1; HRESIMS m/z 430.0156 [M + Na]+ (calcd for C20H1479BrN3O2Na+, 430.0162).
(E)-Coscinamide D (2): yellow amorphous powder (0.5 mg); UV (MeOH) λmax 214, 260, 276, 342 nm; IR (neat film) νmax 3304, 2976, 1674, 1616, 1482, 1438, 1204, 1130, 801, 744 cm−1; 1H and 13C NMR data, see Table 1, Table S2; ESIMS m/z 408/410 [M + H]+ isotopic cluster in the ratio 1:1; HRESIMS m/z 406.0199 [M − H]− (calcd for C20H1379BrN3O2−, 406.0197).
Lamellomorphamide A (3): yellow amorphous powder (1.1 mg); UV (MeOH) λmax 208, 258, 302 nm; IR (neat film) νmax 3198, 2978, 1670, 1616, 1490, 1424, 1238, 1131, 742 cm−1; 1H and 13C NMR data, see Table 2, Table S3; ESIMS m/z 346 [M + H]+; HRESIMS m/z 346.1183 [M + H]+ (calcd for C20H16N3O3+, 346.1186).
Lamellomorphamide B (4): yellow amorphous powder (0.9 mg); UV (MeOH) λmax 208, 246, 268, 310 nm; IR (neat film) νmax 3269, 2977, 1675, 1635, 1616, 1489, 1436, 1202, 1131, 801, 746 cm−1; 1H and 13C NMR data, see Table 2, Table S4; ESIMS m/z 424/426 [M + H]+ isotopic cluster in the ratio 1:1; HRESIMS m/z 446.0108 [M + Na]+ (calcd for C20H1479BrN3O3Na+, 446.0111).
Lamellomorphamide C (5): yellow amorphous powder (2.2 mg); UV (MeOH) λmax 212, 260, 302 nm; IR (neat film) νmax 3349, 2926, 1676, 1634, 1496, 1435, 1202, 1131, 801, 722 cm−1; 1H and 13C NMR data, see Table 3, Table S5; ESIMS m/z 424/426 [M + H]+ isotopic cluster in the ratio 1:1; HRESIMS m/z 446.0108 [M + Na]+ (calcd for C20H1479BrN3O3Na+, 446.0111).
Lamellomorphamide D (6): yellow amorphous powder (1.7 mg); UV (MeOH) λmax 214, 273, 310 nm; IR (neat film) νmax 3291, 2923, 1682, 1615, 1496, 1416, 1202, 1131, 802 cm−1; 1H and 13C NMR data, see Table 3, Table S6; ESIMS m/z 502/504/506 [M + H]+ isotopic cluster in the ratio 1:2:1; HRESIMS m/z 501.9396 [M + H]+ (calcd for C20H1479Br2N3O3+, 501.9396).
3.4. Biological Assays
Primary antimicrobial screening was carried out by whole-cell growth inhibition assays, using the samples at 10 and 20 μM for S. aureus only in DMSO in duplicate (Table S10). The inhibition of growth was measured against methicillin-resistant S. aureus (ATTC 43300), E. coli (ATCC, 25922), K. pneumoniae (MDR, ATCC 700603), A. baumannii (ATCC 19606), P. aeruginosa (ATCC 27853), C. albicans (ATCC 90028), and C. neoformans var. grubii (ATCC 208821). Bacteria were cultured in cation-adjusted Mueller Hinton broth (CAMHB) at 37 °C overnight, and the culture diluted 40-fold with fresh broth and incubated at 37 °C for 1.5–3 h. The resultant mid-log phase cultures were diluted (CFU/mL measured by OD600), then added to each well of the compound containing plates, giving a cell density of 5 × 105 CFU/mL and a total volume of 50 μL. All the plates were covered and incubated at 37 °C for 18 h without shaking. Inhibition of bacterial growth was determined by measuring optical density at 600 nm (OD600), and the percentage of growth inhibition was calculated for each well, using the negative control (media only) and positive control (bacteria without inhibitors) on the same plate as references. Fungi strains were cultured for 3 days on yeast extract–peptone dextrose (YPD) agar at 30 °C. A yeast suspension of 1 × 106 to 5 × 106 CFU/mL (as determined by OD530) was prepared from five colonies. The suspension was subsequently diluted and added to each well of the compound containing plates giving a final cell density of fungi suspension of 2.5 × 103 CFU/mL and a total volume of 50 μL. All plates were covered and incubated at 35 °C for 24 h without shaking. Growth inhibition of C. albicans was determined by measuring absorbance at 530 nm (OD530), while the growth inhibition of C. neoformans was determined by measuring the difference in optical density between 600 and 570 nm (OD600–570), after the addition of resazurin (0.001% final concentration) and incubated at 35 °C for additional 2 h. The absorbance was measured using a Biotek Synergy HTX plate reader. The percentage of growth inhibition was calculated for each well, using the negative control (media only) and positive control (fungi without inhibitors) on the same plate. The significance of the inhibition values was determined by modified Z-scores. Samples with inhibition value above 80% and Z-scores above 2.5 for either replicate were classed as actives. Samples with inhibition values between 50%–80% and Z-scores above 2.5 for either replicate were classed as partial actives. All screening was performed in duplicate using the same microbial cultures, but with replicates on different plates. In addition, two values were used as quality control for individual plates and standard antibiotic controls at different concentrations (>MIC and <MIC). Colistin and vancomycin were used as positive bacterial inhibitor standards for Gram-negative and Gram-positive bacteria, respectively. Fluconazole was used as a positive fungal inhibitor standard for C. albicans and C. neoformans. The plate passes the quality control if the Z′-factor > 0.4 and the standards are active and inactive at highest and lowest concentrations, respectively.
