Novel Insights on the Toxicity of Phycotoxins on the Gut through the Targeting of Enteric Glial Cells

Abstract
:1. Introduction
2. Results
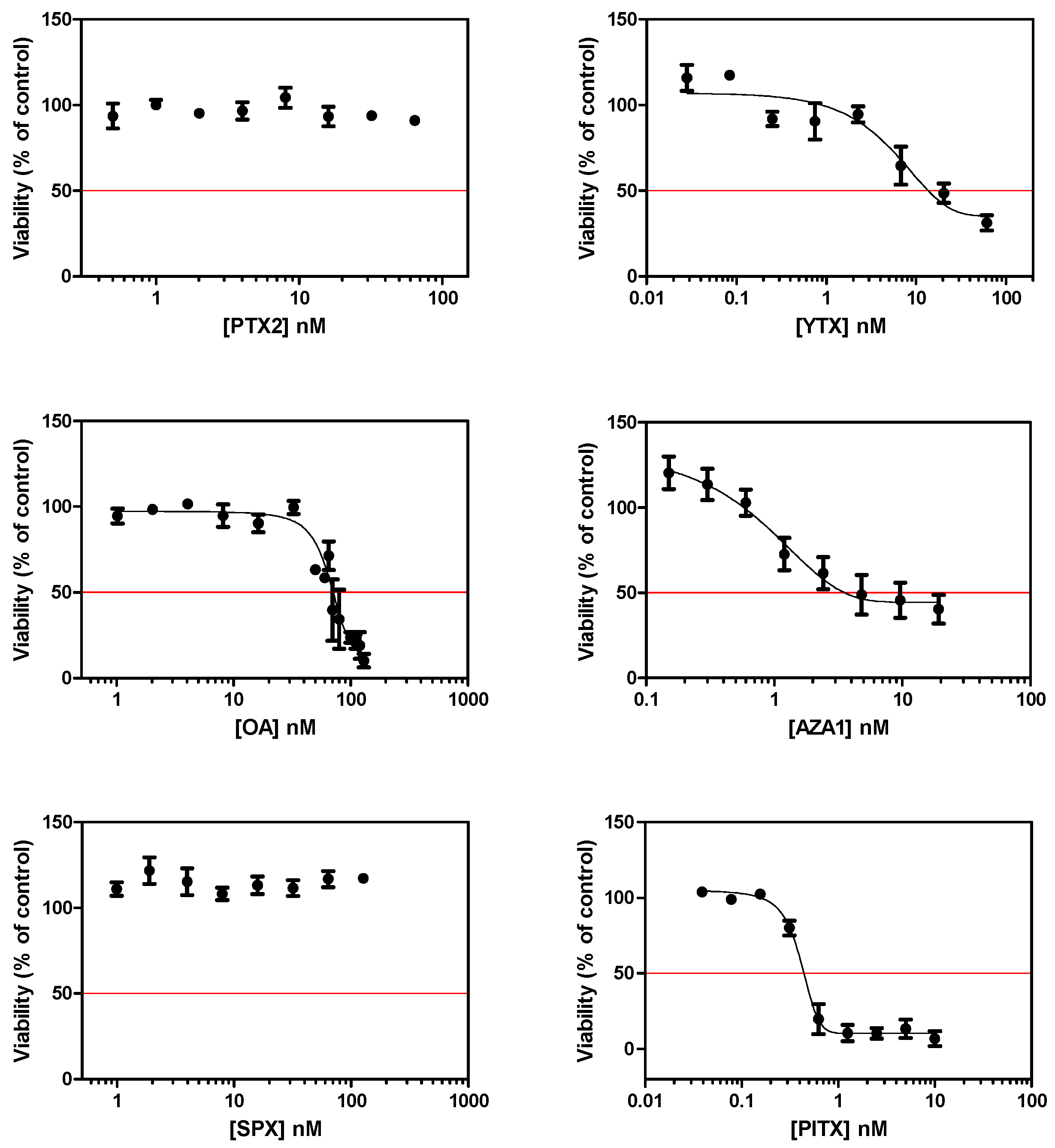
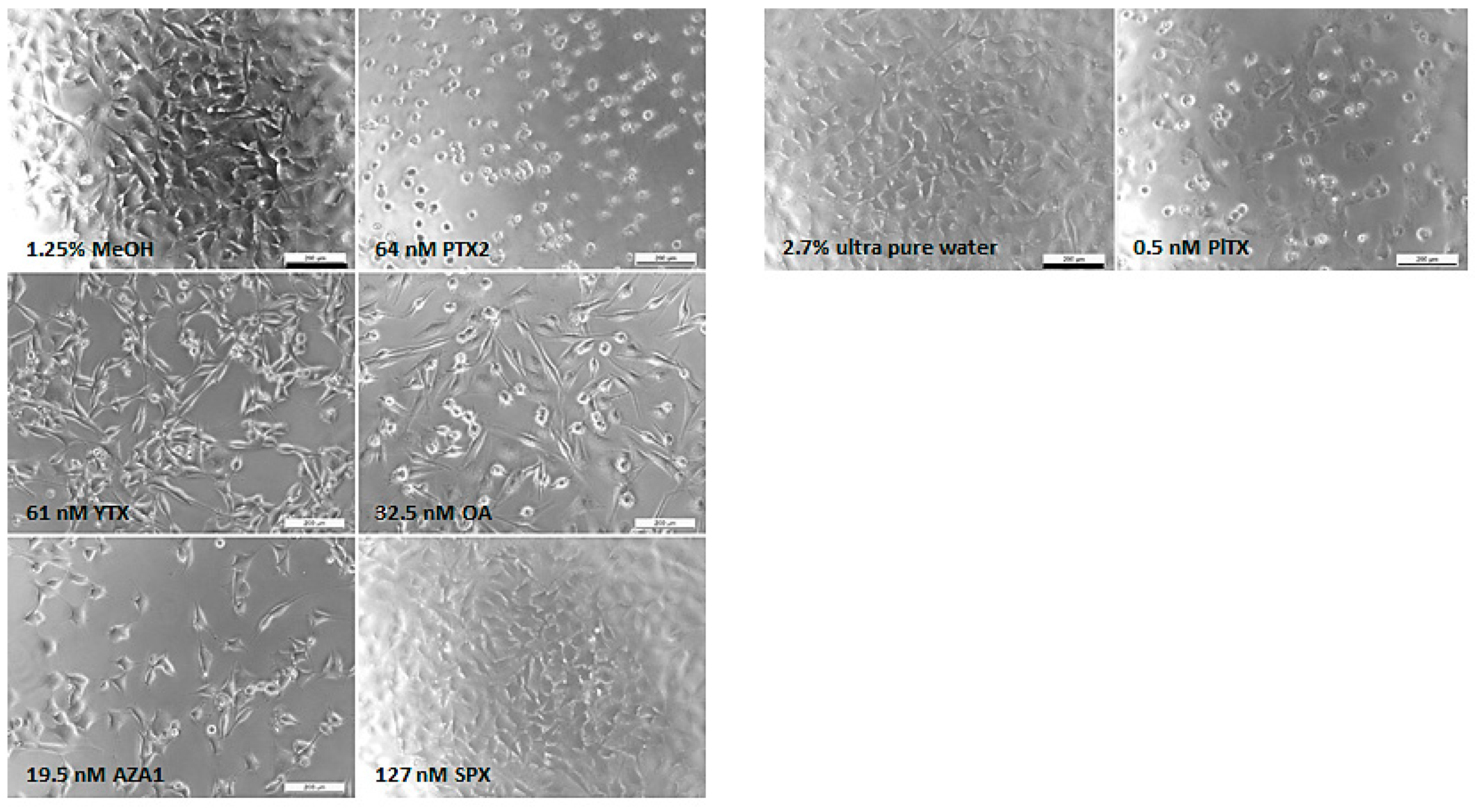
2.1. Cytotoxic and Morphological Effects of Phycotoxins
2.2. Intermediate Filament GFAP and Cell Body Area
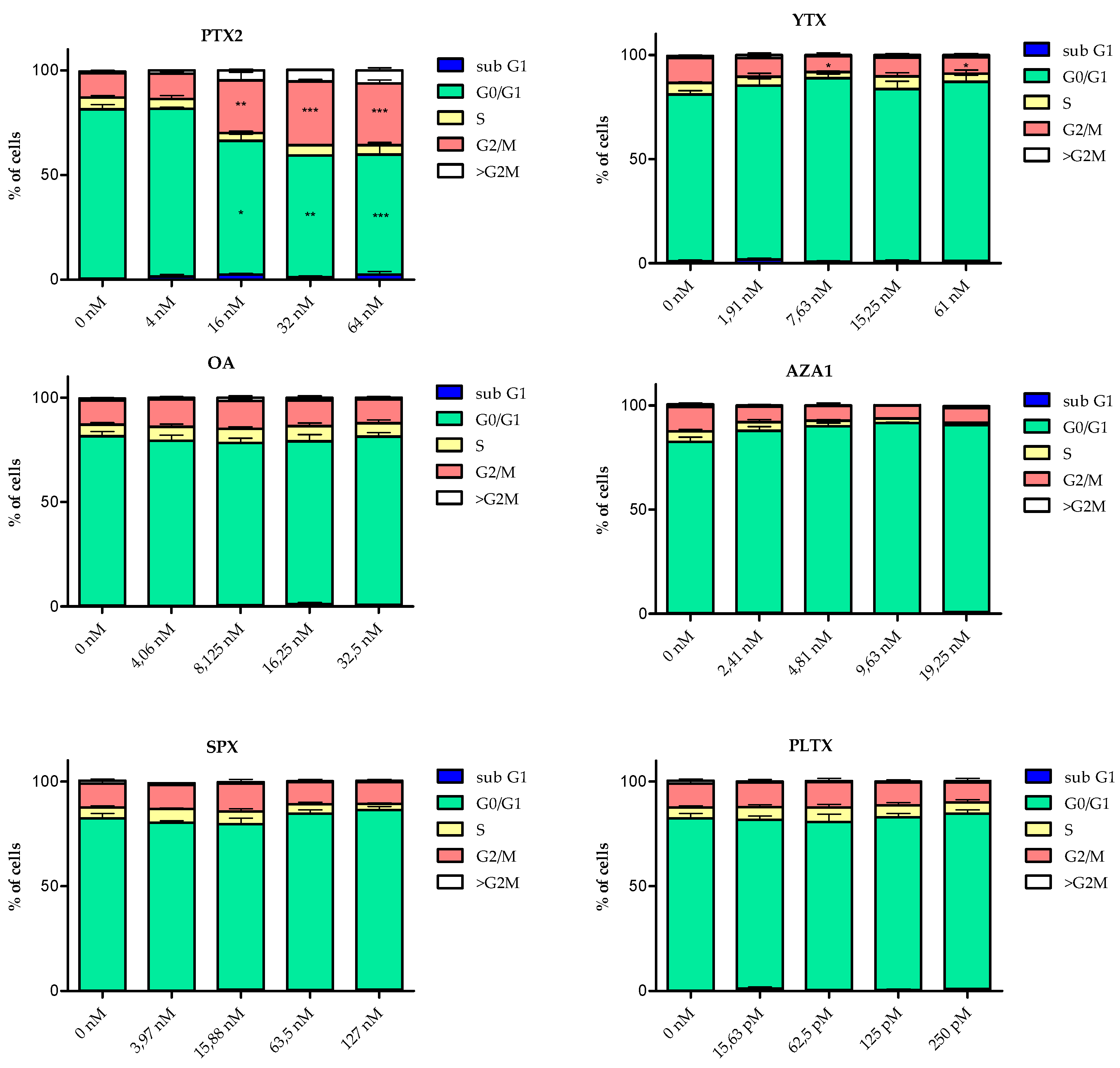
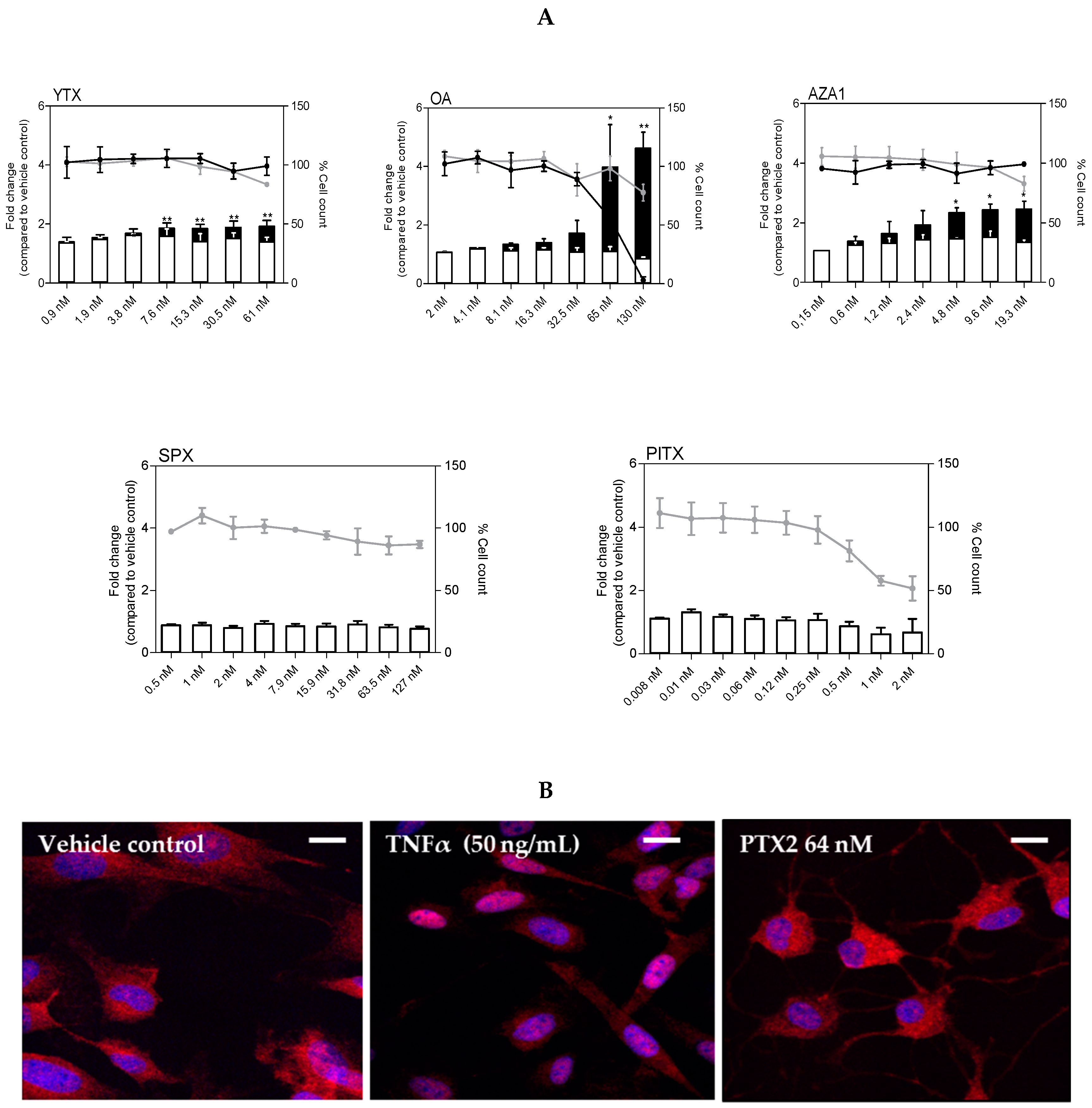
2.3. Cell Cycle Analysis
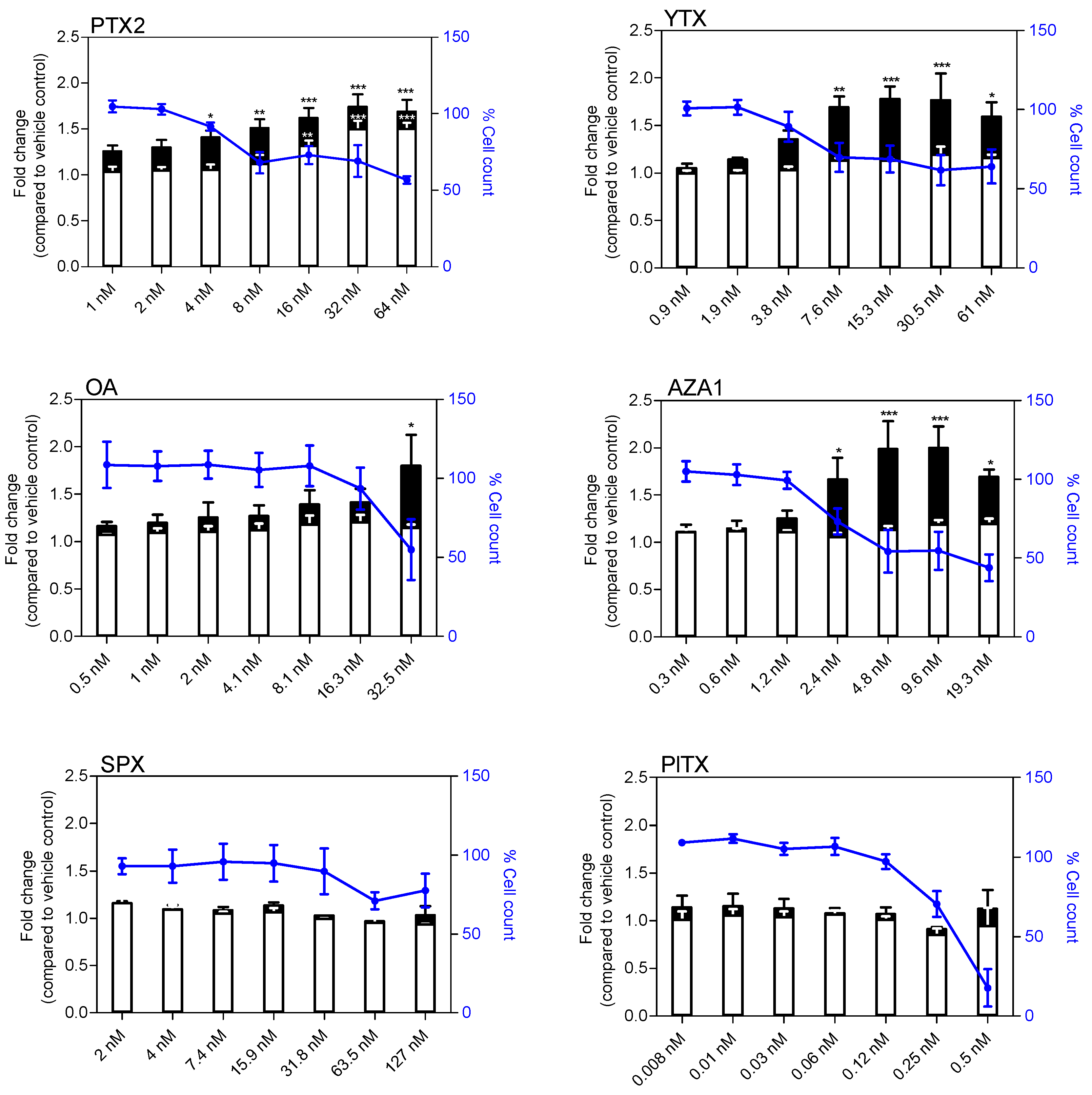
2.4. Apoptosis and Genotoxicity
2.5. NF-κB Nuclear Translocation
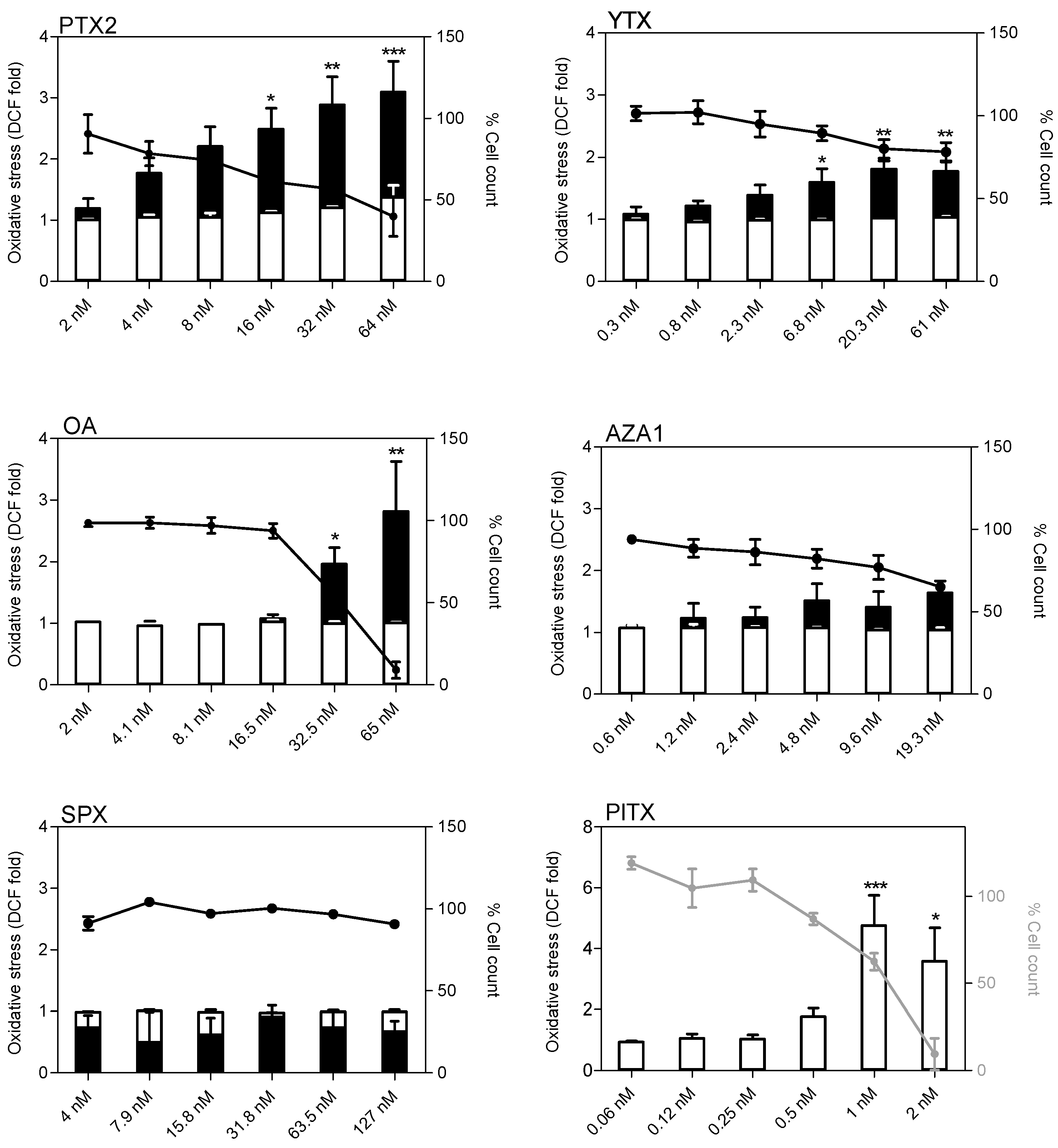
2.6. Oxidative Stress
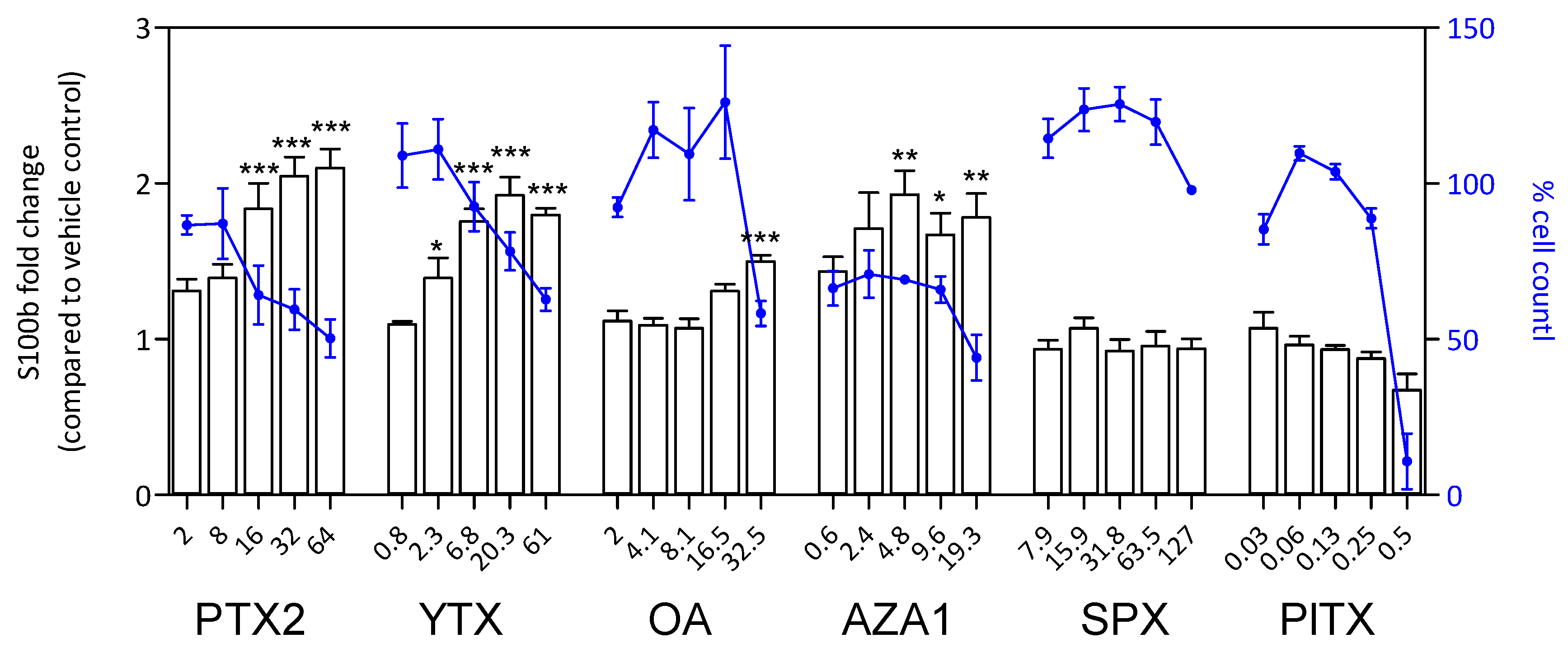
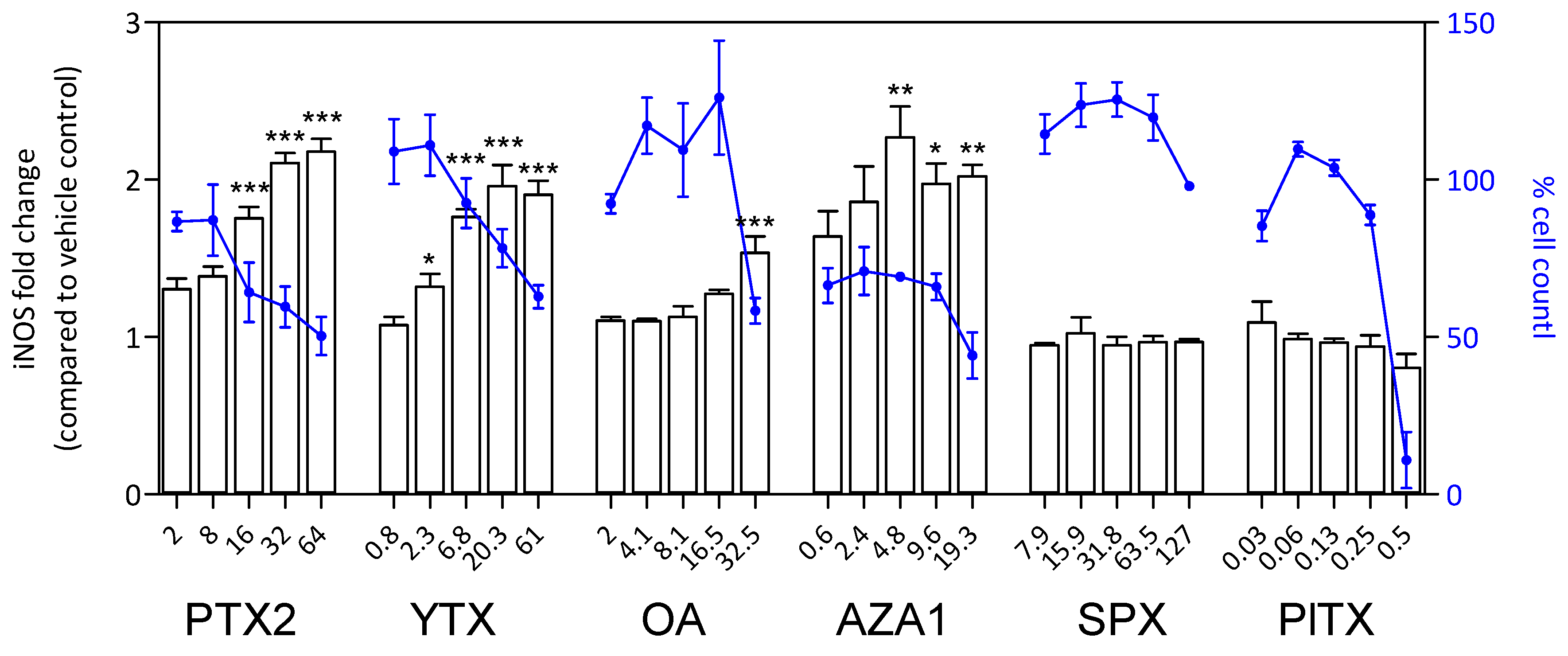
2.7. S100β and iNOS Production
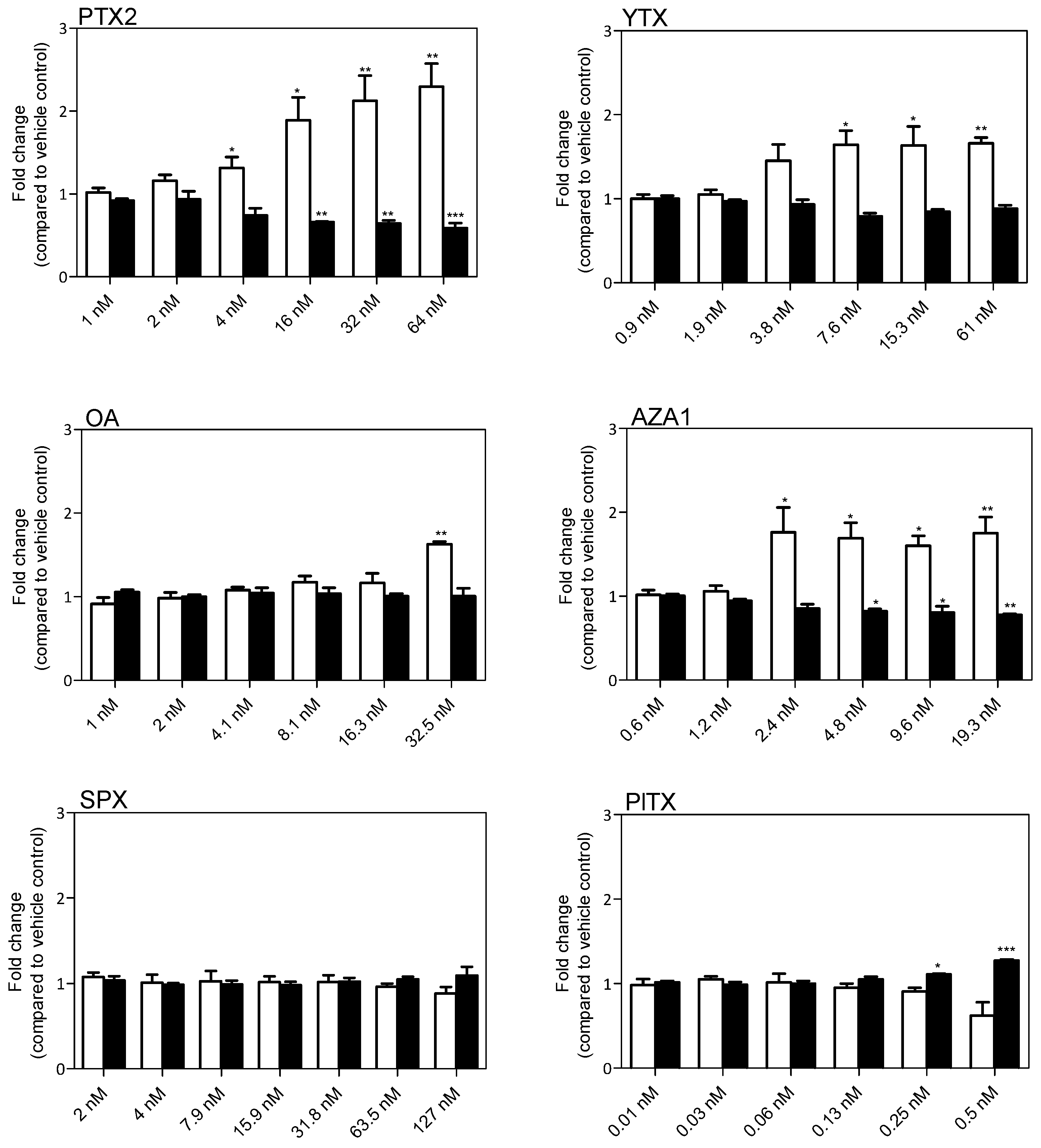
2.8. Modulation of Gene Expression Following Treatment with PTX2, YTX, and AZA1
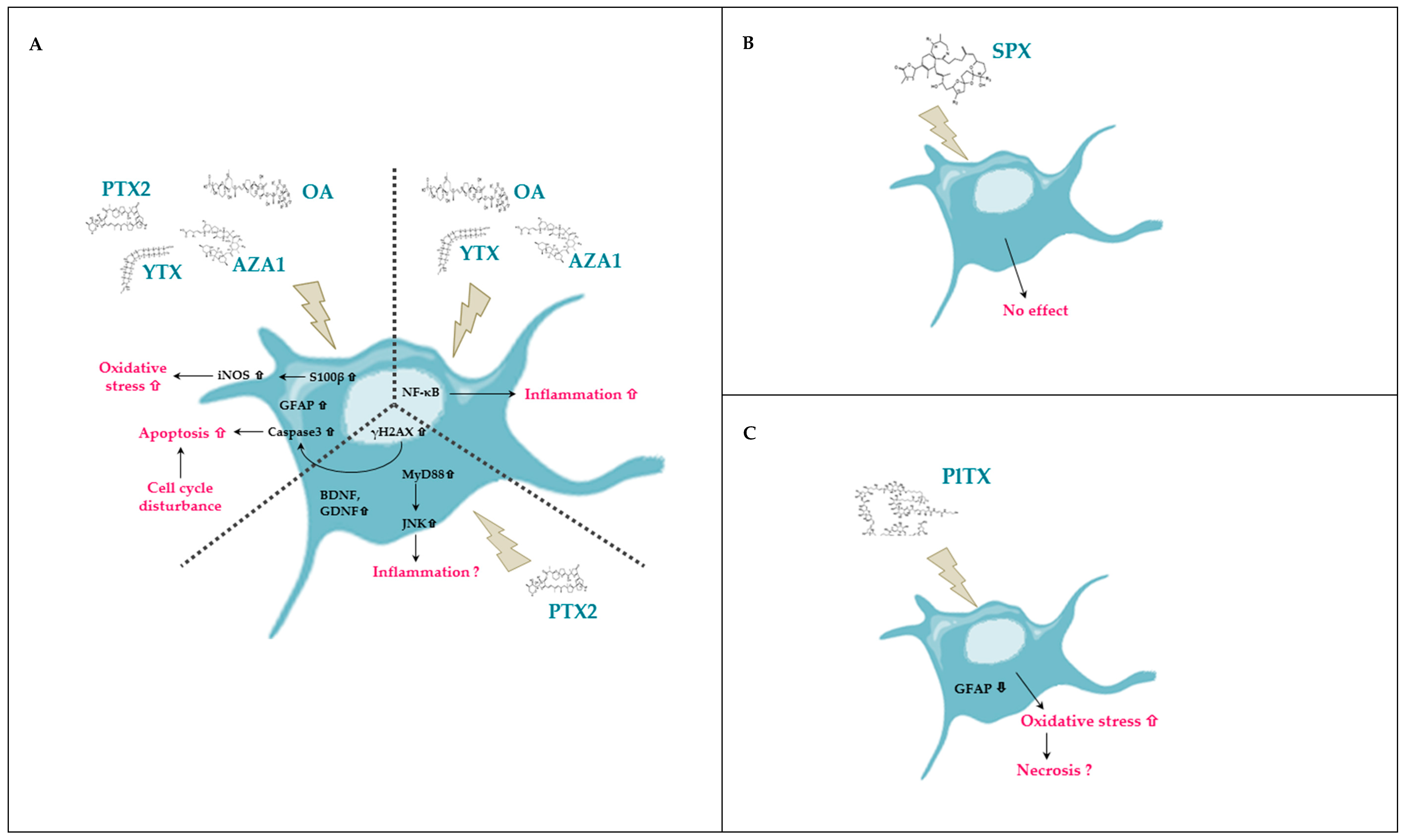
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture and Toxin Exposure
4.3. Cell Morphology and Neutral Red Uptake Assay
4.4. High Content Analysis Multiparametric Toxicity Assays Oxidative Stress
4.5. Cell Cycle Analysis, Inflammation, Genotoxicity, Apoptosis, and Glial Cell Markers
4.6. Confocal Microscopy Imaging
4.7. RT-qPCR
4.8. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Farabegoli, F.; Blanco, L.; Rodriguez, L.P.; Vieites, J.M.; Cabado, A.G. Phycotoxins in marine shellfish: Origin, occurrence and effects on humans. Mar. Drugs 2018, 16, 188. [Google Scholar] [CrossRef] [PubMed]
- Khora, S.S.; Jal, S. Occurrence of natural toxins in seafood. Microb. Contam. Food Degrad. 2018, 177–233. [Google Scholar] [CrossRef]
- EFSA. Marine biotoxins in shellfish—Pectenotoxin group. EFSA J. 2009, 7, 1109. [Google Scholar]
- EFSA. Marine biotoxins in shellfish—Yessotoxin group. EFSA J. 2008, 7, 907. [Google Scholar]
- Özogul, F.; Hamed, I. Marine-based toxins and their health risk. In Handbook of Food Bioengineering, Food Quality: Balancing Health and Disease; Holban, A.M., Grumezescu, A.M., Eds.; Academic Press: Cambridge, MA, USA, 2018; Chapter 3; Volume 13, pp. 109–144. [Google Scholar]
- EFSA. Marine biotoxins in shellfish—Azaspiracid group. EFSA J. 2008, 6, 723. [Google Scholar]
- EFSA. Scientific Opinion on marine biotoxins in shellfish—Cyclic imines (spirolides, gymnodimines, pinnatoxins and pteriatoxins). EFSA J. 2010, 8, 1628. [Google Scholar] [CrossRef]
- EFSA. Scientific Opinion on marine biotoxins in shellfish—Palytoxin group. EFSA J. 2009, 7, 1393. [Google Scholar] [CrossRef]
- Espina, B.; Rubiolo, J.A. Marine toxins and the cytoskeleton: Pectenotoxins, unusual macrolides that disrupt actin. FEBS J. 2008, 275, 6082–6088. [Google Scholar] [CrossRef]
- Twiner, M.J.; Hess, P.; Dechraoui, M.Y.; McMahon, T.; Samons, M.S.; Satake, M.; Yasumoto, T.; Ramsdell, J.S.; Doucette, G.J. Cytotoxic and cytoskeletal effects of azaspiracid-1 on mammalian cell lines. Toxicon 2005, 45, 891–900. [Google Scholar] [CrossRef]
- Takai, A.; Sasaki, K.; Nagai, H.; Mieskes, G.; Isobe, M.; Isono, K.; Yasumoto, T. Inhibition of specific binding of okadaic acid to protein phosphatase 2A by microcystin-LR, calyculin-A and tautomycin: Method of analysis of interactions of tight-binding ligands with target protein. Biochem. J. 1995, 306, 657–665. [Google Scholar] [CrossRef]
- Alexander, J.; Atli Auðunsson, G.A.; Benford, D.; Cockburn, A.; Cravedi, J.P.; Dogliotti, E.; Di Domenico, A.; Fernández-Cruz, M.L.; Fink-Gremmels, J.; Fürst, P.; et al. Marine biotoxins in shellfish—Okadaic acid and analogues, Scientific Opinion of the Panel on Contaminants in the Food chain. EFSA J. 2008, 589, 1–62. [Google Scholar]
- Otero, A.; Chapela, M.J.; Atanassova, M.; Vieites, J.M.; Cabado, A.G. Cyclic imines: Chemistry and mechanism of action: A review. Chem. Res. Toxicol. 2011, 24, 1817–1829. [Google Scholar] [CrossRef] [PubMed]
