Antimicrobial Lavandulylated Flavonoids from a Sponge-Derived Streptomyces sp. G248 in East Vietnam Sea

 ,
,
Abstract
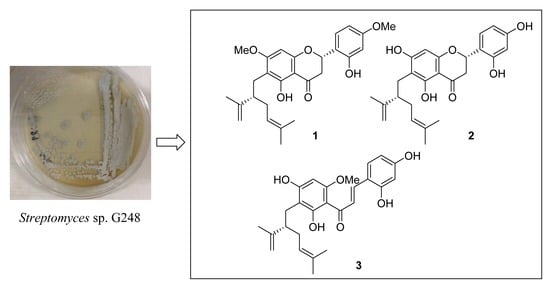
1. Introduction
2. Results and Discussion
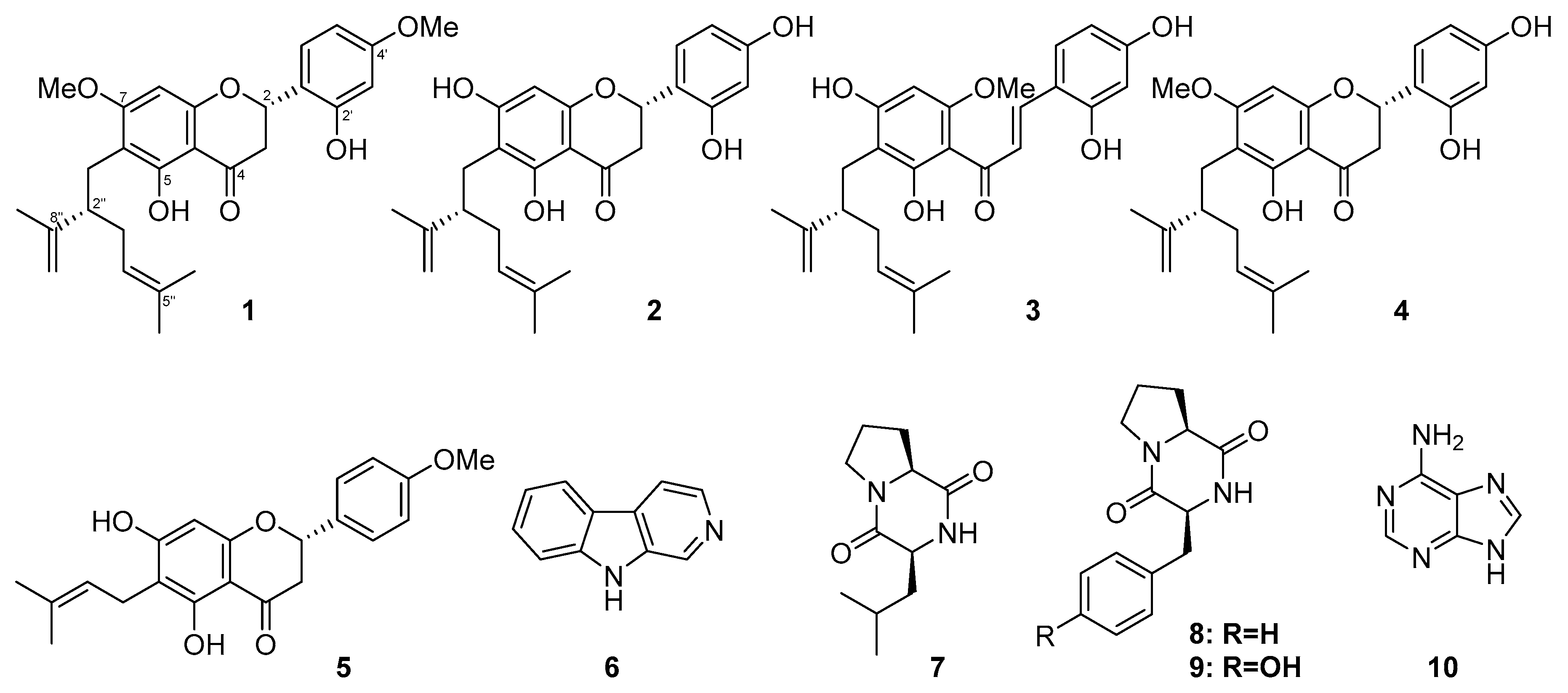
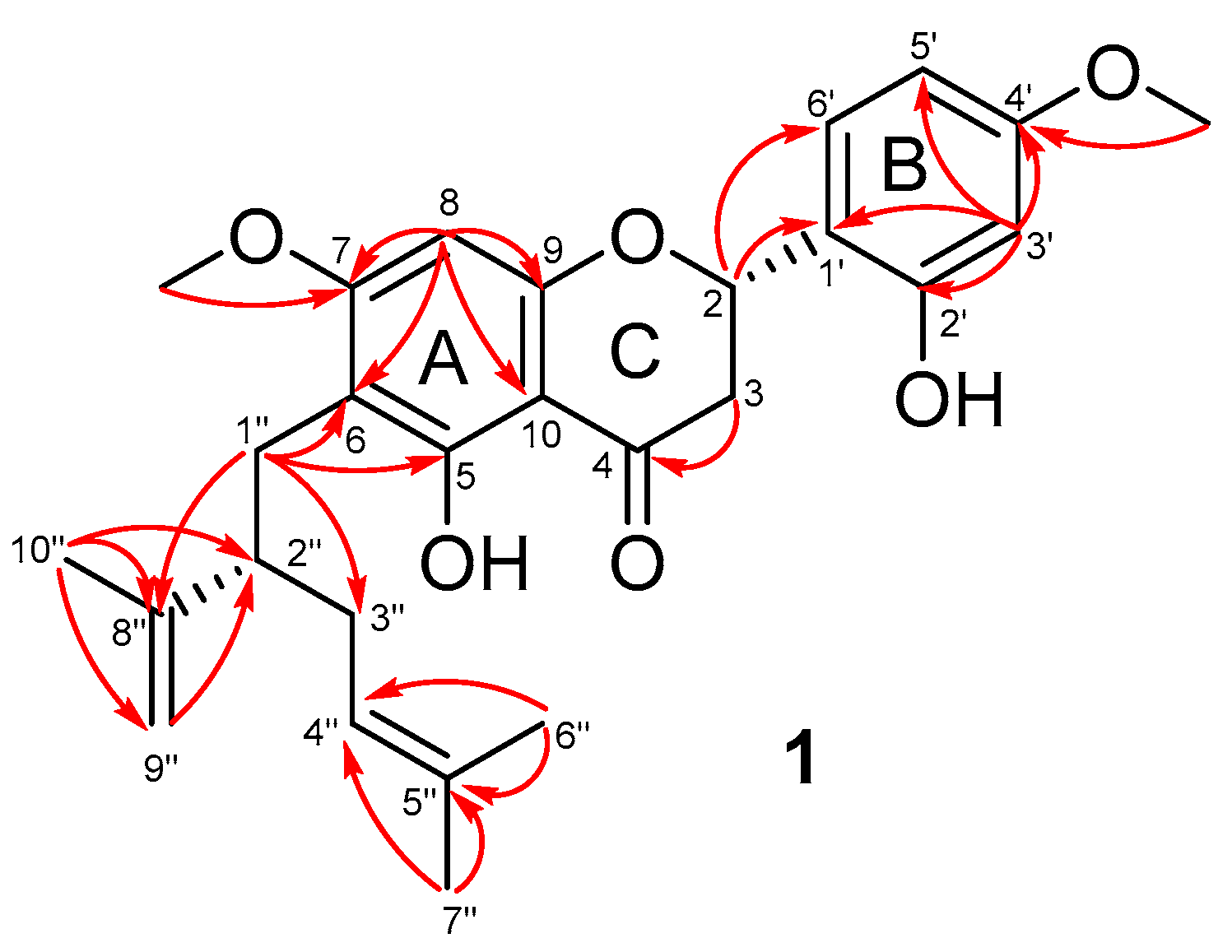
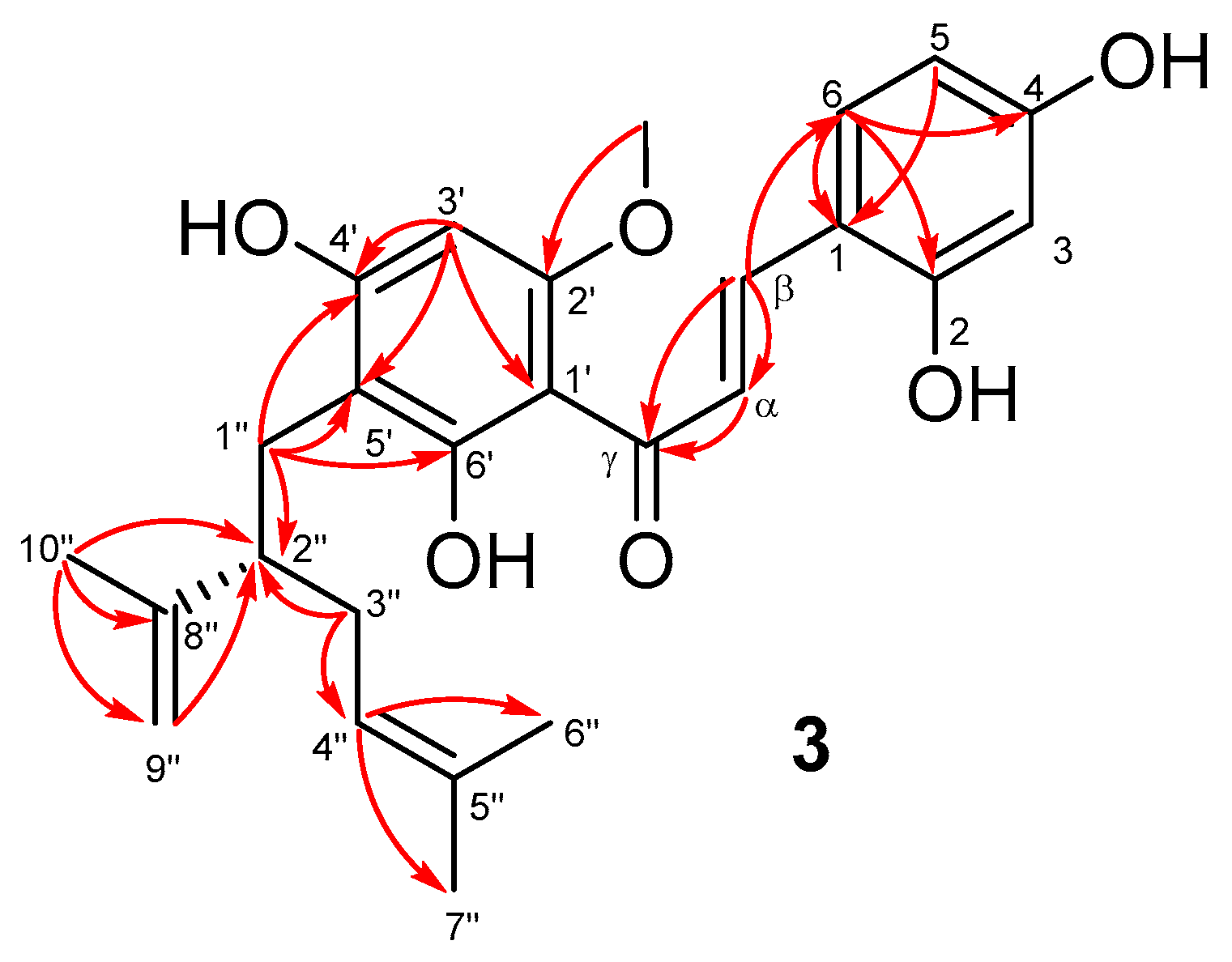
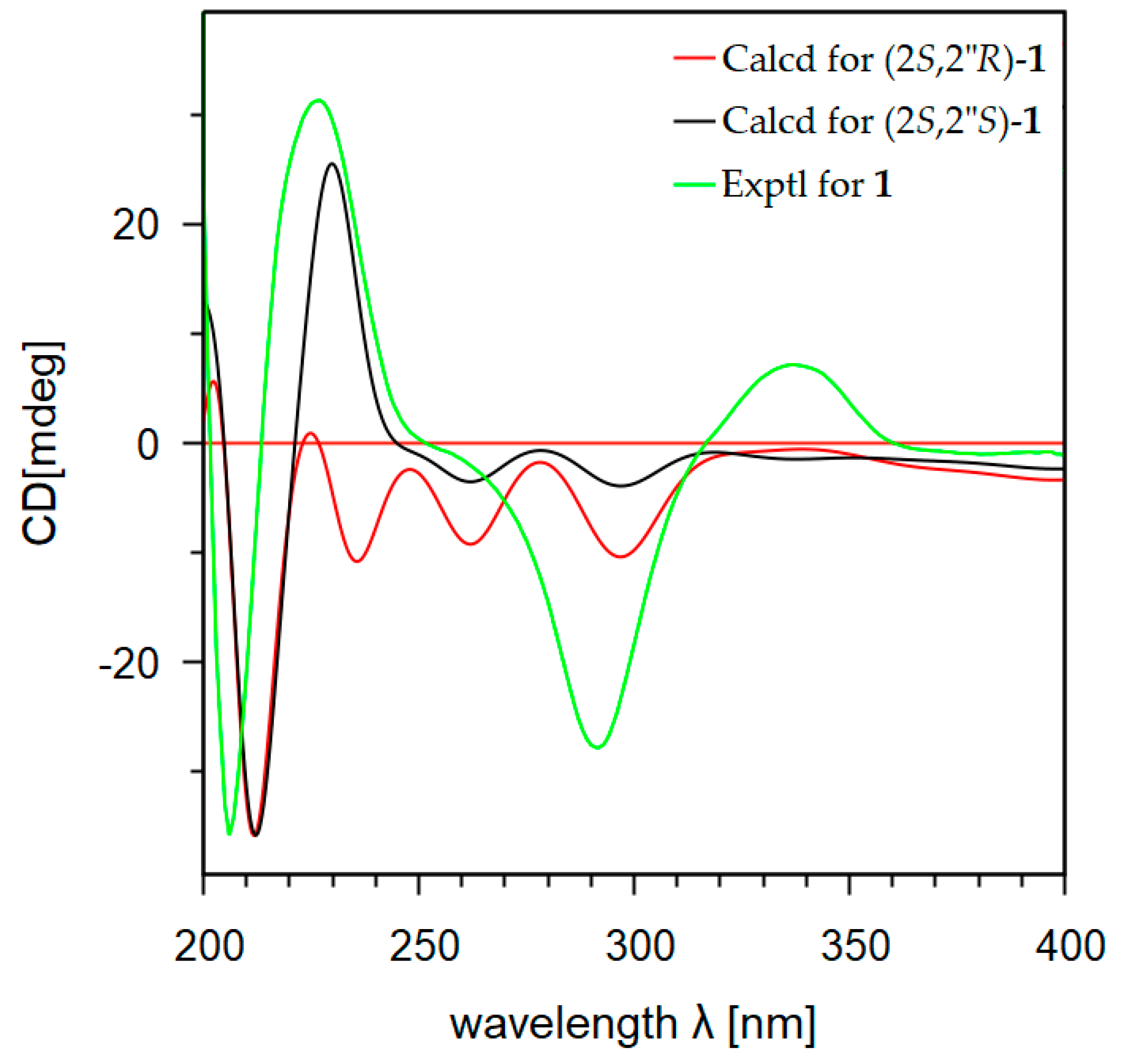
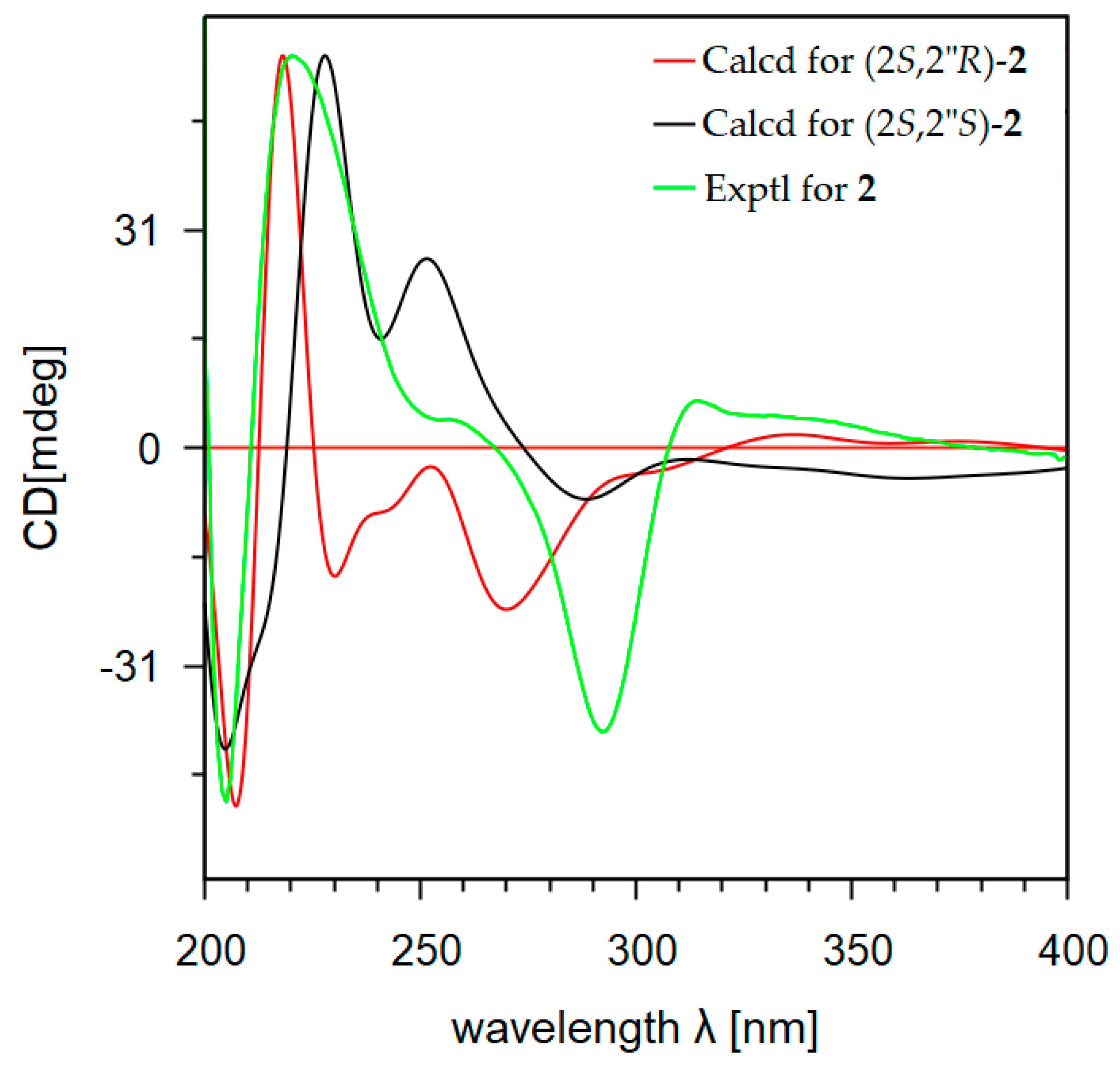
2.1. Isolation and Structural Elucidation
2.2. Biological Activity
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Actinomycete Material
3.3. Fermentation and Extraction
3.4. Isolation and Purification
3.5. Spectral Data
3.6. Antimicrobial Activity Assay
3.7. Anti-Mycobacterial Activity Assay
3.8. Cytotocicity Assay
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Laport, M.S.; Santos, O.C.S.; Muricy, G. Marine sponges: Potential sources of new antimicrobial drugs. Curr. Pharm. Biotechnol. 2009, 10, 86–105. [Google Scholar] [CrossRef] [PubMed]
- El-Demerdash, A.; Tammam, M.A.; Atanasov, A.G.; Hooper, J.N.A.; Al-Mourabit, A.; Kijjoa, A. Chemistry and biological activities of the marine sponges of the genera Mycale (Arenochalina), Biemna and Clathria. Mar. Drugs 2018, 16, 214. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.R.A.; Kavlekar, D.P.; LokaBharathi, P.A. Marine drugs from sponge-microbe association—A review. Mar. Drugs 2010, 8, 1417–1468. [Google Scholar] [CrossRef] [PubMed]
