Abstract
Chemical investigation of secondary metabolites from the endophytic fungus Pseudopestalotiopsis theae led to the isolation of eighteen new polyketide derivatives, pestalotheols I–Q (1–9) and cytosporins O–W (15–23), together with eight known analogs (10–14 and 24–26). The structures of the new compounds were elucidated by HRMS and 1D and 2D NMR data, as well as by comparison with literature data. Modified Mosher’s method was applied to determine the absolute configuration of some compounds. Compound 23 showed significant cytotoxicity against the mouse lymphoma cell line L5178Y with an IC50 value of 3.0 μM. Furthermore, compounds 22 and 23 showed moderate antibacterial activity against drug-resistant Acinetobacter baumannii (ATCC BAA-1605) in combination with sublethal colistin concentrations.
1. Introduction
Since the discovery of antibacterial penicillin from Penicillium notatum, there have been tremendous achievements with regard to the development of fungal drugs [1]. The antifungal medicine griseofulvin, which was initially isolated from Penicillium griseofulvum, has been applied for the treatment of ringworm in skin and nails in animals and humans [2]. Lovastatin, the first commercial statin isolated from a fermentation broth of Aspergillus terreus, has been used to treat high blood cholesterol and reduce the risk of cardiovascular disease [3]. The immunosuppressant agent cyclosporine was isolated from the fungus Tolypocladium inflatum [4]. Endophytic fungi occur in all plants investigated up to now and are important for the fitness and survival of their hosts [5]. The chemically remarkably creative fungal genus Pestalotiopsis contains around 234 described fungal species listed in Index Fungorum, including 44 taxol producers [6,7]. Pestalotiopsis has attracted considerable attention due to the abundance and diversity of secondary products found in members of this genus [8,9]. Pestalachloride A, a new chlorinated benzophenone alkaloid produced by Pestalotiopsis adusta, showed potent antifungal activity against Fusarium culmorum with an IC50 value of 0.89 μM [10]. The first chlorinated pupukeanane derivative, chloropupukeananin, which was isolated from fermentation broth of Pestalotiopsis fici, inhibited HIV-1 replication in C8166 cells with an IC50 value of 14.6 μM [11]. Pestaloquinols A and B, two unique isoprenylated epoxyquinol derivatives obtained from rice cultures of Pestalotiopsis sp., both exhibited cytotoxicity against HeLa cell line with IC50 values of 8.8 μM [12]. Pestalotines A and B, two novel phytotoxic γ-lactonic dimers from Pestalotiopsis sp., showed potent phytotoxicity against the radical growth of Echinochloa crusgalli with IC50 values of 0.19 and 0.25 μM, respectively [13].
In previous studies from our group, cytotoxic 14-membered macrolides were obtained from the mangrove-derived fungus Pestalotiopsis microspora with IC50 values ranging from 0.7 to 5.6 μM [14]. In the present study, the endophytic fungus Pseudopestalotiopsis theae, which was previously reported as Pestalotiopsis theae and taxonomically revised in 2014 [7] was isolated from roots of the mangrove plant Rhizophora racemosa collected around Lagos, Nigeria. Fermentation of this fungus on solid rice medium yielded eighteen new polyketides, namely pestalotheols I–Q (1–9) (Supplementary information) and cytosporins O–W (15–23), in addition to eight known congeners (Figure 1). In this paper, the isolation, structure elucidation, and bioactivity of all isolated compounds are reported.
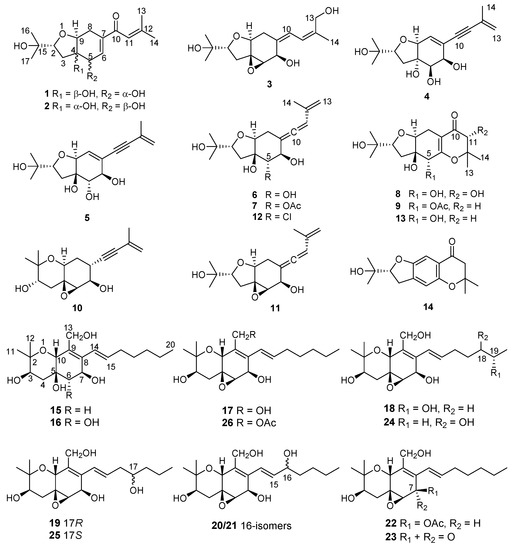
Figure 1.