- Patocka, J.; Nepovimova, E.; Wu, Q.; Kuca, K. Palytoxin congeners. Arch. Toxicol. 2018, 92, 143–156. [Google Scholar] [CrossRef] [PubMed]
- De la Rosa, L.A.; Alfonso, A.; Vilarino, N.; Vieytes, M.R.; Botana, L.M. Modulation of cytosolic calcium levels of human lymphocytes by yessotoxin, a novel marine phycotoxin. Biochem. Pharmacol. 2001, 61, 827–833. [Google Scholar] [CrossRef]
- Paz, B.; Daranas, A.H.; Norte, M.; Riobo, P.; Franco, J.M.; Fernandez, J.J. Yessotoxins, a group of marine polyether toxins: An overview. Mar. Drugs 2008, 6, 73–102. [Google Scholar] [CrossRef]
- Twiner, M.J.; Rehmann, N.; Hess, P.; Doucette, G.J. Azaspiracid shellfish poisoning: A review on the chemistry, ecology, and toxicology with an emphasis on human health impacts. Mar. Drugs 2008, 6, 39–72. [Google Scholar] [CrossRef]
- Terao, K.; Ito, E.; Yanagi, T.; Yasumoto, T. Histopathological studies on experimental marine toxin poisoning. I. Ultrastructural changes in the small intestine and liver of suckling mice induced by dinophysistoxin-1 and pectenotoxin-1. Toxicon 1986, 24, 1141–1151. [Google Scholar] [CrossRef]
- Aune, T.; Espenes, A.; Aasen, J.A.; Quilliam, M.A.; Hess, P.; Larsen, S. Study of possible combined toxic effects of azaspiracid-1 and okadaic acid in mice via the oral route. Toxicon 2012, 60, 895–906. [Google Scholar] [CrossRef]
- Ito, E.; Satake, M.; Ofuji, K.; Higashi, M.; Harigaya, K.; McMahon, T.; Yasumoto, T. Chronic effects in mice caused by oral administration of sublethal doses of azaspiracid, a new marine toxin isolated from mussels. Toxicon 2002, 40, 193–203. [Google Scholar] [CrossRef]
- Fernandez, D.A.; Louzao, M.C.; Vilarino, N.; Espina, B.; Fraga, M.; Vieytes, M.R.; Roman, A.; Poli, M.; Botana, L.M. The kinetic, mechanistic and cytomorphological effects of palytoxin in human intestinal cells (Caco-2) explain its lower-than-parenteral oral toxicity. FEBS J. 2013, 280, 3906–3919. [Google Scholar] [CrossRef]
- Ferron, P.J.; Hogeveen, K.; Fessard, V.; Le Hegarat, L. Comparative analysis of the cytotoxic effects of okadaic acid-group toxins on human intestinal cell lines. Mar. Drugs 2014, 12, 4616–4634. [Google Scholar] [CrossRef] [PubMed]
- Le Hegarat, L.; Jacquin, A.G.; Bazin, E.; Fessard, V. Genotoxicity of the marine toxin okadaic acid, in human Caco-2 cells and in mice gut cells. Environ. Toxicol. 2006, 21, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Neunlist, M.; Van Landeghem, L.; Mahe, M.M.; Derkinderen, P.; des Varannes, S.B.; Rolli-Derkinderen, M. The digestive neuronal-glial-epithelial unit: A new actor in gut health and disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Cabarrocas, J.; Savidge, T.C.; Liblau, R.S. Role of enteric glial cells in inflammatory bowel disease. Glia 2003, 41, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Neunlist, M.; Van Landeghem, L.; Bourreille, A.; Savidge, T. Neuro-glial crosstalk in inflammatory bowel disease. J. Intern. Med. 2008, 263, 577–583. [Google Scholar] [CrossRef] [PubMed]
- Ben-Horin, S.; Chowers, Y. Neuroimmunology of the gut: Physiology, pathology, and pharmacology. Curr. Opin. Pharmcol. 2008, 8, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Carrier, R.L.; March, J.C.; Griffith, L.G. Three dimensional human small intestine models for ADME-Tox studies. Drug Discov. Today 2014, 19, 1587–1594. [Google Scholar] [CrossRef] [PubMed]
- Neunlist, M.; Aubert, P.; Bonnaud, S.; Van Landeghem, L.; Coron, E.; Wedel, T.; Naveilhan, P.; Ruhl, A.; Lardeux, B.; Savidge, T.; et al. Enteric glia inhibit intestinal epithelial cell proliferation partly through a TGF-beta1-dependent pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G231–G241. [Google Scholar] [CrossRef]
- Kermarrec, L.; Durand, T.; Neunlist, M.; Naveilhan, P.; Neveu, I. Enteric glial cells have specific immunosuppressive properties. J. Neuroimmunol. 2016, 295–296, 79–83. [Google Scholar] [CrossRef]
