Genome Sequencing of the Japanese Eel (Anguilla japonica) for Comparative Genomic Studies on tbx4 and a tbx4 Gene Cluster in Teleost Fishes

 , ,
, ,
Abstract
:1. Introduction
2. Results
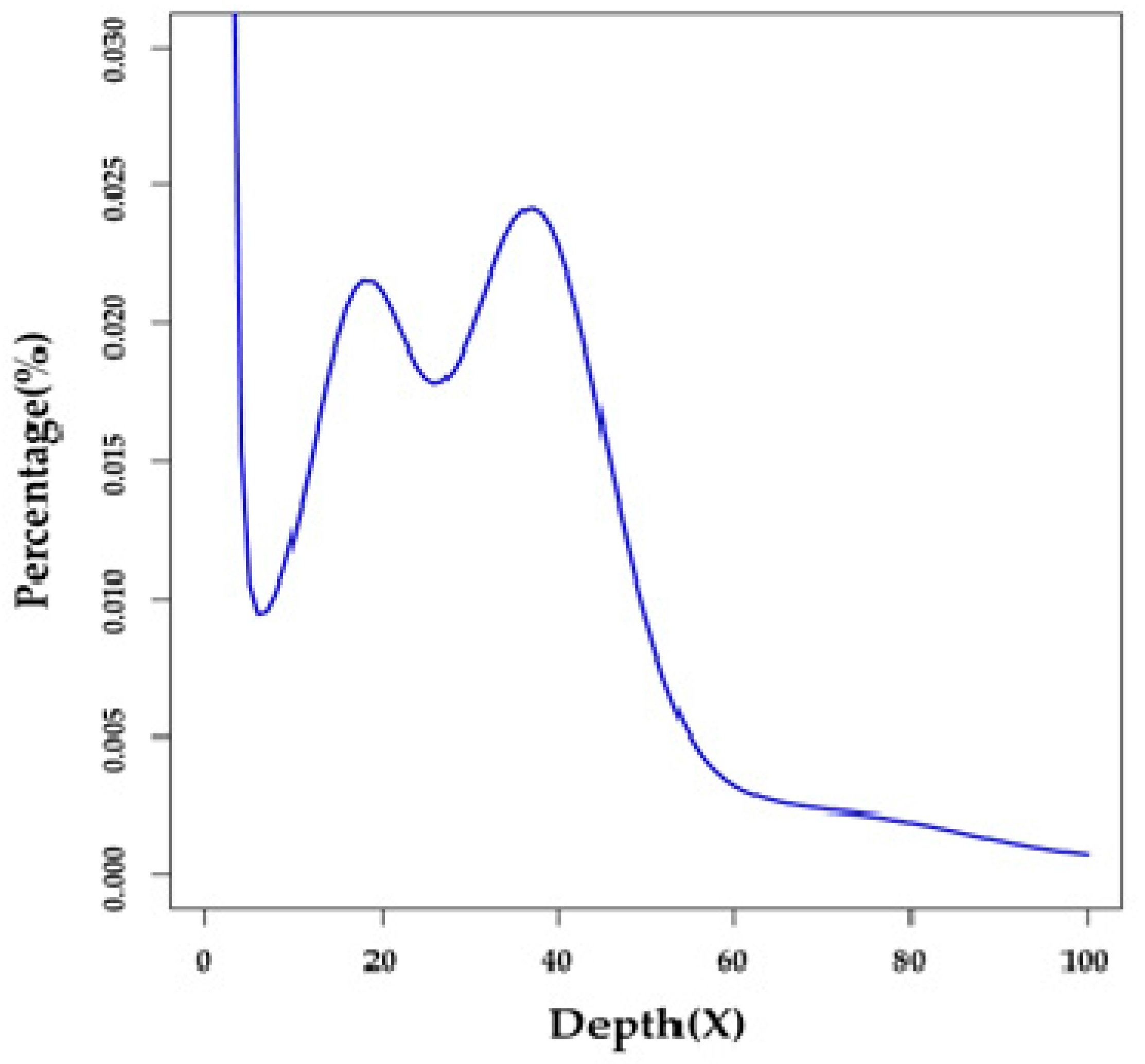
2.1. Summary of Genome Survey and De Novo Assembly
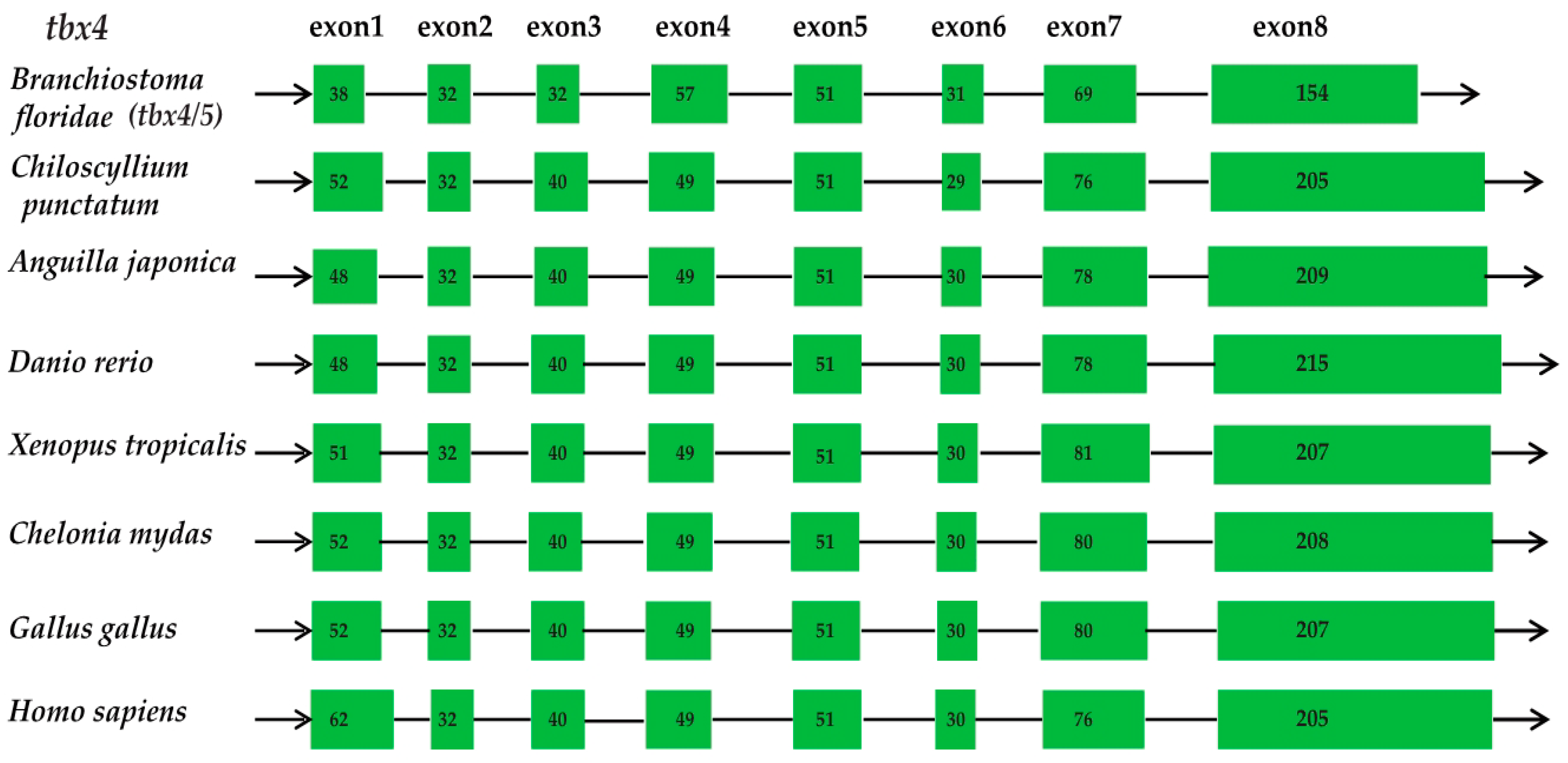
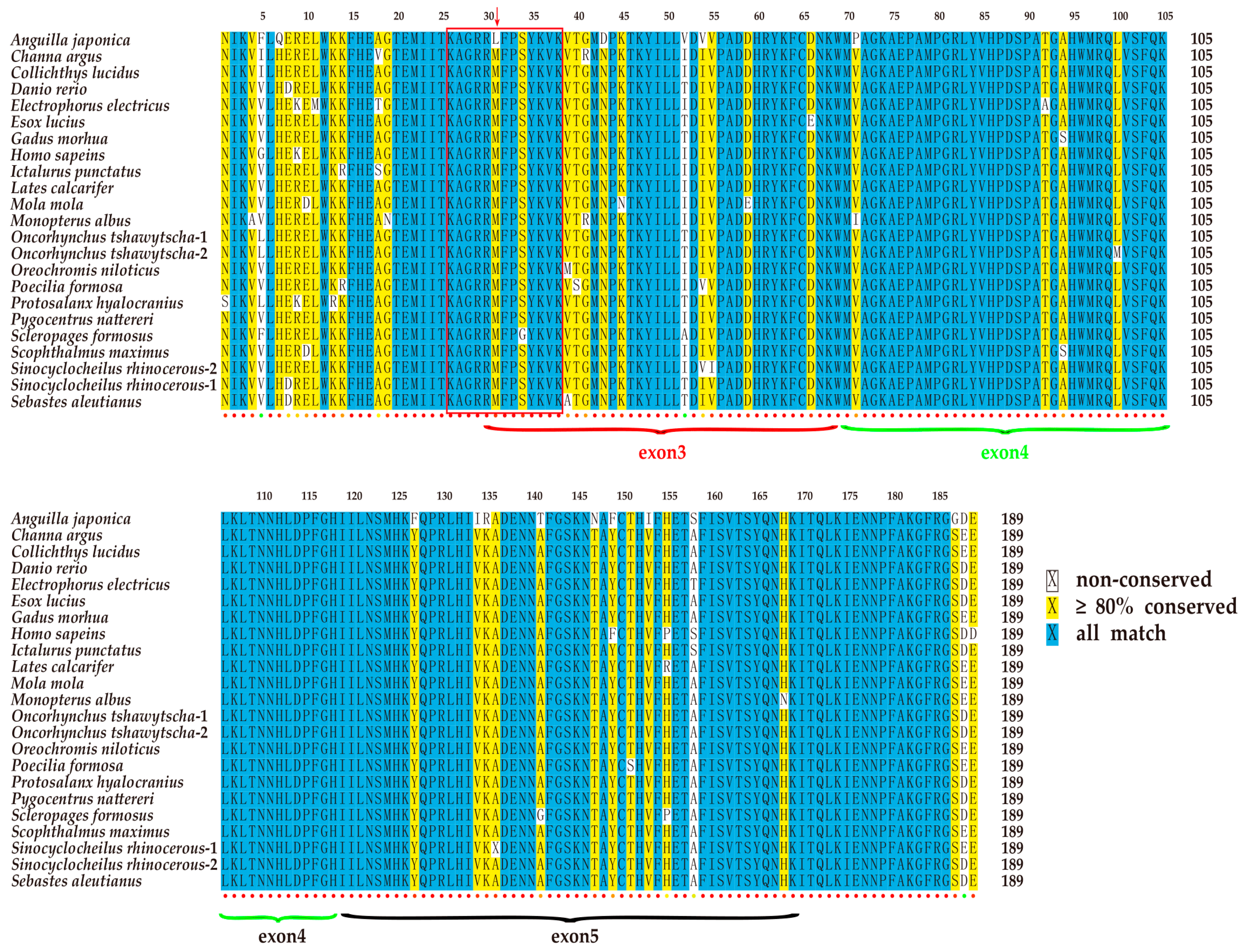
2.2. Conservation of the Vertebrate tbx4 Genes in Gene Structure
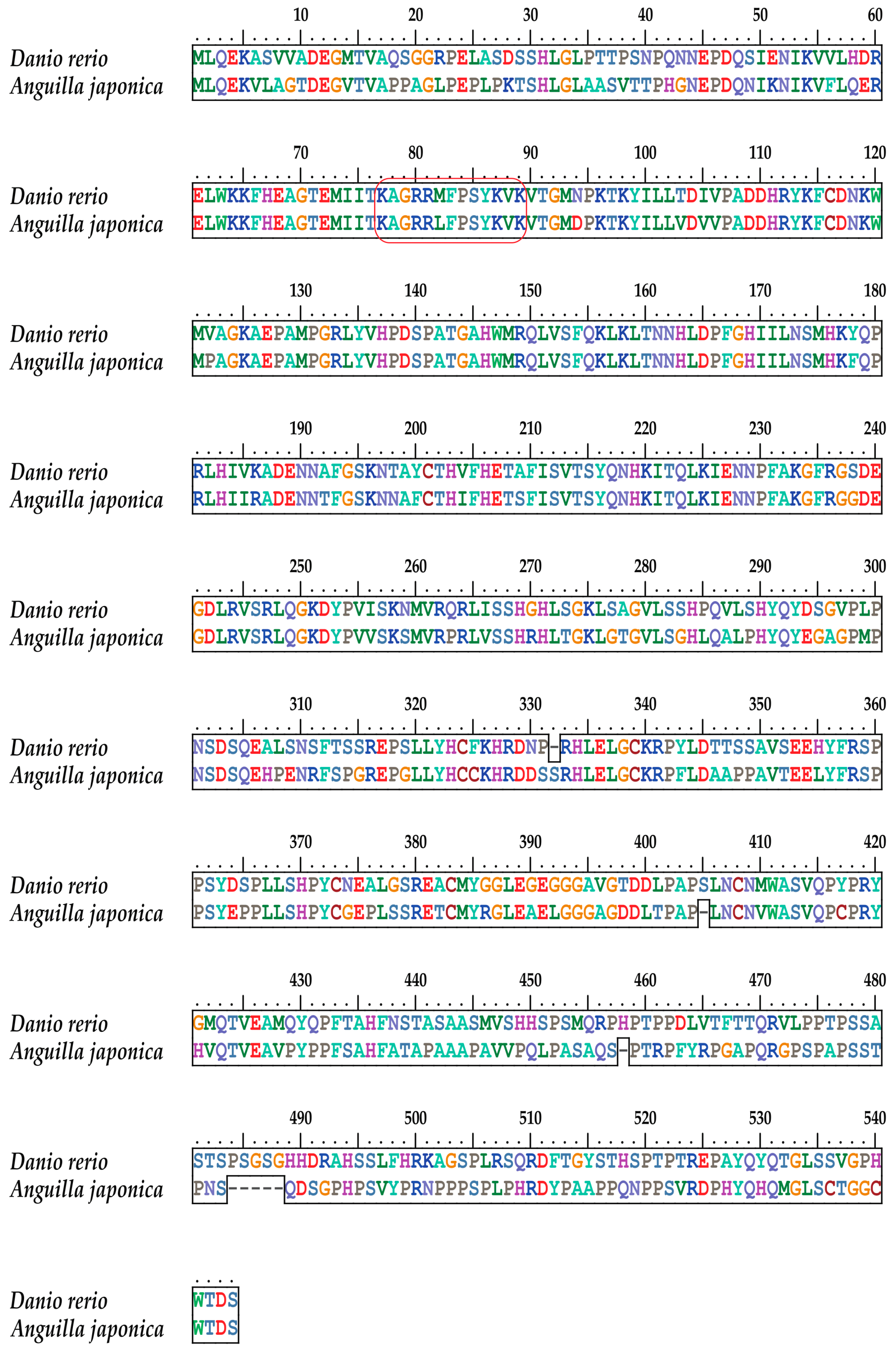
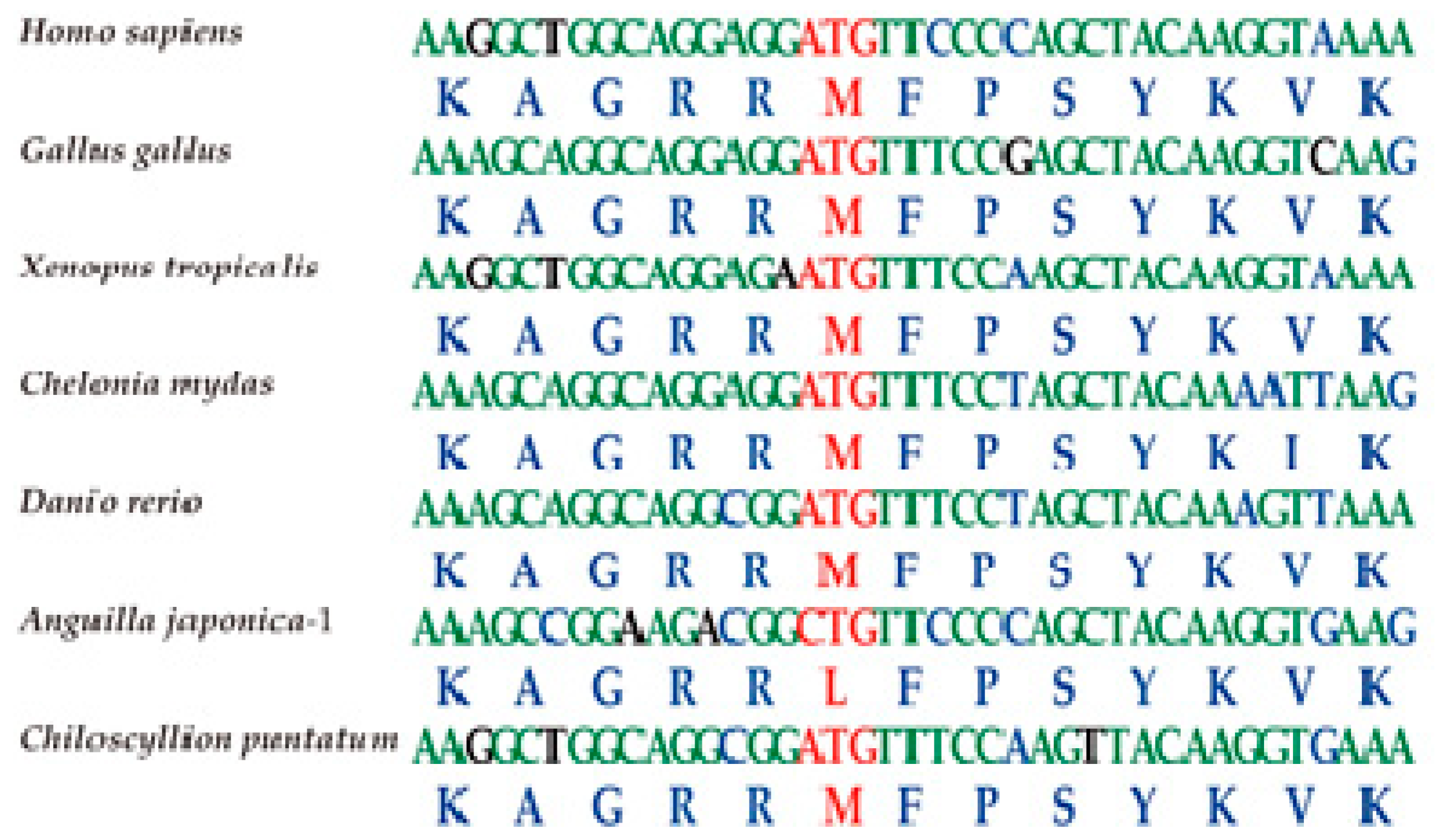
2.3. Conservation of the T-box Region
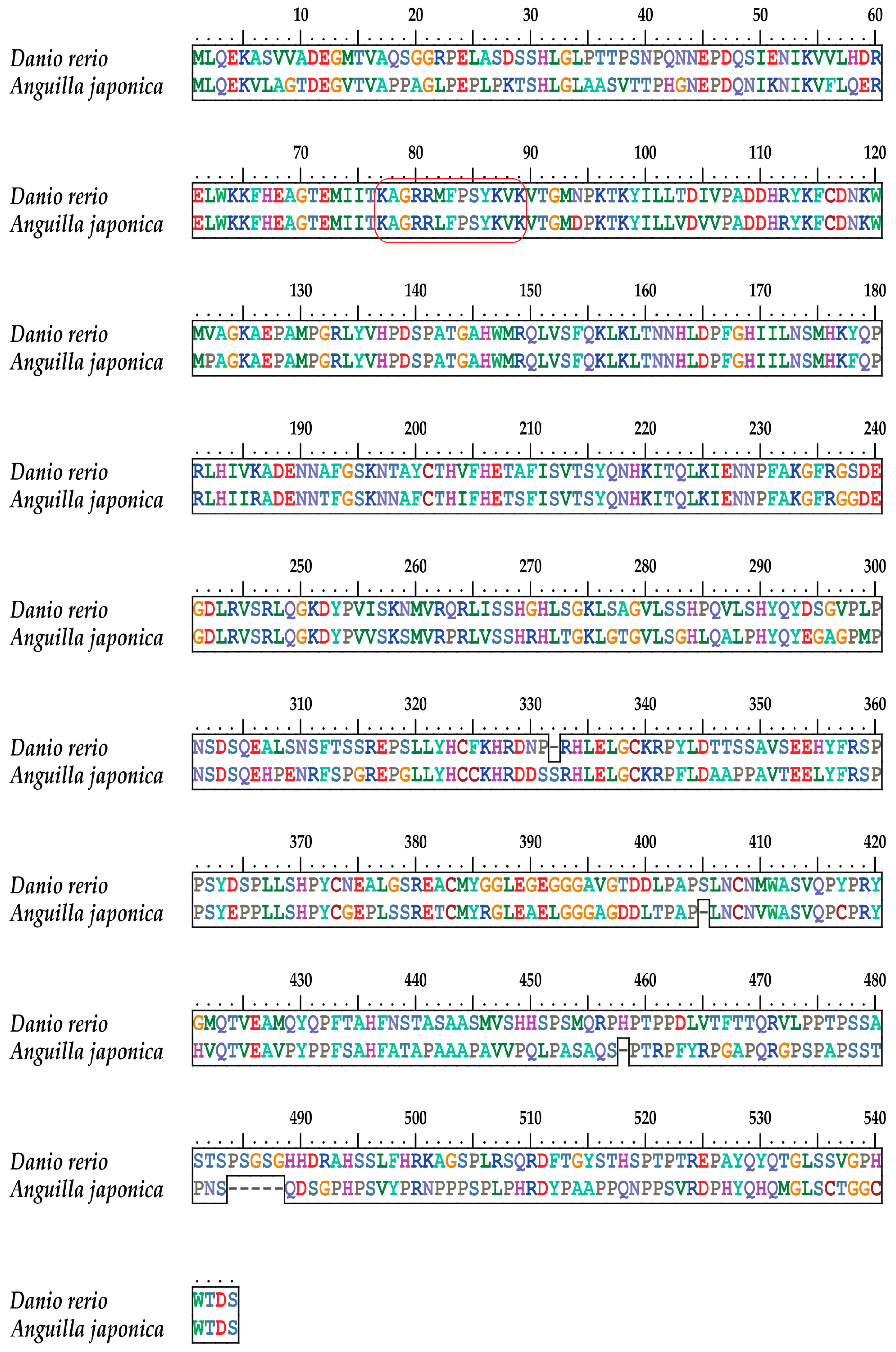
2.4. The Tbx4 Gene of the Japanese Eel
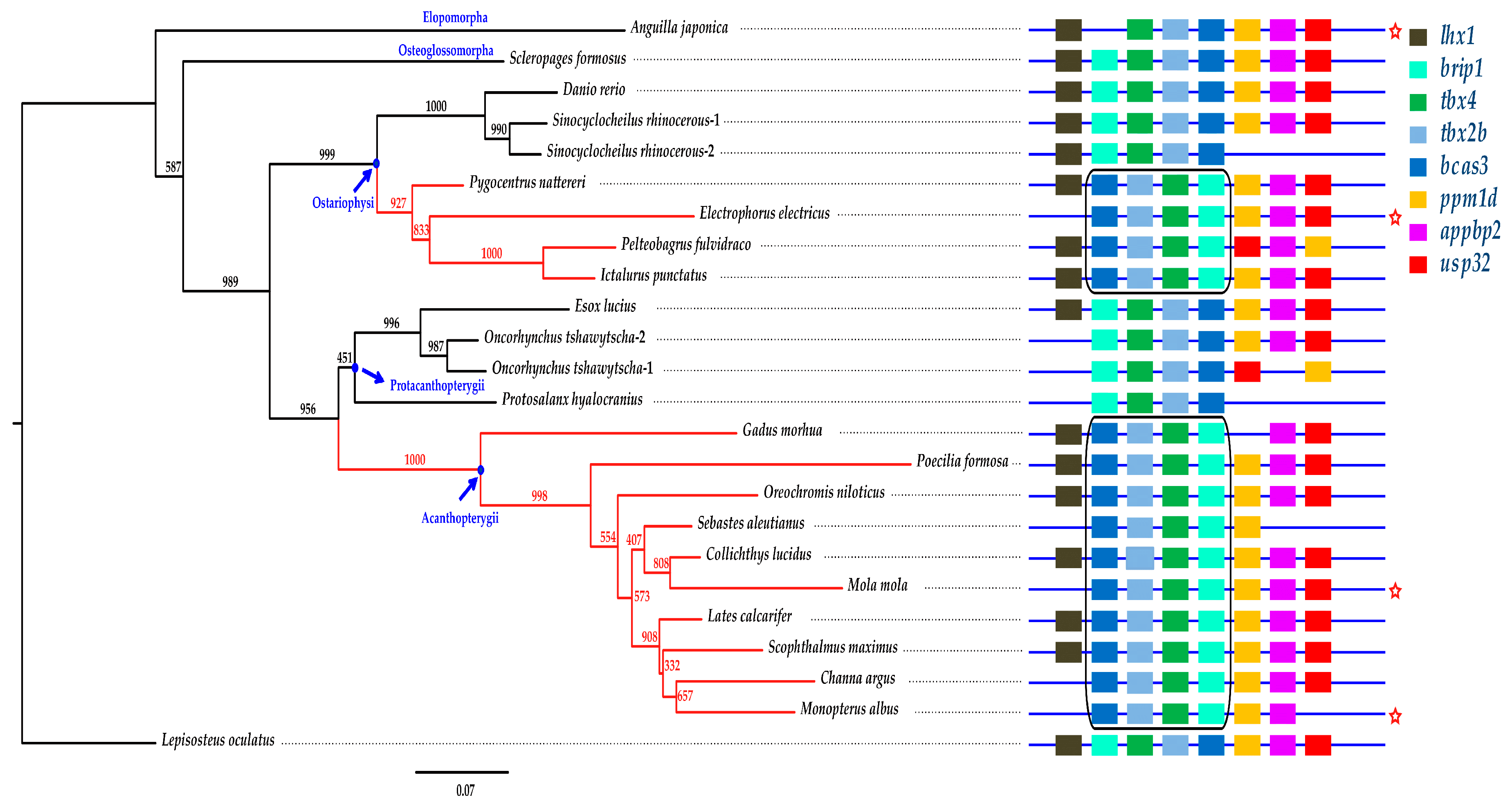
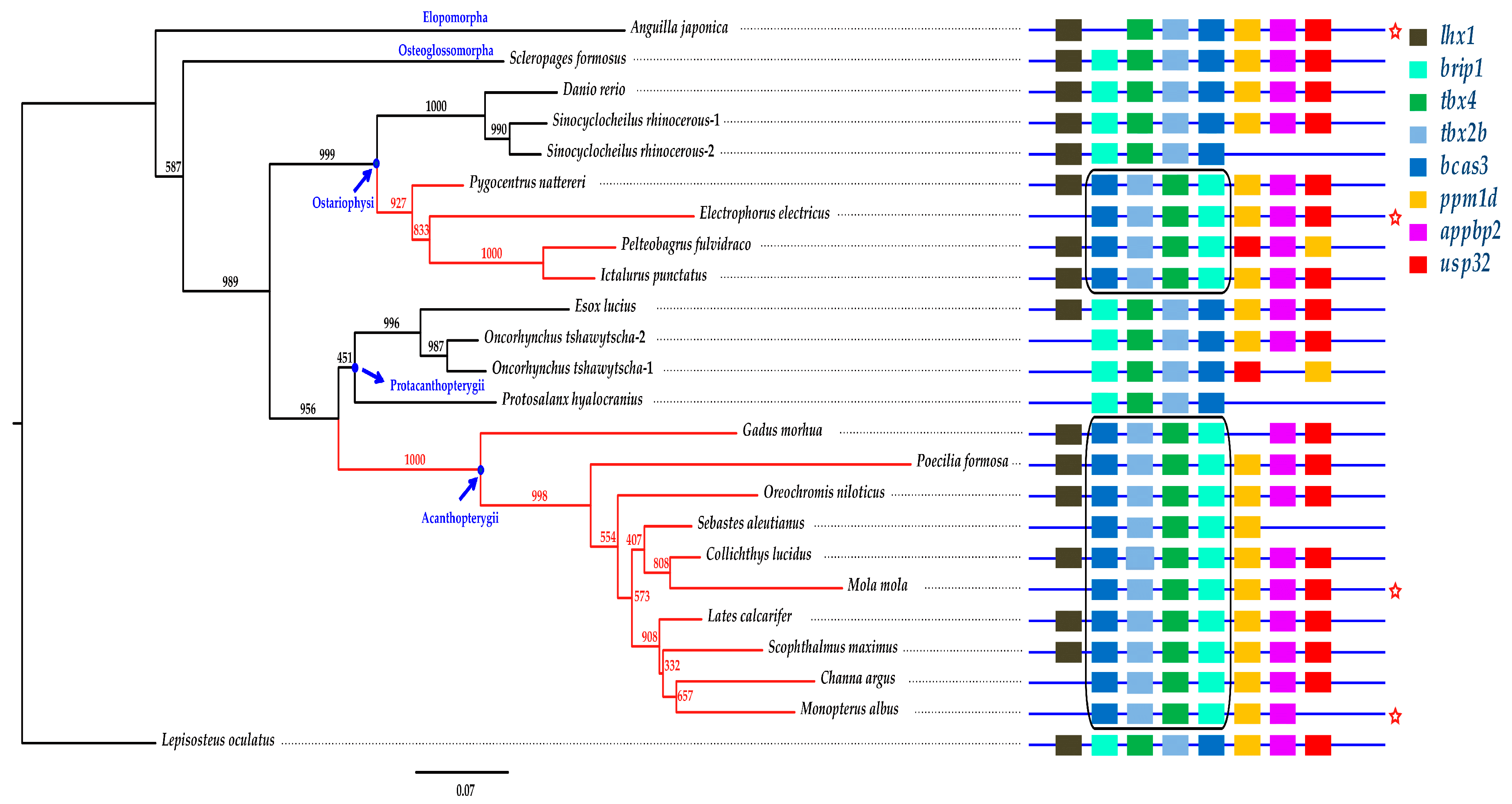
2.5. Phylogenetic Analysis and Synteny Comparison
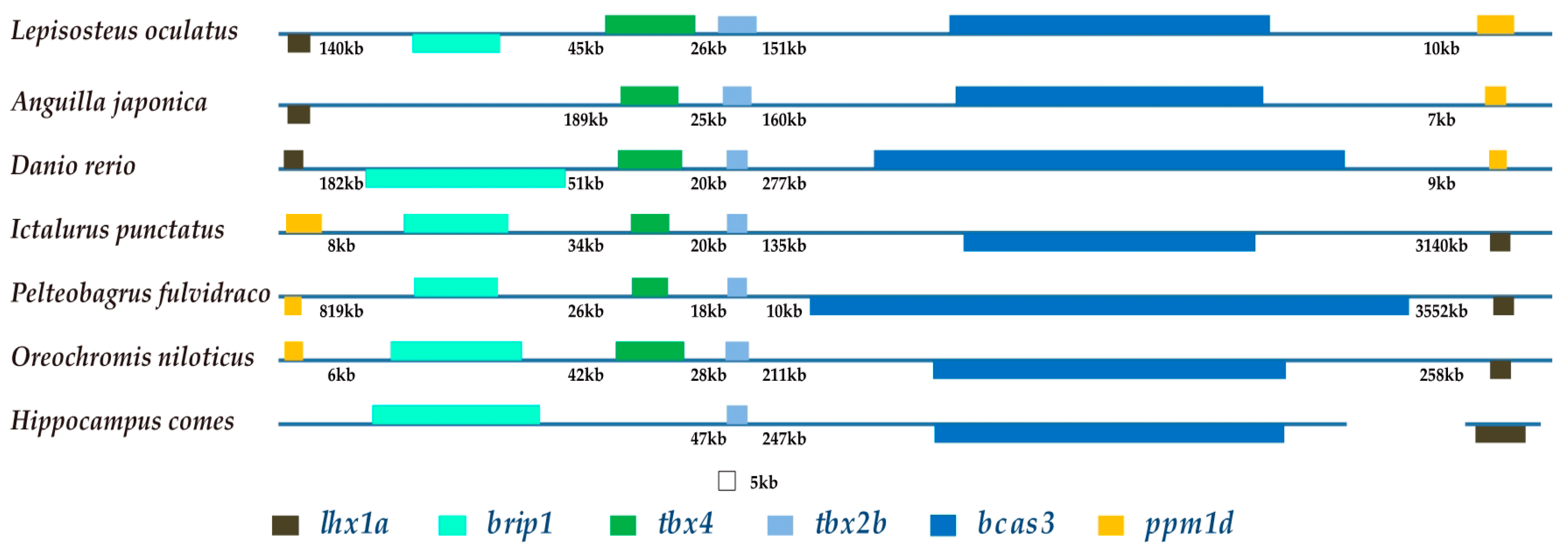
2.6. The Brip1-tbx4-tbx2b-bcas3 Cluster
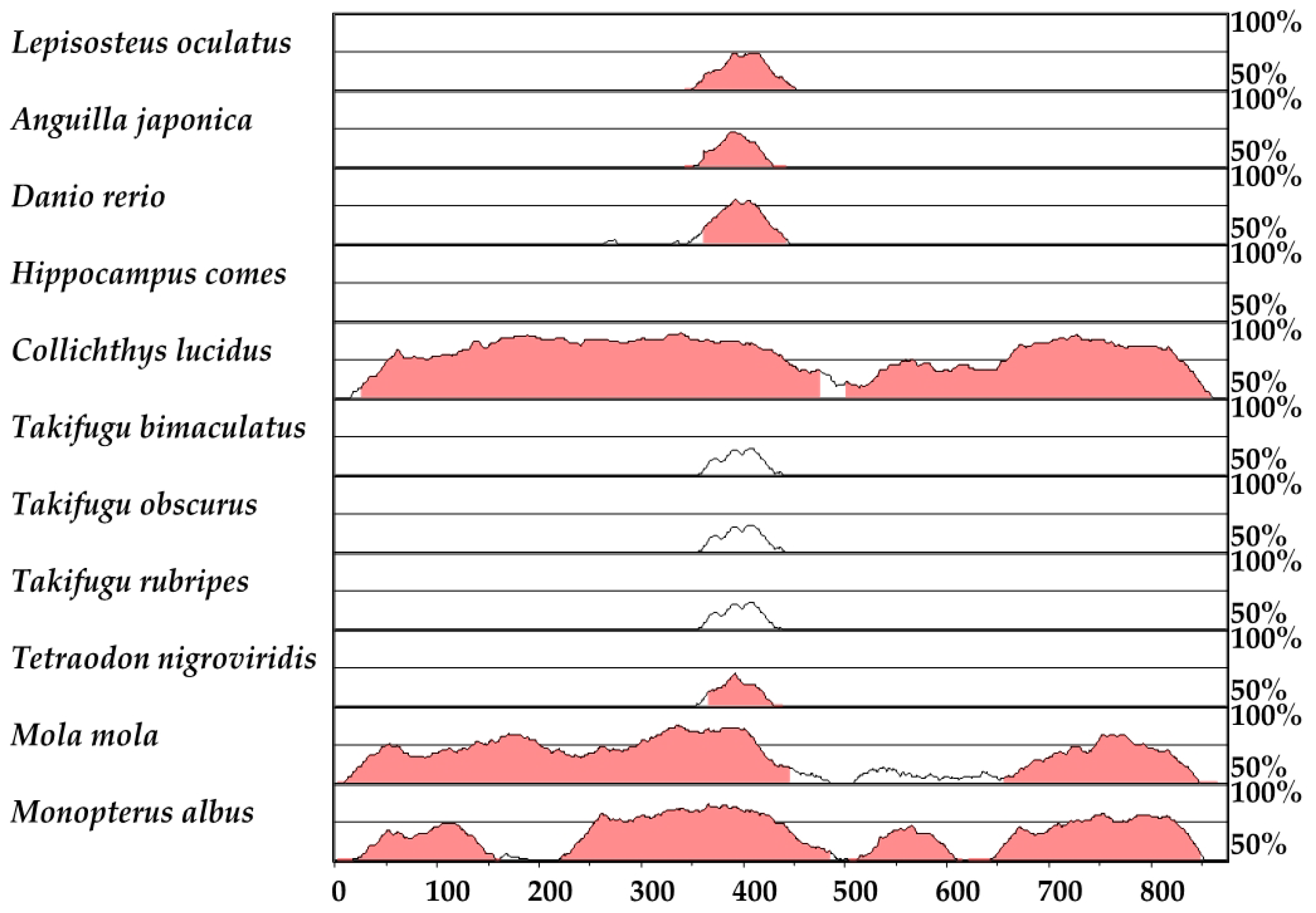
2.7. HLEB
3. Discussion
3.1. Various Genetic Mechanisms for Pelvic Fin Development
3.2. Potential Importance of the Tbx4 Gene Cluster for the Evolutionary Development of Toxic Fin Spines
4. Materials and Methods
4.1. Sample Collection
4.2. Genome Sequencing, Assembling and Annotation
4.3. Collection of the Genome Sequences
4.4. Collection of the Tbx4 Sequences
4.5. Sequence Alignment, Phylogenetic Analysis and Identification of Conserved Synteny
4.6. HLEB Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| appbp2 | amyloid protein-binding protein 2 |
| brip1 | BRCA1 interacting protein C-terminal helicase 1 |
| bcas3 | breast carcinoma amplified sequence 3 |
| eomesa | eomesodermin homolog A |
| eomesb | eomesodermin homolog B |
| fgf family | fibroblast growth factor family |
| HLEA | hindlimb enhancer A |
| HLEB | hindlimb enhancer B |
| hox genes | homeobox genes |
| lhx1a | LIM homeobox transcription factor 1, alpha |
| mga | max gene-associated protein |
| mgal | max gene-associated protein-like |
| NLS | nuclear localization sequence |
| pitx1 | paired-like homeodomain 1 |
| ppm1d | protein phosphatase 1D |
| shh | Sonic hedgehog |
| ta | brachyury homolog A |
| tb | brachyury homolog B |
| tbr1a | T-box brain protein 1A |
| tbr1b | T-box brain protein 1B |
| tbx genes | T-box transcription factors |
| tbx2 | T-Box transcription factor 2 |
| tbx2b | T-Box transcription factor 2B |
| tbx3 | T-Box transcription factor 3 |
| tbx4 | T-Box transcription factor 4 |
| tbx5 | T-Box transcription factor 5 |
| usp32 | ubiquitin specific peptidase 32 |
| vegt | vegetal T-box transcription factor |
| wnt family | wingless-type MMTV integration site family |
References