4. Conclusions
In summary, two new bisindole alkaloids, (Z)-coscinamide D and (E)-coscinamide D, together with lamellomorphamides A–D were isolated for the first time from a natural source. While bisindole alkaloids have been reported from Rhaphisia lacazei, Spongosorites, Hamacantha and Coscinoderma spp, it is of interest to note that this is the first time this class of compound has been isolated from the genus Lamellomorpha, for which there are only two previous reports of unrelated natural products. In addition, we have isolated seven hamacanthin-type alkaloids, three of which appear to be optically pure while the other four are partial racemates, providing another fascinating example of enantiodivergent biosynthesis in marine alkaloids.
Supplementary Materials
The following are available online at https://www.mdpi.com/1660-3397/17/12/683/s1. Supplementary Materials including NMR spectra, tabulated 2D NMR data, HRMS, 1D and 2D NMR spectra, CD spectra.
Author Contributions
K.R. extracted and isolated the compounds and performed the structure elucidation. P.K. aided the structure elucidation and performed the DFT calculations. K.R. and P.K. drafted the manuscript and A.M.P. and P.K. edited the final version.
Funding
This research was funded, in part, by the Australian Research Council (DP130103281 to P.K. and A.M.P.; FT130100142 to A.M.P.) and Macquarie University (iMQRES scholarship to K.R.).
Acknowledgments
The sponge material was donated and taxonomically identified by Patricia R. Bergquist (University of Auckland) and the location of collection and voucher number confirmed by Michelle Kelly (NIWA, New Zealand). We thank M. McKay (APAF, Macquarie University) for the acquisition of HRMS data. Antimicrobial screening was performed by CO-ADD (The Community for Antimicrobial Drug Discovery).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gotsbacher, M.P.; Karuso, P. New antimicrobial bromotyrosine analogues from the sponge Pseudoceratina purpurea and its predator Tylodina corticalis. Mar. Drugs 2015, 13, 1389–1409. [Google Scholar] [CrossRef] [PubMed]
- Hao, E.; Fromont, J.; Jardine, D.; Karuso, P. Natural Products from Sponges of the Genus Agelas—On the Trail of a [2+2]-Photoaddition Enzyme. Molecules 2001, 6, 130–141. [Google Scholar] [CrossRef]
- Ragini, K.; Fromont, J.; Piggott, A.M.; Karuso, P. Enantiodivergence in the Biosynthesis of Bromotyrosine Alkaloids from Sponges? J. Nat. Prod. 2017, 80, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Bergquist, P.R. The Marine Fauna of New Zealand: Porifera, Demospongiae, Part 1 (Tetractinomorpha and Lithistida). NZL Dep. Sci. Industr. Res. Bull. 1968, 37, 1–106. [Google Scholar]
- Dumdei, E.J.; Blunt, J.W.; Munro, M.H.G.; Pannell, L.K. Isolation of calyculins, calyculinamides, and swinholide H from the New Zealand deep-water marine sponge Lamellomorpha strongylata. J. Org. Chem. 1997, 62, 2636–2639. [Google Scholar] [CrossRef]
- Shangxiao, L.; Dumdei, E.J.; Blunt, J.W.; Munro, M.H.G.; Robinson, W.T.; Pannell, L.K. Theonellapeptolide IIIe, a New Cyclic Peptolide from the New Zealand Deep Water Sponge, Lamellomorpha strongylata. J. Nat. Prod. 1998, 61, 724–728. [Google Scholar]
- Miyake, F.Y.; Yakushijin, K.; Horne, D.A. Synthesis of marine sponge bisindole alkaloids dihamacanthin. Org. Lett. 2002, 4, 941–943. [Google Scholar] [CrossRef]
- Kim, A.; Kim, M.J.; Noh, T.H.; Hong, J.; Liu, Y.; Wei, X.; Jung, J.H. Synthesis and antibacterial evaluation of hamacanthin B analogues. Bioorg. Med. Chem. Lett. 2016, 26, 5013–5017. [Google Scholar] [CrossRef]
- Bokesch, H.R.; Pannell, L.K.; McKee, T.C.; Boyd, M.R. Coscinamides A, B and C, three new bis indole alkaloids from the marine sponge Coscinoderma sp. Tetrahedron Lett. 2000, 41, 6305–6308. [Google Scholar] [CrossRef]