Structures of compounds 1–26 isolated from Pseudopestalotiopsis theae.
2. Results and Discussion
Compound 1 was obtained as yellowish gel. The molecular formula of 1 was determined as C16H24O5 by HRESIMS data, indicating five degrees of unsaturation. The 13C NMR data (Table 1) for 1 revealed the presence of a carbonyl at δC 191.9 (C-10), four olefinic carbons at δC 153.7 (C-12), 138.7 (C-7), 138.0 (C-6), and 120.7 (C-11), five oxygenated carbons at δC 84.2 (C-2), 77.8 (C-4), 76.3 (C-9), 70.8 (C-15), and 68.9 (C-5), two methylene groups at δC 34.0 (C-3) and 25.7 (C-8), together with four methyls at δC 27.2 (C-14), 25.9 (C-17), 25.8 (C-16), and 20.5 (C-13), accounting for three degrees of unsaturation. Thus, compound 1 was suggested to be bicyclic in nature. The COSY correlations between H-2/H-3ab, OH-5/H-5/H-6, H-8/H-9, together with the HMBC correlations from H-2 to H-9, from OH-4 to C-3, C-4, C-5, and C-9, from H-5 to C-4 and C-9, from H-6 to C-4 and C-8, and from H-8ab to C-4, C-6, C-7, and C-9, established the presence of a tetrahydrofuranocyclohexene moiety with two hydroxy groups at C-4 and C-5. Additional HMBC correlations from Me-14 to C-11, C-12, and C-13, and from H-11, H-6 and H-8a to C-10 indicated the attachment of a 3-methyl-1-oxobut-2-en-1-yl side chain at C-7. Moreover, the location of a 2-hydroxyisopropyl subunit at C-2 was confirmed by the HMBC correlations from Me-16(17) to C-2, C-15 and C-17(16), and from OH-15 to C-2, C-15, C-16, and C-17. Thus, the planar structure of 1 was established as shown. The relative configuration of 1 was elucidated by coupling constants and NOE correlations (Figure 2). The large values of JH-2/H-3a (9.9 Hz) and JH-9/H-8b (10.7 Hz) suggested trans-diaxial orientation of these protons. The NOE correlations from H-9 to Me-16, H-3a, OH-5, and H-8a, and between H-3a and OH-5 indicated these protons to be on the same side, whereas the NOE correlations from OH-4 to H-2, H-3b, H-5, and H-8b confirmed they were in opposite orientation. Based on the summarized results (Figure 3) of the modified Mosher’s method, the absolute configuration of C-5 was determined to be S, thereby assigning 2R, 4R, 5S, 9S absolute configuration for pestalotheol I (1).

Table 1.
NMR Data of compounds 1–3.
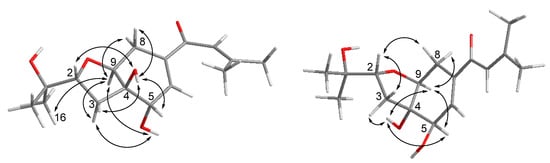
Figure 2.
Key NOE correlations for compound 1 (left) and 2 (right).
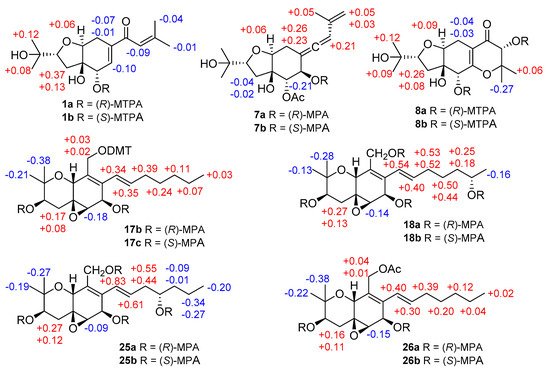
Figure 3.