- Sansom, R.S.; Gabbott, S.E.; Purnell, M.A. Unusual anal fin in a Devonian jawless vertebrate reveals complex origins of paired appendages. Biol. Lett. 2013, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Forey, P.L. Agnathans recent and fossil, and the origin of jawed vertebrates. Rev. Fish Biol. Fisher. 1995, 5, 267–303. [Google Scholar] [CrossRef]
- Zhu, M.; Yu, X.B.; Choo, B.; Wang, J.Q.; Jia, L.T. An antiarch placoderm shows that pelvic girdles arose at the root of jawed vertebrates. Biol. Lett. 2012, 8, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Don, E.K.; Currie, P.D.; Cole, N.J. The evolutionary history of the development of the pelvic fin/hindlimb. J. Anat. 2013, 222, 114–133. [Google Scholar] [CrossRef]
- Blair, J.E.; Hedges, S.B. Molecular phylogeny and divergence times of deuterostome animals. Mol. Biol. Evol. 2005, 22, 2275–2284. [Google Scholar] [CrossRef]
- Lin, Q.; Fan, S.; Zhang, Y.; Xu, M.; Zhang, H.; Yang, Y.; Lee, A.P.; Woltering, J.M.; Ravi, V.; Gunter, H.M.; et al. The seahorse genome and the evolution of its specialized morphology. Nature 2016, 540, 395–399. [Google Scholar] [CrossRef] [Green Version]
- Papaioannou, V.E. T-box genes in development: From hydra to humans. Int. Rev. Cytol. 2001, 207, 1–70. [Google Scholar]
- Horton, A.C.; Mahadevan, N.R.; Minguillon, C.; Osoegawa, K.; Rokhsar, D.S.; Ruvinsky, I.; de Jong, P.J.; Logan, M.P.; Gibson-Brown, J.J. Conservation of linkage and evolution of developmental function within the Tbx2/3/4/5 subfamily of T-box genes: Implications for the origin of vertebrate limbs. Dev. Genes Evol. 2008, 218, 613–628. [Google Scholar] [CrossRef]
- Jackson, L.M.; Fernando, P.C.; Hanscom, J.S.; Balhoff, J.P.; Mabee, P.M. Automated integration of trees and traits: A case study using paired fin loss across teleost fishes. Syst. Biol. 2018, 67, 559–575. [Google Scholar] [CrossRef]
- Bian, C.; Huang, Y.; Li, J.; You, X.; Yi, Y.; Ge, W.; Shi, Q. Divergence, evolution and adaptation in ray-fined fish genomes. Sci. China Life Sci. 2019, 62. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Bian, C.; Luo, Y.; Wang, L.; You, X.; Li, J.; Qiu, Y.; Ma, X.; Zhu, Z.; Ma, L.; et al. Draft genome of the Chinese mitten crab, Eriocheir sinensis. GigaScience 2016, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 15, 578–579. [Google Scholar] [CrossRef]
- Simao, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Zdobnov, E.M.; Tegenfeldt, F.; Kuznetsov, D.; Waterhouse, R.M.; Simão, F.A.; Ioannidis, P.; Seppey, M.; Loetscher, A.; Kriventseva, E.V. OrthoDB v9.1: Cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Res. 2017, 45, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Parfrey, L.W.; Lahr, D.J.; Knoll, A.H.; Katz, L.A. Estimating the timing of early eukaryotic diversification with multigene molecular clocks. Proc. Natl. Acad. Sci. USA 2011, 108, 13624–13629. [Google Scholar] [CrossRef] [Green Version]
