Fusarisolins A–E, Polyketides from the Marine-Derived Fungus Fusarium solani H918




Abstract
:1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Identification, Fermentation, and Extract
3.3. Isolation and Purification
3.4. Preparation of MPA Esters of 1
3.5. Alkaline Hydrolysis of 8
3.6. Antifungal Assay
3.7. ED50 Detection
3.8. Total RNA Isolation
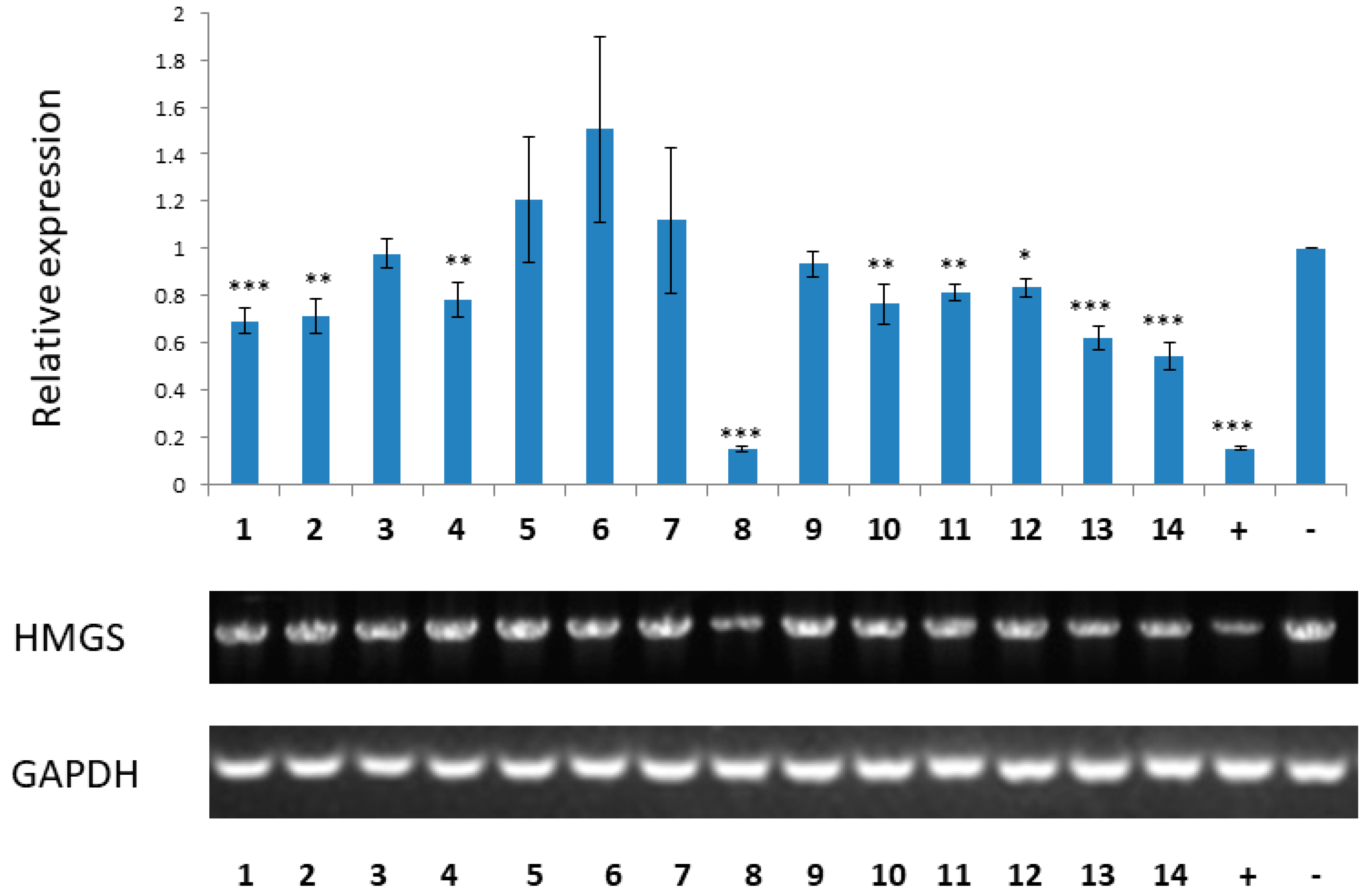
3.9. RT-PCR Analysis of HMG-CoA Synthase Gene Expression
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rateb, M.E.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar] [CrossRef]
- Aldridge, D.C.; Giles, D.; Turner, W.B. Antibiotic 1233A: A fungal β-lactone. J. Chem. Soc. 1971, 23, 3888–3891. [Google Scholar] [CrossRef]
- Greenspan, M.D.; Yudkovitz, J.B.; Lo, C.Y.L.; Chen, J.S.; Alberts, A.W.; Hunt, V.M.; Chang, M.N.; Yang, S.S.; Thompson, K.L.; Chiang, Y.C.P.; et al. Inhibition of hydroxymethylglutaryl-coenzyme A synthase by L-659,699. Proc. Natl. Acad. Sci. USA 1987, 84, 7488–7492. [Google Scholar] [CrossRef]
- Tomoda, H.; Kumagai, H.; Takahashi, Y.; Tanaka, Y.; Iwai, Y.; Omura, S. F-244 (1233A), a specific inhibitor of 3-hydroxy-3-methylglutaryl coenzyme A synthase: Taxonomy of producing strain, fermentation, isolation and biological properties. J. Antibiot. 1988, 41, 247–249. [Google Scholar] [CrossRef]
- Umezawa, H.; Aoyagi, T.; Uotani, K.; Hamada, M.; Takeuchi, T.; Takahashi, S. Ebelactone, an inhibitor of esterase, produced by actinomycetes. J. Antibiot. 1980, 33, 1594–1596. [Google Scholar] [CrossRef]
- Wells, J.S.; Trejo, W.H.; Principe, P.A.; Sykes, R.B. Obafluorin, a novel β-lactone produced by Pseudomonas fluorescens. Taxonomy, fermentation and biological properties. J. Antibiot. 1984, 37, 802–803. [Google Scholar] [CrossRef]
- Asai, A.; Hasegawa, A.; Ochiai, K.; Yamashita, Y.; Mizukami, T. Belactosin A, a novel antitumor antibiotic acting on cyclin/CDK mediated cell cycle regulation, produced by Streptomyces sp. J. Antibiot. 2000, 53, 81–83. [Google Scholar] [CrossRef]
- Weibel, E.K.; Hadvary, P.; Hochuli, E.; Kupfer, E.; Lengsfeld, H. Lipstatin, an inhibitor of pancreatic lipase, produced by Streptomyces toxytricini. I. Producing organism, fermentation, isolation and biological activity. J. Antibiot. 1987, 40, 1081–1085. [Google Scholar] [CrossRef]