- Ochoa-Cortes, F.; Turco, F.; Linan-Rico, A.; Soghomonyan, S.; Whitaker, E.; Wehner, S.; Cuomo, R.; Christofi, F.L. Enteric glial cells: A new frontier in neurogastroenterology and clinical target for inflammatory bowel diseases. Inflamm. Bowel Dis. 2016, 22, 433–449. [Google Scholar] [CrossRef]
- Langness, S.; Kojima, M.; Coimbra, R.; Eliceiri, B.P.; Costantini, T.W. Enteric glia cells are critical to limiting the intestinal inflammatory response after injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G274–G282. [Google Scholar] [CrossRef] [PubMed]
- Gulbransen, B.D. Enteric Glia. Colloquim Ser. Neurogl. Biol. Med. Physiol. Dis. 2014, 1, 1–72. [Google Scholar] [CrossRef]
- Bach-Ngohou, K.; Mahe, M.M.; Aubert, P.; Abdo, H.; Boni, S.; Bourreille, A.; Denis, M.G.; Lardeux, B.; Neunlist, M.; Masson, D. Enteric glia modulate epithelial cell proliferation and differentiation through 15-deoxy-12,14-prostaglandin J2. J. Physiol. 2010, 588 Pt 14, 2533–2544. [Google Scholar] [CrossRef]
- Gulbransen, B.D.; Sharkey, K.A. Novel functional roles for enteric glia in the gastrointestinal tract. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Espina, B.; Otero, P.; Louzao, M.C.; Alfonso, A.; Botana, L.M. 13-Desmethyl spirolide-c and 13,19-didesmethyl spirolide-c trans-epithelial permeabilities: Human intestinal permeability modelling. Toxicology 2011, 287, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, A.; Scholz, J.; These, A.; Hessel, S.; Preiss-Weigert, A.; Lampen, A. Analysis of the passage of the marine biotoxin okadaic acid through an in vitro human gut barrier. Toxicology 2011, 279, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Louzao, M.C.; Fernandez, D.A.; Abal, P.; Fraga, M.; Vilarino, N.; Vieytes, M.R.; Botana, L.M. Diarrhetic effect of okadaic acid could be related with its neuronal action: Changes in neuropeptide Y. Toxicol. Lett. 2015, 237, 151–160. [Google Scholar] [CrossRef]
- Boesmans, W.; Cirillo, C.; Van den Abbeel, V.; Van den Haute, C.; Depoortere, I.; Tack, J.; Vanden Berghe, P. Neurotransmitters involved in fast excitatory neurotransmission directly activate enteric glial cells. Neurogastroenterol. Motil. 2013, 25, e151–e160. [Google Scholar] [CrossRef] [Green Version]
- Le Berre-Scoul, C.; Chevalier, J.; Oleynikova, E.; Cossais, F.; Talon, S.; Neunlist, M.; Boudin, H. A novel enteric neuron-glia coculture system reveals the role of glia in neuronal development. J. Physiol. 2017, 595, 583–598. [Google Scholar] [CrossRef]
- Espina, B.; Louzao, M.C.; Ares, I.R.; Cagide, E.; Vieytes, M.R.; Vega, F.V.; Rubiolo, J.A.; Miles, C.O.; Suzuki, T.; Yasumoto, T.; et al. Cytoskeletal toxicity of pectenotoxins in hepatic cells. Br. J. Pharmol. 2008, 155, 934–944. [Google Scholar] [CrossRef] [Green Version]
- Vilarino, N. Marine toxins and the cytoskeleton: Azaspiracids. FEBS J. 2008, 275, 6075–6081. [Google Scholar] [CrossRef] [PubMed]
- Diogène, G.; Fessard, V.; Dubreuil, A.; Puisseux-Dao, S. Comparative studies of the actin cytoskeleton response to Maitotoxin and Okadaic Acid. Toxicol. In Vitro 1995, 9, 1–10. [Google Scholar] [CrossRef]
- Valverde, I.; Lago, J.; Vieites, J.M.; Cabado, A.G. In vitro approaches to evaluate palytoxin-induced toxicity and cell death in intestinal cells. J. Appl. Toxicol. 2008, 28, 294–302. [Google Scholar] [CrossRef]
- Rossini, G.P.; Hess, P. Phycotoxins: Chemistry, mechanisms of action and shellfish poisoning. Mol. Clin. Environ. Toxicol. 2010, 100, 65–122. [Google Scholar]
- Vilarino, N.; Nicolaou, K.C.; Frederick, M.O.; Cagide, E.; Alfonso, C.; Alonso, E.; Ares, I.R.; Louzao, M.C.; Vieytes, M.R.; Botana, L.M. Cell Growth Inhibition and Actin Cytoskeleton Disorganization Induced by Azaspiracid-1 Structure-Activity Studies. Chem. Res. Toxicol. 2006, 19, 1456–1466. [Google Scholar] [CrossRef]