- Tada, M.; Smith, J.C. T-targets: Clues to understanding the functions of T-box proteins. Dev. Growth Differ. 2001, 43, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, X.; Bai, J.; Fang, D.; Qiu, Y.; Jiang, W.; Yuan, H.; Bian, C.; Lu, J.; He, S.; et al. The Sinocyclocheilus cavefish genome provides insights into cave adaptation. BMC Biol. 2016, 14, 1. [Google Scholar] [CrossRef]
- Christensen, K.A.; Leong, J.S.; Sakhrani, D.; Biagi, C.A.; Minkley, D.R.; Withler, R.E.; Rondeau, E.B.; Koop, B.F.; Devlin, R.H. Chinook salmon (Oncorhynchus tshawytscha) genome and transcriptome. PLoS ONE 2018, 13, e0195461. [Google Scholar] [CrossRef]
- Don, E.K.; de Jong-Curtain, T.A.; Doggett, K.; Hall, T.E.; Heng, B.; Badrock, A.P.; Winnick, C.; Nicholson, G.A.; Guillemin, G.J.; Currie, P.D.; et al. Genetic basis of hindlimb loss in a naturally occurring vertebrate model. Biol. Open 2016, 5, 359–366. [Google Scholar] [CrossRef] [Green Version]
- Collavoli, A.; Hatcher, C.J.; He, J.; Okin, D.; Deo, R.; Basson, C.T. TBX5 nuclear localization is mediated by dual cooperative intramolecular signals. J. Mol. Cell. Cardiol. 2003, 35, 1191–1195. [Google Scholar] [CrossRef]
- Betancur-R, R.; Broughton, R.E.; Wiley, E.O.; Carpenter, K.; López, J.A.; Li, C.; Holcroft, N.I.; Arcila, D.; Sanciangco, M.; Cureton, J.C., II; et al. The tree of life and a new classification of bony fishes. PLoS Curr. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Dos Reis, M.; Thawornwattana, Y.; Angelis, K.; Telford, M.J.; Donoghue, P.C.; Yang, Z. Uncertainty in the timing of origin of animals and the limits of precision in molecular timescales. Curr. Biol. 2015, 25, 2939–2950. [Google Scholar] [CrossRef] [PubMed]
- Inoue, J.G.; Miya, M.; Lam, K.; Tay, B.H.; Danks, J.A.; Bell, J.; Walker, T.I.; Venkatesh, B. Evolutionary origin and phylogeny of the modern holocephalans (Chondrichthyes: Chimaeriformes): A mitogenomic perspective. Mol. Biol. Evol. 2010, 27, 2576–2586. [Google Scholar] [CrossRef] [PubMed]
- Betancur-R, R.; Orti, G.; Pyron, A.R. Fossil-based comparative analyses reveal ancient marine ancestry erased by extinction in ray-finned fishes. Ecol. Lett. 2015, 18, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Infante, C.R.; Park, S.; Mihala, A.G.; Kingsley, D.M.; Menke, D.B. Pitx1 broadly associates with limb enhancers and is enriched on hindlimb cis-regulatory elements. Dev. Biol. 2013, 374, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Menke, D.B.; Guenther, C.; Kingsley, D.M. Dual hindlimb control elements in the Tbx4 gene and region-specific control of bone size in vertebrate limbs. Development 2008, 135, 2543–2553. [Google Scholar] [CrossRef] [PubMed]