- Feling, R.H.; Buchanan, G.O.; Mincer, T.J.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Salinosporamide A: A highly cytotoxic proteasome inhibitor from a novel microbial source, a marine bacterium of the new genus Salinospora. Angew. Chem. Int. Ed. 2003, 42, 355–357. [Google Scholar] [CrossRef]
- Liu, D.Z.; Wang, F.; Liao, T.G.; Tang, J.G.; Steglich, W.; Zhu, H.J.; Liu, J.K. Vibralactone: A lipase inhibitor with an unusual fused β-lactone produced by cultures of the basidiomycete Boreostereum vibrans. Org. Lett. 2006, 8, 5749–5752. [Google Scholar] [CrossRef]
- Morris, B.D.; Smyth, R.R.; Foster, S.P.; Hoffmann, M.P.; Roelofs, W.L.; Franke, S.; Francke, W. Vittatalactone, a β-lactone from the striped cucumber beetle, Acalymma vittatum. J. Nat. Prod. 2005, 68, 26–30. [Google Scholar] [CrossRef]
- Gill, K.A.; Berrue, F.; Arens, J.C.; Carr, G.; Kerr, R.G. Cystargolides, 20S Proteasome Inhibitors Isolated from Kitasatospora cystarginea. J. Nat. Prod. 2015, 78, 822–826. [Google Scholar] [CrossRef]
- Böttcher, T.; Sieber, S.A. β-Lactams and β-lactones as activity-based probes in chemical biology. Med. Chem. Commun. 2012, 3, 408–417. [Google Scholar] [CrossRef]
- De Pascale, G.; Nazi, I.; Harrison, P.H.M.; Wright, G.D. β-lactone natural products and derivatives inactivate homoserine transacetylase, a target for antimicrobial agents. J. Antibiot. 2011, 64, 483–487. [Google Scholar] [CrossRef]
- Pojer, F.; Ferrer, J.L.; Richard, S.B.; Nagegowda, D.A.; Chye, M.L.; Bach, T.J.; Noel, J.P. Structural basis for the design of potent and species-specific inhibitors of 3-hydroxy-3-methylglutaryl CoA synthases. Proc. Natl. Acad. Sci. USA 2006, 103, 11491–11496. [Google Scholar] [CrossRef]
- Guerciolini, R. Mode of action of orlistat. Int. J. Obes. Relat. Metab. Disord. 1997, 21 (Suppl. 3), S12–S23. [Google Scholar]
- Yang, X.W.; Peng, K.; Liu, Z.; Zhang, G.Y.; Li, J.; Wang, N.; Steinmetz, A.; Liu, Y. Strepsesquitriol, a rearranged zizaane-type sesquiterpenoid from the deep-sea-derived actinomycete Streptomyces sp. SCSIO 10355. J. Nat. Prod. 2013, 76, 2360–2363. [Google Scholar] [CrossRef]