- Botana, L.M.; Alfonso, A.; Vale, C.; Vilariño, N.; Rubiolo, J.; Alonso, E.; Cagide, E. The mechanistic complexities of phycotoxins: Toxicology of azaspiracids and yessotoxins. Adv. Mol. Toxicol. 2014, 8, 1–33. [Google Scholar]
- Roman, Y.; Alfonso, A.; Louzao, M.C.; de la Rosa, L.A.; Leira, F.; Vieites, J.M.; Vieytes, M.R.; Ofuji, K.; Satake, M.; Yasumoto, T.; et al. Azaspiracid-1, a potent, nonapoptotic new phycotoxin with several cell targets. Cell. Signal. 2002, 14, 703–716. [Google Scholar] [CrossRef]
- Munday, R.; Quilliam, M.A.; LeBlanc, P.; Lewis, N.; Gallant, P.; Sperker, S.A.; Ewart, H.S.; MacKinnon, S.L. Investigations into the toxicology of spirolides, a group of marine phycotoxins. Toxins 2012, 4, 1–14. [Google Scholar] [CrossRef]
- Ares, I.R.; Louzao, M.C.; Espina, B.; Vieytes, M.R.; Miles, C.O.; Yasumoto, T.; Botana, L.M. Lactone ring of pectenotoxins: A key factor for their activity on cytoskeletal dynamics. Cell. Physiol. Biochem. 2007, 19, 283–292. [Google Scholar] [CrossRef]
- Von Boyen, G.B.T. Proinflammatory cytokines increase glial fibrillary acidic protein expression in enteric glia. Gut 2004, 53, 222–228. [Google Scholar] [CrossRef] [Green Version]
- Rolland, B.; Le Prince, G.; Fages, C.; Nunez, J.; Tardy, M. GFAP turnover during astroglial proliferation and differentiation. Brain Res. Dev. Brain Res. 1990, 56, 144–149. [Google Scholar] [CrossRef]
- Moody, L.; Barrett-Wilt, G.; Sussman, M.; Messing, A. Glial fibrillary acidic protein exhibits altered turnover kinetics in a mouse model of Alexander disease. J. Biol. Chem. 2017, 292, 5814–5824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brahmachari, S.; Fung, Y.K.; Pahan, K. Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J. Neurosci. 2006, 18, 4930–4939. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, C.; Schick, M.A.; Wollborn, J.; Heider, A.; Scholz, C.J.; Cecil, A.; Niesler, B.; Hirrlinger, J.; Walles, H.; Metzger, M. Activation of myenteric glia during acute inflammation in vitro and in vivo. PLoS ONE 2016, 11, e0151335. [Google Scholar] [CrossRef] [PubMed]
- Franchini, A.; Malagoli, D.; Ottaviani, E. Targets and Effects of Yessotoxin, Okadaic Acid and Palytoxin: A Differential Review. Mar. Drugs 2010, 8, 658–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonoda, Y.; Kasahara, T.; Yamaguchi, Y.; Kuno, K.; Matsushima, K.; Mukaida, N. Stimulation of interleukin-8 production by okadaic acid and vanadate in a human promyelocyte cell line, an HL-60 subline. Possible role of mitogen-activated protein kinase on the okadaic acid-induced NF-kappaB activation. J. Biol. Chem. 1997, 272, 15366–15372. [Google Scholar] [CrossRef] [PubMed]
- Rieckmann, P.; Thévenin, C.; Kehrl, J.H. Okadaic acid is a potent inducer of AP-1, NF-kappa B, and tumor necrosis factor-alpha in human B lymphocytes. Biochem. Biophys. Res. Commun. 1992, 187, 51–57. [Google Scholar] [CrossRef]
- Gupta, S.C.; Sundaram, C.; Reuter, S.; Aggarwal, B.B. Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta 2010, 1799, 775–787. [Google Scholar] [CrossRef]
- Nezami, B.G.; Srinivasan, S. Enteric nervous system in the small intestine: Pathophysiology and clinical implications. Curr. Gastroenterol. Rep. 2010, 12, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.O.; Moon, D.O.; Heo, M.S.; Lee, J.D.; Jung, J.H.; Kim, S.K.; Choi, Y.H.; Kim, G.Y. Pectenotoxin-2 abolishes constitutively activated NF-κB, leading to suppression of NF-κB related gene products and potentiation of apoptosis. Cancer Lett. 2008, 271, 25–33. [Google Scholar] [CrossRef]