- Tsujii, S.; Rinehart, K.L.; Gunasekera, S.P.; Kashman, Y.; Cross, S.S.; Lui, M.S.; Pomponi, S.A.; Diaz, M.C. Topsentin, bromotopsentin, and dihydrodeoxybromotopsentin: Antiviral and antitumor bis(indolyl)imidazoles from Caribbean deep-sea sponges of the family Halichondriidae. Structural and synthetic studies. J. Org. Chem. 1988, 53, 5446–5453. [Google Scholar] [CrossRef]
- Shin, J.; Seo, Y.; Cho, K.W.; Rho, J.-R.; Sim, C.J. New bis (indole) alkaloids of the topsentin class from the sponge Spongosorites genitrix. J. Nat. Prod. 1999, 62, 647–649. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.; Jensen, J.; Anthoni, U.; Christophersen, C.; Nielsen, P.H. Structure and synthesis of bromoindoles from the marine sponge Pseudosuberites hyalinus. J. Nat. Prod. 1993, 56, 1553–1558. [Google Scholar] [CrossRef]
- Bao, B.; Zhang, P.; Lee, Y.; Hong, J.; Lee, C.-O.; Jung, J. Monoindole alkaloids from a marine sponge Spongosorites sp. Mar. Drugs 2007, 5, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Sun, Q.; Yao, X.; Hong, J.; Lee, C.-O.; Cho, H.Y.; Jung, J.H. Bisindole alkaloids of the topsentin and hamacanthin classes from a marine sponge Spongosorites sp. J. Nat. Prod. 2007, 70, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yakushijin, K.; Miyake, F.; Horne, D. A concise synthesis of indolic enamides: Coscinamide A, coscinamide B, and igzamide. Tetrahedron Lett. 2009, 50, 4343–4345. [Google Scholar] [CrossRef]
- Bartik, K.; Braekman, J.-C.; Daloze, D.; Stoller, C.; Huysecom, J.; Vandevyver, G.; Ottinger, R. Topsentins, new toxic bis-indole alkaloids from the marine sponge Topsentiagenitrix. Can. J. Chem. 1987, 65, 2118–2121. [Google Scholar] [CrossRef]
- Gunasekera, S.P.; McCarthy, P.J.; Kelly-Borges, M. Hamacanthins A and B, new Antifungal bis indole alkaloids from the deep-water marine sponge, Hamacantha sp. J. Nat. Prod. 1994, 57, 1437–1441. [Google Scholar] [CrossRef]
- Kouko, T.; Matsumura, K.; Kawasaki, T. Total synthesis of marine bisindole alkaloids, (+)-hamacanthins A, B and (−)-antipode of cis-dihydrohamacanthin B. Tetrahedron 2005, 61, 2309–2318. [Google Scholar] [CrossRef]
- Jiang, B.; Yang, C.-G.; Wang, J. Enantioselective synthesis for the (−)-antipode of the pyrazinone marine alkaloid, hamacanthin A. J. Org. Chem. 2001, 66, 4865–4869. [Google Scholar] [CrossRef]
- Bao, B.; Sun, Q.; Yao, X.; Hong, J.; Lee, C.-O.; Sim, C.J.; Im, K.S.; Jung, J.H. Cytotoxic bisindole alkaloids from a marine sponge Spongosorites sp. J. Nat. Prod. 2005, 68, 711–715. [Google Scholar] [CrossRef]
- Yang, C.-G.; Wang, J.; Tang, X.-X.; Jiang, B. Asymmetric aminohydroxylation of vinyl indoles: A short enantioselective synthesis of the bisindole alkaloids dihydrohamacanthin A and dragmacidin A. Tetrahedron Asymmetry 2002, 13, 383–394. [Google Scholar] [CrossRef]
- Jiang, B.; Yang, C.-G.; Wang, J. Enantioselective synthesis of marine indole alkaloid hamacanthin B. J. Org. Chem. 2002, 67, 1396–1398. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Wang, X.; Wang, X.; Rodriguez, R.A.; Moore, C.E.; Gao, S.; Tan, X.; Ma, Y.; Rheingold, A.L.; Baran, P.S.; et al. Asymmetric syntheses of sceptrin and massadine and evidence for biosynthetic enantiodivergence. Science 2014, 346, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Sherman, D.H.; Tsukamoto, S.; Williams, R.M. Comment on “asymmetric syntheses of sceptrin and massadine and evidence for biosynthetic enantiodivergence”. Science 2015, 349, 149b. [Google Scholar] [CrossRef]
- Ma, Z.; Wang, X.; Wang, X.; Rodriguez, R.A.; Moore, C.E.; Gao, S.; Tan, X.; Ma, Y.; Rheingold, A.L.; Baran, P.S.; et al. Response to Comment on “Asymmetric syntheses of sceptrin and massadine and evidence for biosynthetic enantiodivergence”. Science 2015, 349, 149c. [Google Scholar] [CrossRef]
- Johnson, I.S.; Armstrong, J.G.; Gorman, M.; Burnett, J.P. The vinca alkaloids: A new class of oncolytic agents. Cancer Res. 1963, 23, 1390–1427. [Google Scholar]
- Miyake, F.Y.; Yakushijin, K.; Horne, D.A. A concise synthesis of topsentin A and nortopsentins B and D. Org. Lett. 2000, 2, 2121–2123. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).