- Xie, C.L.; Liu, Q.; Xia, J.M.; Gao, Y.; Yang, Q.; Shao, Z.Z.; Liu, G.; Yang, X.W. Anti-allergic compounds from the deep-sea-derived actinomycete Nesterenkonia flava MCCC 1K00610. Mar. Drugs 2017, 15, 71. [Google Scholar] [CrossRef]
- Niu, S.; Zhou, T.T.; Xie, C.L.; Zhang, G.Y.; Yang, X.W. Microindolinone A, a novel 4,5,6,7-tetrahydroindole, from the deep-sea-derived actinomycete Microbacterium sp. MCCC 1A11207. Mar. Drugs 2017, 15, 230. [Google Scholar] [CrossRef]
- Niu, S.; Liu, Q.; Xia, J.; Xie, C.; Luo, Z.; Shao, Z.; Liu, G.; Yang, X. Polyketides from the deep-sea-derived fungus Graphostroma sp. MCCC 3A00421 showed potent antifood allergic activities. J. Agric. Food Chem. 2018, 66, 1369–1376. [Google Scholar] [CrossRef]
- Chiang, Y.C.P.; Yang, S.S.; Heck, J.V.; Chabala, J.C.; Chang, M.N. Total synthesis of L-659,699, a novel inhibitor of cholesterol biosynthesis. J. Org. Chem. 1989, 54, 5708–5712. [Google Scholar] [CrossRef]
- Yasuhara, F.; Yamaguchi, S. Use of shift reagent with MTPA derivatives in 1H NMR spectroscopy. III. Determination of absolute configuration and enantiomeric purity of primary carbinols with chiral center at the C-2 position. Tetrahedron Lett. 1977, 18, 4085–4088. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Tsuyuki, T.; Moriyama, Y.; Takahashi, T. Application of the MTPA method to determination of abusolute stereochemistry. The hydroxymethyl-substituted chiral carbon of carbocycles. Bull. Chem. Soc. Jpn. 1980, 53, 3723–3724. [Google Scholar] [CrossRef]
- Wovkulich, P.M.; Shankaran, K.; Kiegiel, J.; Uskokovic, M.R. Total synthesis of 1233A. J. Org. Chem. 1993, 58, 832–839. [Google Scholar] [CrossRef]
- Franot, C.; Benezra, C.; Lepoittevin, J.P. Synthesis and interaction studies of 13C labeled lactone derivatives with a model protein using 13C NMR. Bioorg. Med. Chem. 1993, 1, 389–397. [Google Scholar] [CrossRef]
- White, J.D.; Johnson, A.T. Synthesis of the aliphatic depside (+)-bourgeanic acid. J. Org. Chem. 1994, 59, 3347–3358. [Google Scholar] [CrossRef]
- Yoshida, W.Y.; Bryan, P.J.; Baker, B.J.; McClintock, J.B. Pteroenone: A defensive metabolite of the abducted Antarctic Pteropod Clione antarctica. J. Org. Chem. 1995, 60, 780–782. [Google Scholar] [CrossRef]
- Nanda, S.; Scott, A.I. Asymmetric synthesis of (E)- and (Z)-3,7-dimethyl-2-octene-1,8-diol and callosobruchusic acid. Tetrahedron Asymmetry 2004, 15, 963–970. [Google Scholar] [CrossRef]