- Farin, H.F.; Lüdtke, T.H.; Schmidt, M.K.; Placzko, S.; Schuster-Gossler, K.; Petry, M.; Christoffels, V.M.; Kispert, A. Tbx2 terminates Shh/Fgf signaling in the developing mouse limb bud by direct repression of Gremlin1. PLoS Genet. 2013, 9, e1003467. [Google Scholar] [CrossRef] [PubMed]
- Bärlund, M.; Monni, O.; Weaver, J.D.; Kauraniemi, P.; Sauter, G.; Heiskanen, M.; Kallioniemi, O.P.; Kallioniemi, A. Cloning of BCAS3 (17q23) and BCAS4 (20q13) genes that undergo amplification, overexpression, and fusion in breast cancer. Genes Chromosomes Cancer 2003, 35, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Chini, C.C.; He, M.; Mer, G.; Chen, J. The BRCT domain is a phospho-protein binding domain. Science 2003, 302, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Kubicek, K.M.; Britz, R.; Conway, K.W. Ontogeny of the catfish pectoral-fin spine (Teleostei: Siluriformes). J. Morphol. 2019, 280, 339–359. [Google Scholar] [CrossRef] [PubMed]
- Souza, C.S.; Costa-Silva, G.J.; Roxo, F.F.; Foresti, F.; Oliveira, C. Genetic and morphological analyses demonstrate that Schizolecis guntheri (Siluriformes: Loricariidae) is likely to be a species complex. Front. Genet. 2018, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Stewart, T.A.; Bonilla, M.M.; Ho, R.K.; Hale, M.E. Adipose fin development and its relation to the evolutionary origins of median fins. Sci. Rep. 2019, 9, 512. [Google Scholar] [CrossRef] [PubMed]
- Marinho, M.M.F.; Bastos, D.A.; Menezes, N.A. New species of miniature fish from Marajó Island, Pará, Brazil, with comments on its relationships (Characiformes: Characidae). Neotrop. Ichthyol. 2013, 11, 739–746. [Google Scholar] [CrossRef]
- Albert, J.; Crampton, W. Diversity and phylogeny of neotropical electric fishes (Gymnotiformes). In Electroreception; Bullock, T.H., Hopkins, C.D., Popper, A.N., Fay, R.R., Eds.; Springer: New York, NY, USA, 2005; pp. 360–409. [Google Scholar]
- Zhang, S.; Li, J.; Qin, Q.; Liu, W.; Bian, C.; Yi, Y.; Wang, M.; Zhong, L.; You, X.; Tang, S.; et al. Whole-genome sequencing of Chinese yellow catfish provides a valuable genetic resource for high-throughput identification of toxin genes. Toxins 2018, 10, 448. [Google Scholar] [CrossRef] [PubMed]
- Galloway, K.A.; Porter, M.E. Mechanical properties of the venomous spines of Pterois volitans and morphology among lionfish species. J. Exp. Biol. 2019, 222, jeb197905. [Google Scholar] [CrossRef] [PubMed]