∆δ = (δS − δR) values (in ppm) for the methoxytrifluoromethylphenylacetic acid (MTPA) esters of 1 and 8, ∆δ = (δR − δS) values (in ppm) for the MPA esters of 7, 17, 18, 25, and 26.
Based on the HRESIMS result as well as 1D and 2D NMR data (Table 1), the gross structure of 2 was elucidated as being identical to that of 1. However, different NOE relationships were observed in the ROESY spectrum of 2 compared to 1 (Figure 2). The NOE correlations between H-9/H-8b, H-9/H-5, H-9/OH-4, OH-4/H-5, OH-4/H-3b indicated that these protons were on the α-face, whereas the NOE correlations from H-2 to H-3a and H-8a indicated they were β-oriented. Thus, the absolute configuration of 2 was tentatively assigned as 2R, 4S, 5R, 9S in comparison to 1.
Pestalotheol K (3) has the molecular formula C16H24O5, containing one additional oxygen atom compared to truncateol Q, a polyketide analog isolated from Truncatella angustata [15]. The NMR data of 3 (Table 1) were similar to those of truncateol Q, except for the replacement of an olefinic methyl by an oxygenated methylene moiety at δC 60.9 and δH 4.21 and 4.16 (CH-13) and a hydroxy group at δH 3.70 (t, OH-13). The COSY correlation between OH-13/H-13ab and the HMBC correlations from H-13ab to C-11 (δC 122.8), C-12 (δC 139.9), and C-14 (δC 22.0) confirmed the location of this additional oxygenated methylene moiety at C-13. The remaining substructure and relative configuration of 3 were elucidated to be identical to truncateol Q after detailed analysis of the 2D NMR spectroscopic data of 3.
The NMR data (Table 2) of pestalotheol L (4) were similar to those of truncateol K, a known polyketide containing a chloride atom at C-4 [16]. The molecular formula of 4 was determined as C16H22O5 by HRESIMS, suggesting the replacement of the chlorine atom at C-4 by a hydroxy group in 4, which was supported by the observation of four hydroxy groups at δH 5.08, 5.05, 4.77, and 4.31 in the 1H NMR spectrum of 4 rather than three signals of hydroxy groups in truncateol K. On the basis of the 2D NMR spectroscopic data of 4, the remaining substructure and relative configuration of 4 were determined to be identical to truncateol K.
Table 2.
NMR Data of compounds 4–6.
Compound 5 has the same planar structure as 4 based on the 1D and 2D NMR spectroscopic data as well as on the HRESIMS data of 5. However, the NOE interactions of 5 differed from those of 4 as also observed between 1 and 2. In the ROESY spectrum of 5, correlations from H-9 to Me-16, H-3a and OH-5, and from OH-5 to H-3a and H-6 were found, indicating they were on the same side. On the other hand, the NOE correlations between H-3b/H-2, H-2/OH-4, OH-4/H-5, H-5/OH-6 placed those protons on the other side of the ring. Hence the relative configuration of 5 was elucidated as 2R*, 4R*, 5S*, 6R*, 9S* whereas that of 4 was 2R*, 4S*, 5R*, 6R*, 9S*.
The molecular formula of pestalotheol N (6) was deduced as C16H24O5 from the HRESIMS data. The NMR data (Table 2) of 6 were compatible with those of the co-isolated known compound truncateol H (12) except for the lack of the chloride atom at C-5 [16]. Instead, the COSY correlation between the D2O changeable proton at δH 5.28 and H-5 (δH 3.83) suggested C-5 to be hydroxylated. Similar NOE interactions were observed for 6 and 12, indicating the relative configuration of 6 to be identical to that of 12, for which the absolute configuration had previously been determined by Mosher’s reaction and by the X-ray data of its 6-epimer [16].