- Hardoim, C.C.P.; Costa, R. Microbial communities and bioactive compounds in marine sponges of the family Irciniidae—A review. Mar. Drugs 2014, 12, 5089–5122. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Mangwani, N. Ocean acidification and marine microorganisms: Responses and consequences. Oceanologia 2015, 57, 349–361. [Google Scholar] [CrossRef]
- Brinkmann, C.M.; Marker, A.; Kurtböke, D.I. An overview on marine sponge-symbiotic bacteria as unexhausted sources for natural product discovery. Diversity 2017, 9, 40. [Google Scholar] [CrossRef]
- Romano, S.; Jackson, S.A.; Patry, S.; Dobson, A.D.W. Extending the “one strain many compounds” (OSMAC) principle to marine microorganisms. Mar. Drugs 2018, 16, 244. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Othman, E.M.; Kampik, D.; Stopper, H.; Hentschel, U.; Ziebuhr, W.; Oelschlaeger, T.A.; Abdelmohsen, U.R. Marine sponge-derived Streptomyces sp. SBT343 extract inhibits staphylococcal biofilm formation. Front. Microbiol. 2017, 8, 236. [Google Scholar] [CrossRef]
- Watve, M.G.; Tickoo, R.; Jog, M.M.; Bhole, B.D. How many antibiotics are produced by the genus Streptomyces. Arch. Microbiol. 2001, 176, 386–390. [Google Scholar] [CrossRef]
- Subramani, R.; Aalbersberg, W. Marine actinomycetes: An ongoing source of novel bioactive metabolites. Microbiol. Res. 2012, 167, 571–580. [Google Scholar] [CrossRef]