- Yamanoue, Y.; Setiamarga, D.H.; Matsuura, K.J. Pelvic fins in teleosts: Structure, function and evolution. Fish Biol. 2010, 77, 1173–1208. [Google Scholar] [CrossRef]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.S.; Dubchak, I. VISTA: Visualizing Global DNA Sequence Alignments of Arbitrary Length. Bioinformatics 2000, 16, 1046. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, 273–279. [Google Scholar] [CrossRef]
- Tanaka, M.; Hale, L.A.; Amores, A.; Yan, Y.L.; Cresko, W.A.; Suzuki, T.; Postlethwait, J.H. Developmental genetic basis for the evolution of pelvic fin loss in the pufferfish Takifugu rubripes. Dev. Biol. 2005, 281, 227–239. [Google Scholar] [CrossRef]
- Kvon, E.Z.; Kamneva, O.K.; Melo, U.S.; Barozzi, I.; Osterwalder, M.; Mannion, B.J.; Tissières, V.; Pickle, C.S.; Plajzer-Frick, I.; Lee, E.A.; et al. Progressive loss of function in a limb enhancer during snake evolution. Cell 2016, 167, 633–642. [Google Scholar] [CrossRef]
- Infante, C.R.; Mihala, A.G.; Park, S.; Wang, J.S.; Johnson, K.K.; Lauderdale, J.D.; Menke, D.B. Shared enhancer activity in the limbs and phallus and functional divergence of a limb-genital cis-regulatory element in snakes. Dev. Cell 2015, 35, 107–119. [Google Scholar] [CrossRef]
- Lettice, L.A.; Heaney, S.J.; Purdie, L.A.; Li, L.; de Beer, P.; Oostra, B.A.; Goode, D.; Elgar, G.; Hill, R.E.; de Graaff, E. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum. Mol. Genet. 2003, 12, 1725–1735. [Google Scholar] [CrossRef]
- Lettice, L.A.; Williamson, I.; Wiltshire, J.H.; Peluso, S.; Devenney, P.S.; Hill, A.E.; Essafi, A.; Hagman, J.; Mort, R.; Grimes, G.; et al. Opposing functions of the ETS factor family define Shh spatial expression in limb buds and underlie polydactyly. Dev. Cell 2012, 22, 459–467. [Google Scholar] [CrossRef]
- Sagai, T.; Hosoya, M.; Mizushina, Y.; Tamura, M.; Shiroishi, T. Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development 2005, 132, 797–803. [Google Scholar] [CrossRef]
- Lettice, L.A.; Hill, A.E.; Devenney, P.S.; Hill, R.E. Point mutations in a distant sonic hedgehog cis-regulator generate a variable regulatory output responsible for preaxial polydactyly. Hum. Mol. Genet. 2008, 17, 978–985. [Google Scholar] [CrossRef]
- Lettice, L.A.; Williamson, I.; Devenney, P.S.; Kilanowski, F.; Dorin, J.; Hill, R.E. Development of five digits is controlled by a bipartite long-range cis-regulator. Development 2014, 141, 1715–1725. [Google Scholar] [CrossRef]
- Sagai, T.; Masuya, H.; Tamura, M.; Shimizu, K.; Yada, Y.; Wakana, S.; Gondo, Y.; Noda, T.; Shiroishi, T. Phylogenetic conservation of a limb-specific, cis-acting regulator of Sonic hedgehog (Shh). Mamm. Genom. 2004, 15, 23–34. [Google Scholar] [CrossRef]