Pestalotheol O (7) was obtained as colorless oil. The molecular formula of 7 was determined as C18H26O6 by HRESIMS, containing an additional acetyl group when compared to 6, which was further supported by the presence of a carbonyl carbon at δC 169.2 and a methyl group at δC 20.5 and δH 2.09 in the NMR data of 7 (Table 3). The location of this additional acetyl group at C-5 was evident from the HMBC correlation from H-5 (δH 5.01) to the carbonyl carbon of the acetyl group. Analysis of 1D and 2D NMR data of 7 concluded its remaining substructure to be the same as 6. The absolute configuration of C-6 in 7 was determined as R by comparison of chemical shifts between its methoxyphenylacetic acid (MPA) esters (Figure 3).
Table 3.
NMR Data of compounds 7–9.
Based on the HRESIMS data, the molecular formula of compound 8 was deduced as C16H25O7, containing one additional oxygen atom when compared to the coisolated known derivative, pestalotheol A (13) [17]. The COSY correlation between the D2O changeable proton at δH 4.30 and H-11 (δH 4.13) and the HMBC correlations from H-11 to C-10, C-12, C-13, and C-14 confirmed the replacement of the methylene group by a hydroxy group at C-11 in 8. Compounds 8 and 13 share the same relative configuration at C-2, C-4, C-5, and C-9 as evident from their similar coupling constants and NOE relationships. Moreover, Mosher’s method was applied to determine the absolute configuration at C-5 and C-11 of 8. Due to the influence of multiple MPA moieties, anomalous ∆δ values were observed for Me-13 (−0.27) and Me-14 (+0.06). The absolute configuration at C-5 and C-11 of 8 was tentatively assigned as R (Figure 3). Therefore, the 2R, 4R, 5R, 9S, and 11R absolute configuration was assigned for pestalotheol P (8).
The HRESIMS of pestalotheol Q (9) showed the pseudomolecular ion peak at 355.1754 [M + H]+, indicating the molecular formula of C18H27O7. The UV spectrum and the NMR data of 9 suggested it to be an analog of pestalotheol A [17], with addition of an acetyl group as evident from signals in the 1H and 13C NMR spectroscopic data at δH 2.09, δC 169.5 and 20.5. The HMBC correlations from H-5 (δH 5.28) to the carbonyl carbon of the acetyl group indicated the attachment of the acetyl group at C-5. Based on the similar coupling constants and NOE relationships, the relative configuration of 9 was suggested to be identical to that of 13. Thus, pestalotheol Q (9) was elucidated to be 5-acetoxy derivative of pestalotheol A (13).
Cytosporin O (15) was obtained as yellowish gel. The molecular formula of 15 was established as C19H32O5 from the HRESIMS data, indicating four degrees of unsaturation. The 13C NMR data (Table 4) displayed signals of four olefinic carbons at δC 133.8 (C-8), 132.1 (C-15), 132.0 (C-9), and 126.0 (C-14), accounting for two degrees of unsaturation. Therefore, 15 was suggested to contain a bicyclic skeleton. In addition, six oxygenated carbons at δC 74.9 (C-2), 70.6 (C-3), 70.0 (C-10), 69.3 (C-5), 63.9 (C-7), and 57.7 (C-13), and three methyls at δC 27.9 (C-11), 16.1 (C-12), and 13.9 (C-20) were observed. Upon analysis of the 1D and 2D NMR data, 15 was suggested to be a derivative of cytosporin D [18] except for the replacement of signals of the epoxy ring at C5/C6 by an additional D2O exchangeable proton at δH 5.07 (s, OH-5) and an additional methylene group at δC 35.7, and δH 2.00, 1.54 (CH2-6). The HMBC correlations from OH-5 to C-4, C-5, C-6, and C-10, together with the COSY correlations between H-6ab and H-7, indicated a hydroxy group at C-5 and a methylene group at C-6. Thus, the planar structure of 15 was established as shown. The large coupling constant (15.7 Hz) revealed E-geometry of the double bond at C-15/C-16. The NOE interactions between H-10/OH-5, OH-5/H-4b, H-4b/H-10, H-10/Me-12, Me-12/H-4b indicated those protons to be orientated on the same side, whereas the NOE correlations between Me-11/H-3, H-3/H-4a, H-3/H-6a, H-6a/H-7 supported them to be on the opposite side (Figure 4).