- Assia, M.; Hasnaa, A.; Sara, M.; Jamal, M.; Menggad, M. Physico-chemical characterization of a pink red-like pigments produced by five new bacterial soil strains identified as Streptomyces coelicoflavus. Am. J. Microbiol. Res. 2018, 6, 67–72. [Google Scholar] [CrossRef]
- Igarashi, Y.; Rakicidin, D. An inhibitor of tumor cell invasion from marine-derived Streptomyces sp. J. Antibiot. 2010, 63, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento-Vizcaino, A.; Espadas, J.; Martin, J.; Brana, A.F.; Reyes, F.; Garcia, L.A.; Blanco, G. Atmospheric precipitations, hailstone and rainwater, as a novel source of Streptomyces producing bioactive natural products. Front. Microbiol. 2018, 9, 773. [Google Scholar] [CrossRef] [PubMed]
- Ha, T.J.; Yang, M.S.; Jang, D.S.; Choi, S.U.; Park, K.H. Inhibitory activities of flavanone derivatives isolated from Sophora flavescens for melanogenesis. Bull. Korean Chem. Soc. 2001, 22, 97–99. [Google Scholar]
- Jung, M.J.; Kang, S.S.; Jung, H.A.; Kim, G.J.; Choi, J.S. Isolation of flavonoids and a cerebroside from the stem bark of Albizzia julibrissin. Arch. Pharmacal Res. 2004, 27, 593–599. [Google Scholar] [CrossRef]
- Berova, N.; Di-Bari, L.; Pescitelli, G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem. Soc. Rev. 2007, 36, 914–931. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, G.; Bruhn, T.; Maksimenka, K.; Hemberger, Y. The assignment of absolute stereostructures through quantum chemical circular dichroism calculations. Eur. J. Org. Chem. 2009, 2717–2727. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision C.01, Gaussian: Wallingford, CT, USA, 2009.
- Gaffield, W. Circular dichroism, optical rotatory dispersion and absolute configuration of flavanones, 3-hydroxyflavanones and their glycosides: Determination of aglycone chirality in flavanone glycosides. Tetrahedron 1970, 26, 4093–4108. [Google Scholar] [CrossRef]
- Cao, D.D.; Do, T.Q.; Doan, T.M.H.; Vu, T.Q.; Le, T.H.M.; Tran, D.T.; Chau, V.M.; Do, C.T.; Pham, V.C. Antimicrobial lavandulylated flavonoids from a sponge-derived actinomycete. Nat. Prod. Res. 2018. [Google Scholar] [CrossRef]
- Parsons, I.C.; Gray, A.I.; Waterman, P.G. New triterpenes and flavonoids from the leaves of Bosistoa brasii. J. Nat. Prod. 1993, 56, 46–53. [Google Scholar] [CrossRef]
- Yomosa, K.; Hirota, A.; Sakai, H.; Isogai, A. Isolation of harman and norharman from Nocardia sp. and their inhibitory activity against plant seedlings. Agric. Biol. Chem. 1987, 51, 921–922. [Google Scholar] [CrossRef][Green Version]
- Rosa, E.B. 13C NMR spectra of substituted carbazoles and azacarbazoles (β-carbolines). Magn. Reson. Chem. 1988, 26, 1109–1113. [Google Scholar]