- Kornsakulkarn, J.; Dolsophon, K.; Boonyuen, N.; Boonruangprapa, T.; Rachtawee, P.; Prabpai, S.; Kongsaeree, P.; Thongpanchang, C. Dihydronaphthalenones from endophytic fungus Fusarium sp. BCC14842. Tetrahedron 2011, 67, 7540–7547. [Google Scholar] [CrossRef]
- Kimura, Y.; Shimada, A.; Nakajima, H.; Hamasaki, T. Structures of naphthoquinones produced by the fungus, Fusarium sp., and their biological activity toward pollen germination. Agric. Biol. Chem. 1988, 52, 1253–1259. [Google Scholar] [CrossRef]
- Chilton, W.S. Isolation and structure of norjavanicin. J. Org. Chem. 1968, 33, 4299–4301. [Google Scholar] [CrossRef]
- Tatum, J.H.; Baker, R.A. Naphthoquinones produced by Fusarium solani isolated from citrus. Phytochemistry 1983, 22, 543–547. [Google Scholar] [CrossRef]
- Kurobane, I.; Zaita, N.; Fukuda, A. New metabolites of Fusarium martii related to dihydrofusarubin. J. Antibiot. 1986, 39, 205–214. [Google Scholar] [CrossRef]
- Arsenault, G.P. Fungal metabolites. III. Quinones from Fusarium solani D2 purple and structure of (+)-solaniol. Tetrahedron 1968, 24, 4745–4749. [Google Scholar] [CrossRef]
- Hashimoto, J.; Motohashi, K.; Sakamoto, K.; Hashimoto, S.; Yamanouchi, M.; Tanaka, H.; Takahashi, T.; Takagi, M.; Shin-ya, K. Screening and evaluation of new inhibitors of hepatic glucose production. J. Antibiot. 2009, 62, 625–629. [Google Scholar] [CrossRef] [Green Version]
- Alex, D.; Bach, T.J.; Chye, M.L. Expression of Brassica juncea 3-hydroxy-3-methylglutaryl CoA synthase is developmentally regulated and stress-responsive. Plant J. 2001, 22, 415–426. [Google Scholar] [CrossRef]
- Woo, J.H.; Kitamura, E.; Myouga, H.; Kamei, Y. An antifungal protein from the marine bacterium Streptomyces sp. Strain AP77 is specific for pythium porphyrae, a causative agent of red rot disease in Porphyra spp. Appl. Environ. Microb. 2002, 68, 2666–2675. [Google Scholar] [CrossRef]
- Kundu, A.; Saha, S.; Walia, S.; Shakil, N.A.; Kumar, J.; Annapurna, K. Cadinene sesquiterpenes from Eupatorium adenophorum and their antifungal activity. J. Environ. Sci. Health B 2013, 48, 516–522. [Google Scholar] [CrossRef]

 ), HMBC (
), HMBC (  ), and NOESY (
), and NOESY (  ) correlations of 1.
) correlations of 1.