- Zeller, R.; Zuniga, A. Shh and Gremlin1 chromosomal landscapes in development and disease. Curr. Opin. Genet. Dev. 2007, 17, 428–434. [Google Scholar] [CrossRef]
- Thompson, A.C.; Capellini, T.D. A novel enhancer near the Pitx1 gene influences development and evolution of pelvic appendages in vertebrates. eLife 2018, 7, 38555. [Google Scholar] [CrossRef]
- Logan, M.; Tabin, C.J. Role of Pitx1 upstream of Tbx4 in specification of hindlimb identity. Science 1999, 283, 1736–1739. [Google Scholar] [CrossRef]
- Marcil, A.; Dumontier, E.; Chamberl, M.; Camper, S.A.; Drouin, J. Pitx1 and Pitx2 are required for development of hindlimb buds. Development 2003, 130, 45–55. [Google Scholar] [CrossRef]
- Shapiro, M.D.; Marks, M.E.; Peichel, C.L.; Blackman, B.K.; Nereng, K.S.; Jónsson, B.; Schluter, D.; Kingsley, D.M. Genetic and developmental basis of evolutionary pelvic reduction in threespine sticklebacks. Nature 2004, 428, 717–723. [Google Scholar] [CrossRef]
- Shapiro, M.D.; Bell, M.A.; Kingsley, D.M. Parallel genetic origins of pelvic reduction in vertebrates. Proc. Natl. Acad. Sci. USA 2006, 103, 13753–13758. [Google Scholar] [CrossRef] [Green Version]
- Xie, B.; Li, X.; Lin, Z.; Ruan, Z.; Wang, M.; Liu, J.; Tong, T.; Li, J.; Huang, Y.; Wen, B.; et al. Prediction of toxin genes from Chinese yellow catfish based on transcriptomic and proteomic sequencing. Int. J. Mol. Sci. 2016, 17, 556. [Google Scholar] [CrossRef]
- Xie, B.; Huang, Y.; Baumann, K.; Fry, B.G.; Shi, Q. From marine venoms to drugs: Efficiently supported by a combination of transcriptomics and proteomics. Mar. Drugs 2017, 15, 103. [Google Scholar] [CrossRef]
- Xu, Z.; Wang, H. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007, 35, 265–268. [Google Scholar] [CrossRef]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 25, 4–10. [Google Scholar]
- Jurka, J.; Kapitonov, V.V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, 435–439. [Google Scholar] [CrossRef]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef] [Green Version]
- Mount, D.W. Using the basic local alignment search tool (blast). Cold Spring Harb. Protoc. 2007, 2007. [Google Scholar] [CrossRef]
- Birney, E.; Durbin, R. Using genewise in the drosophila annotation experiment. Genome Res. 2000, 10, 547–548. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 27, 29–34. [Google Scholar] [CrossRef]
- Bairoch, A.; Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res. 2000, 28, 45–48. [Google Scholar] [CrossRef]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L.; et al. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009, 37, 211–215. [Google Scholar] [CrossRef]