- Fdhila, F.; Vazquez, V.; Sanchez, J.L.; Riguera, R. Diketopiperazines. Antibiotics active against Vibrio anguillarum isolated from marine bacteria associated with cultures of Pecten maximus. J. Nat. Prod. 2003, 66, 1299–1301. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Dai, S.; Chen, M.; Wu, H.; Xie, L.; Luo, X.; Li, X. Two diketopiperazine cyclo(Pro-Phe) isomers from marine bacterium Bacillus subtilis sp. 13–2. Chem. Nat. Compd. 2010, 46, 583–585. [Google Scholar] [CrossRef]
- Wang, L.; Zheng, C.D.; Li, X.J.; Gao, J.M.; Zhang, X.C.; Wei, G.H. Cyclo(Pro-Tyr) from an endophytic rhizobium isolated from Glycyrrhiza uralensis. Chem. Nat. Compd. 2012, 47, 1040–1042. [Google Scholar] [CrossRef]
- Komin, S.; Gossens, C.; Tavernelli, I.; Rothlisberger, U.; Sebastiani, D. NMR solvent shifts of adenine in aqueous solution from hybrid QM/MM molecular dynamics simulations. J. Phys. Chem. B 2007, 111, 5225–5232. [Google Scholar] [CrossRef] [PubMed]
- Andrews, J.M. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 2001, 48, 5–16. [Google Scholar] [CrossRef]
- Cho, S.; Lee, H.S.; Franzblau, S. Microplate alamar blue assay (MABA) and low oxygen recovery assay (LORA) for Mycobacterium tuberculosis. Methods Mol. Biol. 2015, 1285, 281–292. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | ||||
|---|---|---|---|---|---|---|
| N | δC | δH mult. (J in Hz) | δC | δH mult. (J in Hz) | δC | δH mult. (J in Hz) |
| 1 | - | - | - | - | 116.3 | - |
| 2 | 75.0 | 5.55 dd (2.5, 13.5) | 75.8 | 5.58 dd (3.0, 13.0) | 160.3 | - |