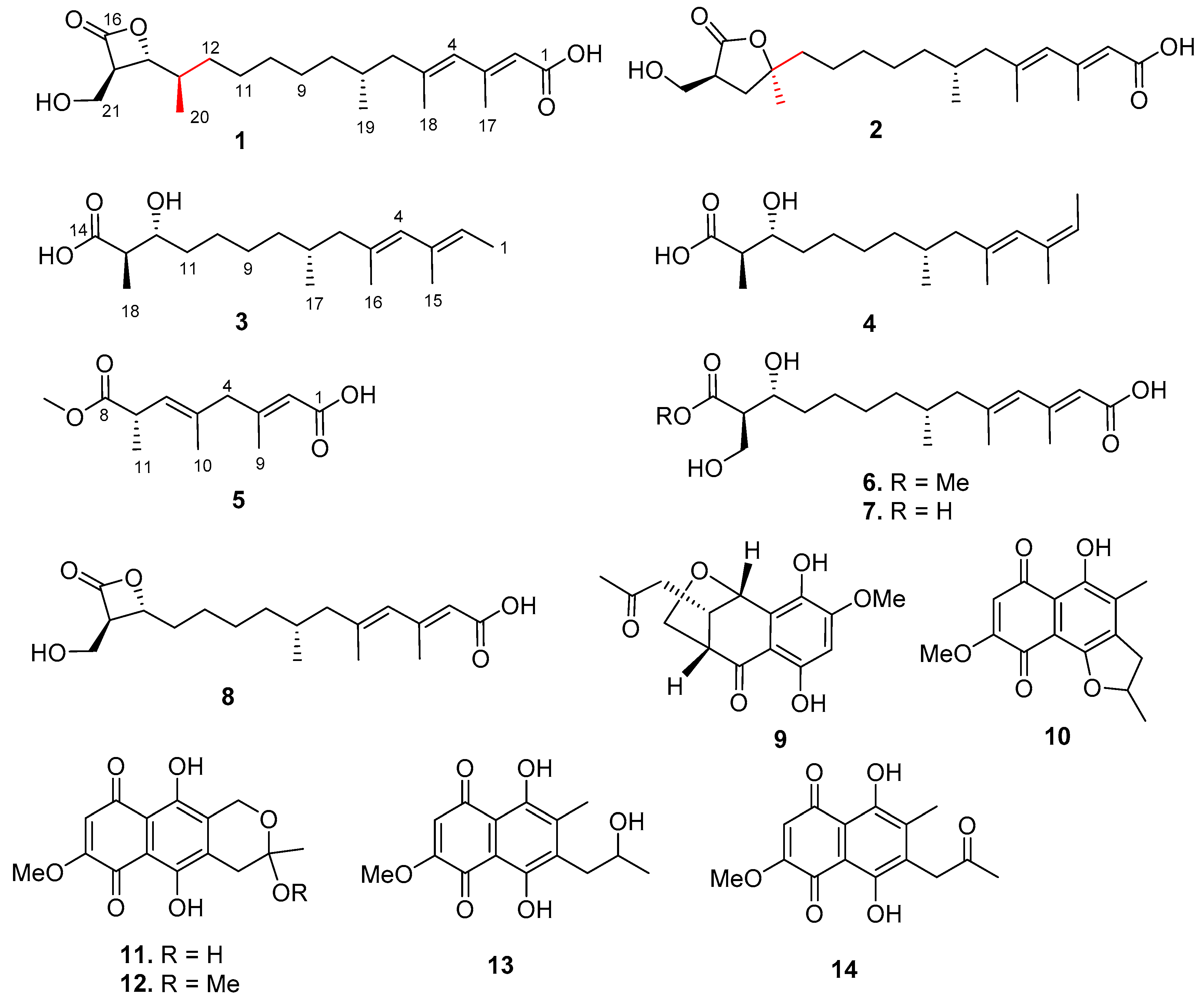{kind=link}
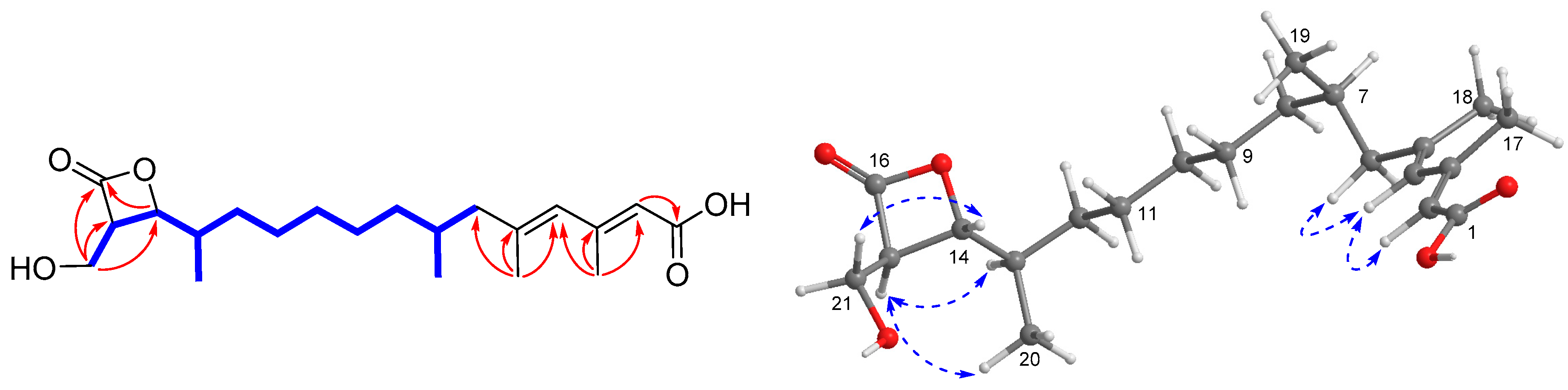{kind=link}
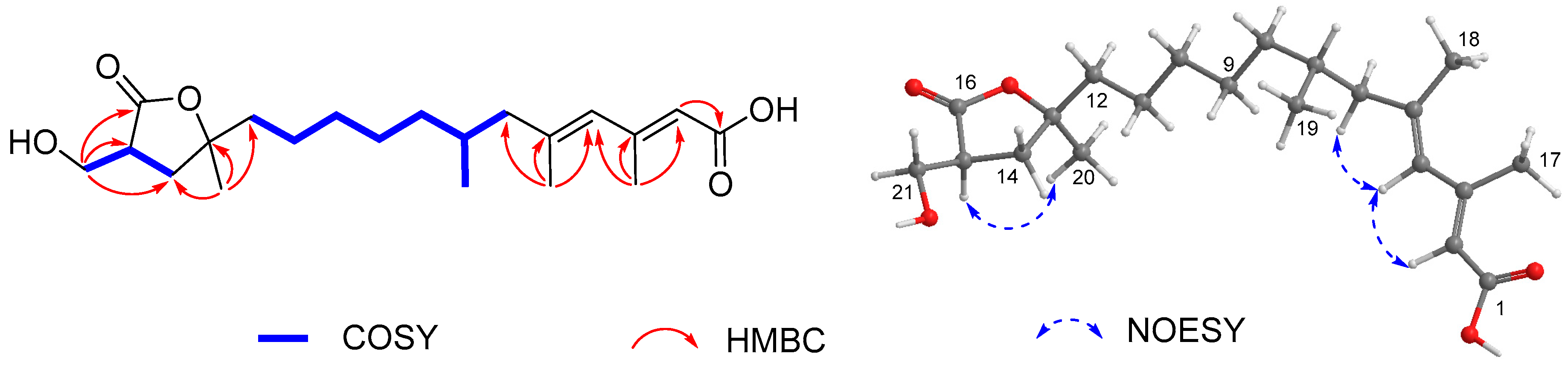{kind=link}
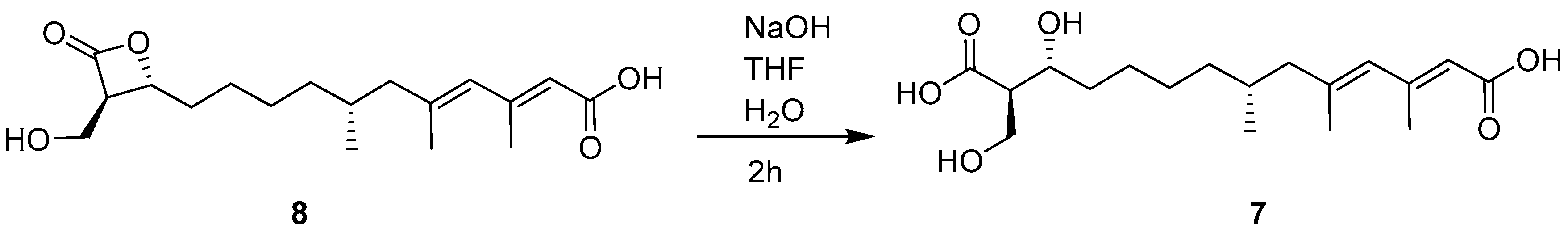{kind=link}
{kind=link}
| No. | 1 a | 2 a | 3 a | 4 a | 5 b | 6 a |
|---|---|---|---|---|---|---|
| 1 | 1.67, d (7.0) | 1.50, dm (6.8) | ||||
| 2 | 5.65, br s | 5.65, br s | 5.29, q (7.0) | 5.30, qq (6.8, 1.2) | 5.74, br s | 5.65, br s |
| 4 | 5.76, br s | 5.76, br s | 5.59, br s | 5.53, br s | 2.85, s | 5.76, br s |
| 6 | 2.13, dd (13.2, 6.2) | 2.14, dd (13.2, 7.2) | 2.03, dd (12.8, 6.4) | 2.09, dd (13.2, 6.3) | 5.32, d (8.7) | 2.14, dd (13.1, 6.4) |
| 1.88, dd (13.2, 8.1) | 1.89, dd (13.2, 8.1) | 1.79, m | 1.86, ddd (13.2, 8.3, 0.8) | 1.89, dd (13.1, 8.2) | ||
| 7 | 1.70, m | 1.71, m | 1.65, m | 1.66, m | 3.39, m | 1.71, m |
| 8 | 1.35, m; 1.15, m | 1.34, m; 1.15, m | 1.36, m; 1.12, m | 1.38, m; 1.15, m | 1.36, m; 1.16, m | |
| 9 | 1.44, m | 1.34, m | 1.36, m | 1.38, m | 2.12, s | 1.37, m |
| 10 | 1.33, m | 1.34, m | 1.49, m; 1.37, m | 1.50, m; 1.38, m | 1.63, s | 1.48, m; 1.37, m |