- Esposito, G.; Capoccia, E.; Turco, F.; Palumbo, I.; Lu, J.; Steardo, A.; Cuomo, R.; Sarnelli, G.; Steardo, L. Palmitoylethanolamide improves colon inflammation through an enteric glia/toll like receptor 4-dependent PPAR-alpha activation. Gut 2014, 63, 1300–1312. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, C.; Sarnelli, G.; Turco, F.; Mango, A.; Grosso, M.; Aprea, G.; Masone, S.; Cuomo, R. Proinflammatory stimuli activates human-derived enteroglial cells and induces autocrine nitric oxide production. Neurogastroenterol. Motil. 2011, 23, e372–e382. [Google Scholar] [CrossRef] [PubMed]
- Grubisic, V.; Gulbransen, B.D. Enteric glia: The most alimentary of all glia. J. Physiol. 2017, 595, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, C.; Sarnelli, G.; Esposito, G.; Turco, F.; Steardo, L.; Cuomo, R. S100B protein in the gut: The evidence for enteroglial-sustained intestinal inflammation. World J. Gastroenterol. 2011, 17, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Donato, R. Functional roles of S100 proteins, calcium-binding proteins of the EF-hand type. Biochim. Et Biophys. Acta 1999, 1450, 191–231. [Google Scholar] [CrossRef] [Green Version]
- Frizzo, K.J.; Tramontina, F.; Bortoli, E.; Gottfried, C.; Leal, R.B.; Lengyel, I.; Donato, R.; Dunkley, P.R.; Gonçalves, C.A. S100B-mediated inhibition of phosphorylation of GFAP is prevented by TRTK-12. Neurochem. Res. 2004, 29, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Donato, R.; Sorci, G.; Riuzzi, F.; Arcuri, C.; Bianchi, R.; Brozzi, F.; Tubaro, C.; Giambanco, I. S100B’s double life: Intracellular regulator and extracellular signal. Biochim. Biophys. Acta 2009, 1793, 1008–1022. [Google Scholar] [CrossRef] [PubMed]
- Chow, A.K.; Gulbransen, B.D. Potential roles of enteric glia in bridging neuroimmune communication in the gut. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G145–G152. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, W.; Chen, W.; Sun, L.; Li, X.; Zhang, C.; Yang, H. GDNF is involved in the barrier-inducing effect of enteric glial cells on intestinal epithelial cells under acute ischemia reperfusion stimulation. Mol. Neurobiol. 2014, 50, 274–289. [Google Scholar] [CrossRef]
- Xiao, W.D.; Chen, W.; Sun, L.H.; Wang, W.S.; Zhou, S.W.; Yang, H. The protective effect of enteric glial cells on intestinal epithelial barrier function is enhanced by inhibiting inducible nitric oxide synthase activity under lipopolysaccharide stimulation. Mol. Cell. Neurosci. 2011, 46, 527–534. [Google Scholar] [CrossRef]
- Razafimanjato, H.; Garmy, N.; Guo, X.J.; Varini, K.; Di Scala, C.; Di Pasquale, E.; Taieb, N.; Maresca, M. The food-associated fungal neurotoxin ochratoxin A inhibits the absorption of glutamate by astrocytes through a decrease in cell surface expression of the excitatory amino-acid transporters GLAST and GLT-1. Neurotoxicology 2010, 31, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Kim, S.F.; Hester, L.; Snyder, S.H. S-nitrosylation/activation of COX-2 mediates NMDA neurotoxicity. PNAS 2008, 105, 10537–10540. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Sekhon, B.; Giri, S.; Jatana, M.; Gilg, A.G.; Ayasolla, K.; Elango, C.; Singh, A.K.; Singh, I. S-Nitrosoglutathione reduces inflammation and protects brain against focal cerebral ischemia in a rat model of experimental stroke. J. Cereb. Blood Flow Metab. 2005, 25, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Franchini, A.; Marchesini, E.; Poletti, R.; Ottaviani, E. Acute toxic effect of the algal yessotoxin on Purkinje cells from the cerebellum of Swiss CD1 mice. Toxicon 2004, 43, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.Y.; Kim, G.Y.; Kim, N.D.; Jung, J.H.; Kim, S.K.; Kang, H.S.; Choi, Y.H. Induction of apoptosis by pectenotoxin-2 is mediated with the induction of DR4/DR5, Egr-1 and NAG-1, activation of caspases and modulation of the Bcl-2 family in 53-deficient Hep3B hepatocellular carcinoma cells. Oncol. Rep. 2008, 19, 517–526. [Google Scholar] [CrossRef]