| 3 | 45.4 | 2.89 dd (13.5, 17.0) 2.67 dd (2.5, 17.0) | 43.3 | 2.99 dd (13.0, 17.0) 2.75 dd (3.0, 17.0) | 103.7 | 6.35 br.s |
| 4 | 193.6 | - | 198.9 | - | 162.4 | - |
| 5 | 164.7 | - | 162.6 | - | 109.0 | 6.36 dd (2.0, 8.0) |
| 6 | 109.6 | - | 108.7 | - | 131.6 | 7.41 d (8.0) |
| 7 | 165.4 | - | 167.0 | - | - | - |
| 8 | 93.6 | 6.12 s | 96.4 | 5.93 s | - | - |
| 9 | 161.9 | - | 163.2 | - | - | - |
| 10 | 105.7 | - | 103.2 | - | - | - |
| 1′ | 119.7 | - | 118.4 | - | 106.5 | - |
| 2′ | 160.1 | - | 156.7 | - | 162.3 | - |
| 3′ | 99.8 | 6.49 d (2.3) | 103.4 | 6.36 d (2.5) | 91.6 | 6.02 s |
| 4′ | 159.0 | - | 159.6 | - | 164.1 | - |
| 5′ | 108.1 | 6.47 dd (2.3, 8.5) | 107.7 | 6.37 dd (2.0, 8.5) | 108.9 | - |
| 6′ | 128.5 | 7.38 d (8.5) | 128.6 | 7.32 d (8.5) | 166.6 | - |
| 1″ | 28.2 | 2.63 m | 28.0 | 2.60 m | 28.2 | 2.65 m |
| 2″ | 48.2 | 2.50 m | 48.4 | 2.49 m | 48.0 | 2.57 m |
| 3″ | 32.3 | 2.02 m | 32.3 | 2.01 m | 32.4 | 2.09 m |
| 4″ | 124.8 | 4.96 t (5.5) | 124.8 | 4.99 t (5.5) | 125.0 | 5.06 td (1.0, 6.5) |
| 5″ | 132.0 | - | 132.0 | - | 131.8 | - |
| 6″ | 17.8 | 1.48 s | 17.8 | 1.50 s | 17.9 | 1.58 s |
| 7″ | 25.8 | 1.57 s | 25.8 | 1.59 s | 25.9 | 1.65 s |
| 8″ | 149.8 | - | 149.8 | - | 149.9 | - |
| 9″ | 111.2 | 4.52 s 4.59 s | 111.1 | 4.54 br s 4.60 br s | 111.1 | 4.61 br s 4.55 br s |
| 10″ | 19.1 | 1.64 s | 19.2 | 1.65 s | 19.1 | 1.72 s |