| 11 | 1.33, m | 1.42, m | 1.52, m; 1.40, m | 1.53, m; 1.40, m | 1.26, d (6.8) | 1.53, m; 1.45, m |
| 12 | 1.44, m; 1.18, m | 1.72, m | 3.71, t (6.9) | 3.70, t (6.9) | 3.79, m | |
| 13 | 1.82, m | 2.48, m | 2.48, m | 2.67, ddd (8.5, 6.3, 5.4) | ||
| 14 | 4.31, dd (8.4, 4.2) | 2.16, br d (10.3) | ||||
| 15 | 3.53, q (4.2) | 3.02, m | 1.69, s | 1.70, m | 2.21, d (1.2) | |
| 16 | 1.71, s | 1.54, d (1.3) | 1.82, d (1.2) | |||
| 17 | 2.20, s | 2.21, s | 0.84, d (6.6) | 0.88, d (6.6) | 0.87, d (6.6) | |
| 18 | 1.82, s | 1.82, s | 1.13, d (6.9) | 1.13, d (7.0) | 3.82, dd (10.9, 8.7) | |
| 3.73, dd (10.9, 5.3) | ||||||
| 19 | 0.87, d (6.6) | 0.87, d (6.5) | ||||
| 20 | 1.04, d (6.5) | 1.38, s | ||||
| 21 | 3.91, dd (11.9, 4.5) | 3.87, dd (11.0, 4.6) | ||||
| 3.77, dd (11.9, 3.8) | 3.72, dd (11.0, 3.6) | |||||
| OMe | 3.70, s | 3.71, s |
| No. | 1 a | 2 a | 3 a | 4 a | 5 b | 6 a |
|---|---|---|---|---|---|---|
| 1 | 171.0, C | 172.1, C | 13.7, CH3 | 15.3, CH3 | 171.7, C | 170.6, C |
| 2 | 119.1, CH | 119.1, CH | 123.7, CH | 121.8, CH | 116.6, CH | 118.8, CH |
| 3 | 155.2, C | 155.0, C | 134.9, C | 135.3, C | 160.1, C | 155.5, C |