- Leira, F.; Alvarez, C.; Vieites, J.M.; Vieytes, M.R.; Botana, L.M. Characterization of distinct apoptotic changes induced by okadaic acid and yessotoxin in the BE(2)-M17 neuroblastoma cell line. Toxicol. In Vitro 2002, 16, 23–31. [Google Scholar] [CrossRef]
- Malaguti, C.; Ciminiello, P.; Fattorusso, E.; Rossini, G.P. Caspase activation and death induced by yessotoxin in HeLa cells. Toxicol. In Vitro 2002, 16, 357–363. [Google Scholar] [CrossRef]
- Rossini, G.P.; Sgarbi, N.; Malaguti, C. The toxic responses induced by okadaic acid involve processing of multiple caspase isoformsinvolve caspase. Toxicon 2001, 39, 763–770. [Google Scholar] [CrossRef]
- Kitazumi, I.; Maseki, Y.; Nomura, Y.; Shimanuki, A.; Sugita, Y.; Tsukahara, M. Okadaic acid induces DNA fragmentation via caspase-3-dependent and caspase-3-independent pathways in Chinese hamster ovary (CHO)-K1 cells. FEBS J. 2010, 277, 404–412. [Google Scholar] [CrossRef]
- Cao, Z.; LePage, K.T.; Frederick, M.O.; Nicolaou, K.C.; Murray, T.F. Involvement of caspase activation in azaspiracid-induced neurotoxicity in neocortical neurons. Toxicol. Sci. 2010, 114, 323–334. [Google Scholar] [CrossRef]
- Vilarino, N.; Nicolaou, K.C.; Frederick, M.O.; Vieytes, M.R.; Botana, L.M. Irreversible cytoskeletal disarrangement is independent of caspase activation during in vitro azaspiracid toxicity in human neuroblastoma cells. Biochem. Pharmol. 2007, 74, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Shin, I.K.; Ahn, Y.T.; Kim, Y.; Kim, J.M.; An, W.G. Actin disruption agents induce phosphorylation of histone H2AX in human breast adenocarcinoma MCF-7 cells. Oncol. Rep. 2011, 25, 1313–1319. [Google Scholar] [PubMed]
- Valdiglesias, V.; Méndez, J.; Pásaro, E.; Cemelic, E.; Anderson, D.; Laffon, B. Assessment of okadaic acid effects on cytotoxicity, DNA damage and DNA repair in human cells. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2010, 689, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Korsnes, M.S.; Korsnes, R. Mitotic catastrophe in BC3H1 CELLS following yessotoxin exposure. Front. Cell Dev. Biol. 2017, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.-O.; Kim, M.-O.; Kang, S.-H.; Lee, K.-J.; Heo, M.-S.; Choi, K.-S.; Choi, Y.H.; Kim, G.-Y. Induction of G2/M arrest, endoreduplication, and apoptosis by actin depolymerization agent pextenotoxin-2 in human leukemia cells, involving activation of ERK and JNK. Biochem. Pharmacol. 2008, 76, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.O.; Kim, M.O.; Nam, T.J.; Kim, S.K.; Choi, Y.H.; Kim, G.Y. Pectenotoxin-2 induces G2/M phase cell cycle arrest in human breast cancer cells via ATM and Chk1/2-mediated phosphorylation of cdc25C. Oncol. Rep. 2010, 24, 271–276. [Google Scholar] [PubMed] [Green Version]
- Hori, M.; Yazama, F.; Matsuura, Y.; Yoshimoto, R.; Kaneda, T.; Yasumoto, T.; Ozaki, H.; Karaki, H. Inhibition of actin polymerization by marine toxin pectenotoxin-2. J. Vet. Med. Sci. 2018, 80, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.Y.; Kim, W.J.; Choi, Y.H. Pectenotoxin-2 from marine sponges: A potential anti-cancer agent—A review. Mar. Drugs 2011, 9, 2176–2187. [Google Scholar] [CrossRef] [PubMed]
- Martin-Lopez, A.; Gallardo-Rodriguez, J.J.; Sanchez-Miron, A.; Garcia-Camacho, F.; Molina-Grima, E. Cytotoxicity of yessotoxin and okadaic acid in mouse T lymphocyte cell line EL-4. Toxicon 2012, 60, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, J.; Rockwell, P. Okadaic acid induces Akt hyperphosphorylation and an oxidative stress-mediated cell death in serum starved SK-N-SH human neuroblastoma cells that are augmented by rapamycin. Neurosci. Lett. 2012, 531, 74–79. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, K.N.; Traenckner, E.B.M.; Meier, B.; Baeuerle, P.A. Induction of oxidative stress by okadaic acid is required for activation of transcription factor NF-κB. J. Biol. Chem. 1995, 270, 27136–27142. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, J.; Gupta, N.; Agrawal, M.; Bala Bhaskar, A.S.; Lakshmana Rao, P.V. Modulation of ROS/MAPK signaling pathways by okadaic acid leads to cell death via, mitochondrial mediated caspase-dependent mechanism. Apoptosis 2011, 16, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Vale, C.; Nicolaou, K.C.; Frederick, M.O.; Vieytes, M.R.; Botana, L.M. Cell volume decrease as a link between azaspiracid-induced cytotoxicity and c-Jun-N-terminal kinase activation in cultured neurons. Toxicol. Sci. 2010, 113, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Hjornevik, L.V.; Froyset, A.K.; Gronset, T.A.; Rungruangsak-Torrissen, K.; Fladmark, K.E. Algal toxin azaspiracid-1 induces early neuronal differentiation and alters peripherin isoform stoichiometry. Mar. Drugs 2015, 13, 7390–7402. [Google Scholar] [CrossRef] [PubMed]
- Meir, M.; Flemming, S.; Burkard, N.; Bergauer, L.; Metzger, M.; Germer, C.T.; Schlegel, N. Glial cell line-derived neurotrophic factor promotes barrier maturation and wound healing in intestinal epithelial cells in vitro. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G613–G624. [Google Scholar] [CrossRef] [Green Version]
- Von Boyen, G.B.; Steinkamp, M.; Geerling, I.; Reinshagen, M.; Schafer, K.H.; Adler, G.; Kirsch, J. Proinflammatory cytokines induce neurotrophic factor expression in enteric glia: A key to the regulation of epithelial apoptosis in Crohn’s disease. Inflamm. Bowel Dis. 2006, 12, 346–354. [Google Scholar] [CrossRef]
- Steinkamp, M.; Gundel, H.; Schulte, N.; Spaniol, U.; Pflueger, C.; Zizer, E.; von Boyen, G.B. GDNF protects enteric glia from apoptosis: Evidence for an autocrine loop. BMC Gastroenterol. 2012, 12, 6. [Google Scholar] [CrossRef]
- Steinkamp, M.; Schulte, N.; Spaniol, U.; Pflueger, C.; Hartmann, C.; Kirsch, J.; von Boyen, G.B. Brain derived neurotrophic factor inhibits apoptosis in enteric glia during gut inflammation. Med. Sci. Monit. 2012, 18, 117–122. [Google Scholar]
- Neunlist, M.; Rolli-Derkinderen, M.; Latorre, R.; Van Landeghem, L.; Coron, E.; Derkinderen, P.; De Giorgio, R. Enteric glial cells: Recent developments and future directions. Gastroenterology 2014, 147, 1230–1237. [Google Scholar] [CrossRef]
- Yoo, B.B.; Mazmanian, S.K. The Enteric Network: Interactions between the Immune and Nervous Systems of the Gut. Immunity 2017, 46, 910–926. [Google Scholar] [CrossRef] [Green Version]
- Pelin, M.; Sosa, S.; Pacor, S.; Tubaro, A.; Florio, C. The marine toxin palytoxin induces necrotic death in HaCaT cells through a rapid mitochondrial damage. Toxicol. Lett. 2014, 229, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Pelin, M.; Ponti, C.; Sosa, S.; Gibellini, D.; Florio, C.; Tubaro, A. Oxidative stress induced by palytoxin in human keratinocytes is mediated by a H+-dependent mitochondrial pathway. Toxicol. Appl. Pharmol. 2013, 266, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Z. Neurotoxins from marine dinoflagellates: A brief review. Mar. Drugs 2008, 6, 349–371. [Google Scholar] [CrossRef] [PubMed]
- Delvalle, N.M.; Fried, D.E.; Rivera-Lopez, G.; Gaudette, L.; Gulbransen, B.D. Cholinergic activation of enteric glia is a physiological mechanism that contributes to the regulation of gastrointestinal motility. Gastrointest. Liver Physiol. 2018, 315, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Wandscheer, C.B.; Vilarino, N.; Espina, B.; Louzao, M.C.; Botana, L.M. Human muscarinic acetylcholine receptors are a target of the marine toxin 13-desmethyl C spirolide. Chem. Res. Toxicol. 2010, 23, 1753–1761. [Google Scholar] [CrossRef]