| OMe | 55.9 | 3.82 s | - | - | - | - |
| OMe | 55.9 | 3.83 s | - | - | 56.1 | 3.91 |
| α | - | - | - | - | 125.4 | 7.95 d (16.0) |
| β | - | - | - | - | 139.8 | 8.02 d (16.0) |
| γ | - | - | - | - | 194.8 | - |
| Compd. | E. faecalis | S. aureus | B. cereus | E. coli | P. aeruginosa | S. enterica | C. albicans | M. tuberculosis |
|---|---|---|---|---|---|---|---|---|
| 1 | 8 | 8 | 8 | >256 | 8 | 8 | 16 | 48.0 |
| 2 | 1 | 1 | 1 | >256 | 1 | 8 | 1 | >50 |
| 3 | 8 | 8 | 8 | 4 | 8 | 8 | 16 | >50 |
| 4 | 32 | 32 | 16 | 128 | 32 | 32 | 32 | 6.0 |
| 5 | >256 | >256 | >256 | >256 | >256 | >256 | >256 | 11.1 |
| 6 | >256 | >256 | >256 | 128 | >256 | >256 | >256 | >50 |
| 7 | >256 | >256 | >256 | >256 | >256 | >256 | >256 | >50 |
| 8 | >256 | >256 | >256 | >256 | >256 | >256 | >256 | >50 |
| 9 | >256 | >256 | >256 | >256 | >256 | >256 | >256 | >50 |
| 10 | 32 | nt | nt | 128 | nt | nt | 64 | >50 |
| Strep | 256 | 256 | 128 | 32 | 256 | 128 | nt | nt |
| Cyclohex | nt | nt | nt | nt | nt | nt | 32 | nt |
| Compd | KB | Hep-G2 | Lu-1 | MCF-7 |
|---|---|---|---|---|
| 1 | 59.7 | 32.0 | 80.0 | 71.7 |
| 2 | 118.4 | >128 | >128 | >128 |
| 3 | >128 | >128 | >128 | >128 |
| 4 | 4.8 | 2.27 | 4.0 | 14.5 |
| 5 | 2.0 | 2.0 | 4.8 | 11.8 |
| 6 | 47.3 | 78.4 | 71.5 | 60.0 |
| 7 | >128 | 56.0 | >128 | >128 |
| 8 | >128 | >128 | >128 | >128 |
| 9 | >128 | >128 | >128 | >128 |