| 4 | 130.6, CH | 130.6, CH | 131.6, CH | 126.8, CH | 51.1, CH2 | 130.6, CH |
| 5 | 142.2, C | 142.1, C | 135.1, C | 137.2, C | 133.5, C | 142.3, C |
| 6 | 49.9, CH2 | 49.9, CH2 | 49.8, CH2 | 48.8, CH2 | 128.0, CH | 50.0, CH2 |
| 7 | 32.1, CH | 32.2, CH | 32.1, CH | 32.1, CH | 38.9, CH | 32.1, CH |
| 8 | 37.8, CH2 | 37.9, CH2 | 38.0, CH2 | 38.0, CH2 | 175.7, C | 37.9, CH2 |
| 9 | 27.9, CH2 | 28.9, CH2 | 28.2, CH2 | 28.1, CH2 | 17.9, CH3 | 32.1, CH2 |
| 10 | 27.8, CH2 | 31.2, CH2 | 27.0, CH2 | 27.0, CH2 | 15.9, CH3 | 26.9, CH2 |
| 11 | 31.0, CH2 | 24.8, CH2 | 35.0, CH2 | 35.0, CH2 | 18.4, CH3 | 36.1, CH2 |
| 12 | 32.4, CH2 | 42.6, CH2 | 74.1, CH | 74.1, CH | 71.0, CH | |
| 13 | 38.1, CH | 86.5, C | 47.5, CH | 47.5, CH | 56.2, CH | |
| 14 | 80.2, CH | 36.5, CH2 | 179.7, C | 179.5, C | 175.1, C | |
| 15 | 58.2, CH | 44.6, CH | 17.0, CH3 | 24.2, CH3 | 19.9, CH3 | |
| 16 | 172.1, C | 179.5, C | 17.9, CH3 | 17.8, CH3 | 18.5, CH3 | |
| 17 | 19.8, CH3 | 19.8, CH3 | 20.0, CH3 | 19.9, CH3 | 19.8, CH3 | |
| 18 | 18.5, CH3 | 18.5, CH3 | 13.9, CH3 | 13.8, CH3 | 61.6, CH2 | |