- Liu, K.; Xu, D.; Li, J.; Bian, C.; Duan, J.; Zhou, Y.; Zhang, M.; You, X.; You, Y.; Chen, J.; et al. Whole genome sequencing of Chinese clearhead icefishe, Protosalanx hyalocranius. GigaScience 2017, 6, 1–6. [Google Scholar] [CrossRef]
- Xu, J.; Bian, C.; Chen, K.; Liu, G.; Jiang, Y.; Luo, Q.; You, X.; Peng, W.; Li, J.; Huang, Y.; et al. Draft genome of the northern snakehead, Channa argus. Gigascience 2017, 6, 1–5. [Google Scholar] [CrossRef]
- Nomura, K.; Fujiwara, A.; Iwasaki, Y.; Nishiki, I.; Matsuura, A.; Ozaki, A.; Sudo, R.; Tanaka, H. Genetic parameters and quantitative trait loci analysis associated with body size and timing at metamorphosis into glass eels in captive-bred Japanese eels (Anguilla japonica). PLoS ONE 2018, 13, e0201784. [Google Scholar] [CrossRef]
- Slater, G.S.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef]
- Edgar, R.C. Muscle: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef]
- Beitz, E. Texshade: Shading and labeling of multiple sequence alignments using latex2 epsilon. Bioinformatics 2000, 16, 135–139. [Google Scholar] [CrossRef]
- Posada, D.; Buckley, T.R. Model selection and model averaging in phylogenetics: Advantages of akaike information criterion and bayesian approaches over likelihood ratio tests. Syst. Biol. 2004, 53, 793–808. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Guindon, S.; Delsuc, F.; Dufayard, J.F.; Gascuel, O. Estimating Maximum Likelihood Phylogenies with PhyML. Methods Mol. Biol. 2009, 537, 113–137. [Google Scholar] [Green Version]
- Brudno, M.; Do, C.B.; Cooper, G.M.; Kim, M.F.; Davydov, E.; Green, E.D.; Sidow, A.; Batzoglou, S. NISC Comparative Sequencing Program. LAGAN and Multi-LAGAN: Efficient Tools for Large-Scale Multiple Alignment of Genomic DNA. Genome Res. 2003, 13, 721–731. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Software | Contig N50 (bp) | Scaffold N50 (bp) | Contig number | Scaffold number | Total length (bp) |
|---|---|---|---|---|---|---|
| Primary assembling | SOAPdenovo | 1,999 | 383,798 | 1,227,464 | 462,272 | 1,167,219,893 |
| Gap filling | krskgf | 3,868 | 375,823 | 850,121 | 462,272 | 1,150,479,312 |
| Gapclose1.12 | 5,372 | 376,296 | 761,523 | 462,272 | 1,154,146,689 | |
| Gapclose1.10 | 10,215 | 376,491 | 624,151 | 462,272 | 1,154,798,407 | |
| Scaffold extending Filtering | SSPACE --- | 10,236 11,468 | 858,288 1,033,285 | 608,352 256,649 | 351,879 41,687 | 1,228,736,536 1,132,698,062 |
| Parameter | Number | Percentage (%) |
|---|---|---|
| Complete BUSCOs (C) | 3847 | 83.9 |
| Complete and single-copy BUSCOs (S) | 3346 | 73.0 |
| Complete and duplicated BUSCOs (D) | 501 | 10.9 |
| Fragmented BUSCOs (F) | 380 | 8.3 |