| Ellipticine | 0.3 ± 0.05 | 0.3 ± 0.05 | 0.4 ± 0.05 | 0.5 ± 0.05 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, D.D.; Trinh, T.T.V.; Doan Thi Mai, H.; Vu, V.N.; Le, H.M.; Vu Thi, Q.; Nguyen, M.A.; Duong, T.T.; Tran, D.T.; Chau, V.M.; et al. Antimicrobial Lavandulylated Flavonoids from a Sponge-Derived Streptomyces sp. G248 in East Vietnam Sea. Mar. Drugs 2019, 17, 529. https://doi.org/10.3390/md17090529
Cao DD, Trinh TTV, Doan Thi Mai H, Vu VN, Le HM, Vu Thi Q, Nguyen MA, Duong TT, Tran DT, Chau VM, et al. Antimicrobial Lavandulylated Flavonoids from a Sponge-Derived Streptomyces sp. G248 in East Vietnam Sea. Marine Drugs. 2019; 17(9):529. https://doi.org/10.3390/md17090529
Chicago/Turabian StyleCao, Duc Danh, Thi Thanh Van Trinh, Huong Doan Thi Mai, Van Nam Vu, Hong Minh Le, Quyen Vu Thi, Mai Anh Nguyen, Thu Trang Duong, Dang Thach Tran, Van Minh Chau, and et al. 2019. "Antimicrobial Lavandulylated Flavonoids from a Sponge-Derived Streptomyces sp. G248 in East Vietnam Sea" Marine Drugs 17, no. 9: 529. https://doi.org/10.3390/md17090529
APA StyleCao, D. D., Trinh, T. T. V., Doan Thi Mai, H., Vu, V. N., Le, H. M., Vu Thi, Q., Nguyen, M. A., Duong, T. T., Tran, D. T., Chau, V. M., Ma, R., Shetye, G., Cho, S., Murphy, B. T., & Pham, V. C. (2019). Antimicrobial Lavandulylated Flavonoids from a Sponge-Derived Streptomyces sp. G248 in East Vietnam Sea. Marine Drugs, 17(9), 529. https://doi.org/10.3390/md17090529