| 19 | 19.9, CH3 | 19.9, CH3 | ||||
| 20 | 15.3, CH3 | 25.5, CH3 | ||||
| 21 | 58.3, CH2 | 61.0, CH2 | ||||
| OMe | 51.9, CH3 | 52.0, CH3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, S.; Tang, X.-X.; Fan, Z.; Xia, J.-M.; Xie, C.-L.; Yang, X.-W. Fusarisolins A–E, Polyketides from the Marine-Derived Fungus Fusarium solani H918. Mar. Drugs 2019, 17, 125. https://doi.org/10.3390/md17020125
Niu S, Tang X-X, Fan Z, Xia J-M, Xie C-L, Yang X-W. Fusarisolins A–E, Polyketides from the Marine-Derived Fungus Fusarium solani H918. Marine Drugs. 2019; 17(2):125. https://doi.org/10.3390/md17020125
Chicago/Turabian StyleNiu, Siwen, Xi-Xiang Tang, Zuowang Fan, Jin-Mei Xia, Chun-Lan Xie, and Xian-Wen Yang. 2019. "Fusarisolins A–E, Polyketides from the Marine-Derived Fungus Fusarium solani H918" Marine Drugs 17, no. 2: 125. https://doi.org/10.3390/md17020125
APA StyleNiu, S., Tang, X. -X., Fan, Z., Xia, J. -M., Xie, C. -L., & Yang, X. -W. (2019). Fusarisolins A–E, Polyketides from the Marine-Derived Fungus Fusarium solani H918. Marine Drugs, 17(2), 125. https://doi.org/10.3390/md17020125