- Araoz, R.; Ouanounou, G.; Iorga, B.I.; Goudet, A.; Alili, D.; Amar, M.; Benoit, E.; Molgo, J.; Servent, D. The neurotoxic effect of 13,19-didesmethyl and 13-desmethyl spirolide C phycotoxins is mainly mediated by nicotinic rather than muscarinic acetylcholine receptors. Toxicol. Sci. 2015, 147, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.; Benford, D.; Boobis, A.; Ceccatelli, S.; Cravedi, J.P.; Di Domenico, A.; Doerge, D.; Dogliotti, E.; Edler, L.; Farmer, P.; et al. Marine biotoxins in shellfish—Summary on regulated marine biotoxins, Scientific opinion of the panel on contaminants in the food chain. EFSA J. 2009, 1306, 1–23. [Google Scholar]
- Ferron, P.J.; Hogeveen, K.; De Sousa, G.; Rahmani, R.; Dubreil, E.; Fessard, V.; Le Hegarat, L. Modulation of CYP3A4 activity alters the cytotoxicity of lipophilic phycotoxins in human hepatic HepaRG cells. Toxicol. In Vitro 2016, 33, 136–146. [Google Scholar] [CrossRef]
- Huguet, A.; Hatton, A.; Villot, R.; Quenault, H.; Blanchard, Y.; Fessard, V. Modulation of chromatin remodelling induced by the freshwater cyanotoxin cylindrospermopsin in human intestinal Caco-2 cells. PLoS ONE 2014, 9, e99121. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Biological Function | PTX2 (nM) | YTX (nM) | AZA1 (nM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | 1 | 2 | 4 | 1 | 2 | 4 | 0.38 | 0.75 | 1.5 | |||
| Viability | BNIP3 | ** | * | |||||||||
| CASP3 | *** | |||||||||||
| GABARAP | * | |||||||||||
| FOS | * | * | * | * | ||||||||
| BCL2 | * | |||||||||||
| Morphology | GFAP | a | a | a | ||||||||
| Cell cycle | CDK1 | * | *** | *** | * | ** | ||||||
| CDK2 | * | ** | ** | ** | ||||||||
| CCNA2 | ** | *** | *** | * | ||||||||
| Inflammation | IL1R1 | ** | ||||||||||
| MAPK8 | * | * | ||||||||||
| CCL2 | * | * | * | |||||||||
| MYD88 | ** | |||||||||||
| RHOA | * | |||||||||||
| TLR4 | * | |||||||||||
| FZD4 | * | |||||||||||
| Oxidative stress | CAT | |||||||||||
| NFE2L2 | ** | |||||||||||
| Gliomediator | BDNF | |||||||||||
| GDNF | * | |||||||||||
| Channel and receptor | GJA1 | ** | ||||||||||
| GFRA1 | * | * | * | * | * | * | ||||||
| LPA1 | * | |||||||||||
| Toxins | MORPHOLOGY | VIABILITY | CELL CYCLE | OXIDATIVE STRESS | INFLAMMATION | GLIOMEDIATORS | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Microscopy | GFAP | IC50 (nM) | Caspase-3 | γH2AX | Phases | Oxidative marker | NF-κB | S100β | iNOS | Gliomediator genes | |
| PTX2 | neurites alteration | ++ | n.e. | ++ | ++ | subG1 ↗ G2/M ↗ | +++ (24 h) | n.e. (3 h) | ++ | ++ | BDNF ↗ GDNF ↗ |
| YTX | neurites alteration | ++ | 14.5 | ++ | n.e. | subG1 ↗ G2/M ↘ | ++ (24 h) | + (8 h) | ++ | ++ | GDNF ↗ |
| OA | cell rounding | + | 75.9 | + | n.e. | subG1 ↗ | + (24 h) | ++ (8 h) | + | + | n.t. |
| AZA1 | neurites alteration | ++ | 7.0 | ++ | n.e. | subG1 ↗ G2/M ↘ | + (24 h) | ++ (8 h) | ++ | ++ | n.e. |
| SPX | n.e | n.e. | n.e. | n.e. | n.e. | n.e. | n.e. | n.e. (3 h) | n.e. | n.e. | n.t. |
| PlTX | blebbing | - | 0.4 | n.e. | n.e. | n.e. | ++ (4 h) | n.e. (3 h) | n.e. | n.e. | n.t. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reale, O.; Huguet, A.; Fessard, V. Novel Insights on the Toxicity of Phycotoxins on the Gut through the Targeting of Enteric Glial Cells. Mar. Drugs 2019, 17, 429. https://doi.org/10.3390/md17070429
Reale O, Huguet A, Fessard V. Novel Insights on the Toxicity of Phycotoxins on the Gut through the Targeting of Enteric Glial Cells. Marine Drugs. 2019; 17(7):429. https://doi.org/10.3390/md17070429
Chicago/Turabian StyleReale, Océane, Antoine Huguet, and Valérie Fessard. 2019. "Novel Insights on the Toxicity of Phycotoxins on the Gut through the Targeting of Enteric Glial Cells" Marine Drugs 17, no. 7: 429. https://doi.org/10.3390/md17070429
APA StyleReale, O., Huguet, A., & Fessard, V. (2019). Novel Insights on the Toxicity of Phycotoxins on the Gut through the Targeting of Enteric Glial Cells. Marine Drugs, 17(7), 429. https://doi.org/10.3390/md17070429