| Missing BUSCOs (M) | 357 | 7.8 |
| Total BUSCO groups searched (n) | 4584 | --- |
| Gene | Species Name | Accession Number |
|---|---|---|
| eomesa | Danio rerio | AAH67719.1 |
| eomesb | D. rerio | NP_001077044.1 |
| mgal | D. rerio | XP_021324416.1 |
| mga | D. rerio | ADA61227.1 |
| ta | D. rerio | Q07998.1 |
| tb | D. rerio | XP_001343633.3 |
| tbr1a | D. rerio | XP_693121.1 |
| tbr1b | D. rerio | AAG48249.1 |
| tbx15 | D. rerio | AAM54074.1 |
| tbx16 | D. rerio | AAI65213.1 |
| tbx18 | D. rerio | AAI63460.1 |
| tbx19 | D. rerio | XP_003198807.1 |
| tbx11 | D. rerio | XP_017206601.2 |
| tbx1 | D. rerio | Q8AXX2.1 |
| tbx20 | D. rerio | AAF64322.1 |
| tbx21 | D. rerio | NP_001164070.1 |
| tbx22 | D. rerio | ACU00296.1 |
| tbx2a | D. rerio | AAH68364.1 |
| tbx2b | D. rerio | Q7ZTU9.4 |
| tbx3a | D. rerio | NP_001095140.2 |
| tbx3b | D. rerio | XP_002662050.2 |
| tbx4 | D. rerio | AAI62554.1 |
| tbx5a | D. rerio | Q9IAK8.2 |
| tbx5b | D. rerio | ADX53331.1 |
| tbx6l | D. rerio | P79742.1 |
| tbx6 | D. rerio | Q8JIS6.2 |
| vegt | Fundulus heteroclitus | JAQ45978.1 |
| lhx1a | D. rerio | Q90476.1 |
| brip1 | Aphyosemion striatum | SBP21433.1 |
| bcas3 | Nothobranchius furzeri | SBP60348.1 |
| ppm1da | N. furzeri | SBP60348.1 |
| appbp2 | N. furzeri | SBP60348.1 |
| usp32 | N. kuhntae | SBP60348.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.; Bian, C.; You, X.; Li, J.; Ye, L.; Wen, Z.; Lv, Y.; Zhang, X.; Xu, J.; Yang, S.; et al. Genome Sequencing of the Japanese Eel (Anguilla japonica) for Comparative Genomic Studies on tbx4 and a tbx4 Gene Cluster in Teleost Fishes. Mar. Drugs 2019, 17, 426. https://doi.org/10.3390/md17070426
Chen W, Bian C, You X, Li J, Ye L, Wen Z, Lv Y, Zhang X, Xu J, Yang S, et al. Genome Sequencing of the Japanese Eel (Anguilla japonica) for Comparative Genomic Studies on tbx4 and a tbx4 Gene Cluster in Teleost Fishes. Marine Drugs. 2019; 17(7):426. https://doi.org/10.3390/md17070426
Chicago/Turabian StyleChen, Weiwei, Chao Bian, Xinxin You, Jia Li, Lizhen Ye, Zhengyong Wen, Yunyun Lv, Xinhui Zhang, Junmin Xu, Shaosen Yang, and et al. 2019. "Genome Sequencing of the Japanese Eel (Anguilla japonica) for Comparative Genomic Studies on tbx4 and a tbx4 Gene Cluster in Teleost Fishes" Marine Drugs 17, no. 7: 426. https://doi.org/10.3390/md17070426
APA StyleChen, W., Bian, C., You, X., Li, J., Ye, L., Wen, Z., Lv, Y., Zhang, X., Xu, J., Yang, S., Gu, R., Lin, X., & Shi, Q. (2019). Genome Sequencing of the Japanese Eel (Anguilla japonica) for Comparative Genomic Studies on tbx4 and a tbx4 Gene Cluster in Teleost Fishes. Marine Drugs, 17(7), 426. https://doi.org/10.3390/md17070426