Table 4.
NMR Data of compounds 15–17.
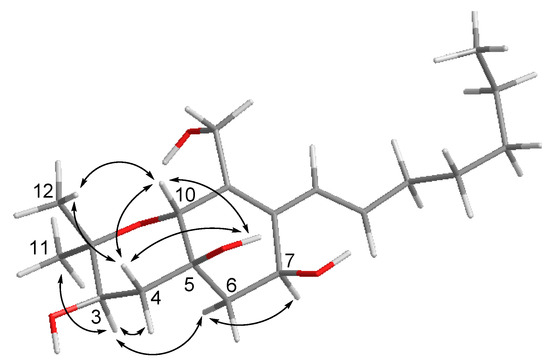
Figure 4.
Key NOE correlations for compound 15.
Compound 16 has the molecular formula C19H32O6 as deduced from the sodium adduct ion at m/z 379.2094 [M + Na] + in HRESIMS, containing an additional oxygen atom when compared to 15. The NMR data (Table 4) of 16 resembled those of 15 except for the replacement of a methylene group by an oxygenated methine at δC 75.3 and δH 3.55 (CH-6). The presence of an additional D2O exchangeable proton at δH 3.98 and its COSY correlation with H-6 indicated a hydroxy group at C-6. Therefore, 16 was elucidated as 6-hydroxylated derivative of cytosporin O (15). The relative configuration at C-3, C-5, C-7, and C-10 in 16 was determined to be identical to that of 15 by comparison of their coupling constants and NOE relationships. In addition, the NOE correlations from OH-6 to H-3 and H-7, and from H-6 to 5-OH and 7-OH confirmed α-orientation of OH-6 in 16.
The HRESIMS as well as 1D and 2D NMR data of cytosporin Q (17) suggested that it possessed the same planar structure as the known cytosporin D bearing an epoxy ring at C-5/C-6 [18]. The NOE correlations observed in 17 between H-10/Me-12, Me-12/H-4a, H-4a/H-10, and between Me-11/H-3, H-3/H-4b, H-4b/H-6, indicated that 17 shared the same relative configuration at C-3, C-5, C-6, and C-10 as cytosporin D. However, the relative configuration at C-7 could not be determined by NOE relationships or J values. Therefore, the modified Mosher’s method was applied to determine the absolute configuration at C-3 and C-7 of 17. In order to exclude the influence of the primary alcohol at C-13, 4,4′-dimethoxytrytil chloride (DMT-Cl) was used for protection before Mosher’s reaction, yielding compound 17a. Meanwhile, Mosher’s reaction was also conducted with the coisolated known compound cytosporin F (26) [19], bearing an acetyl group attached to C-13. The differences in chemical shifts of (R)- and (S)-MPA esters assigned 3R and 7R absolute configuration for both 17a and cytosporin F (26) (Figure 3). Thus, 17 was identified as 7-epimer of cytosporin D and has the absolute configuration of 3R, 5S, 6R, 7R, and 10R. Cytosporin D had been firstly reported from the fungus Eutypella scoparia derived from a marine pulmonate mollusk [18], while cytosporins F–K were produced by Pestalotiopsis theae, an endophyte from leaves of Turraeanthus longipes [19]. From biogenetic considerations and the results of Mosher’s reaction, it is reasonable to revise the absolute configurations of cytosporins F–K [19] as 3R, 5S, 6R, 7R, and 10R, the same as 17 rather than that of cytosporin D.
The molecular formula of cytosporin R (18) was determined as C19H30O6 by HRESIMS, with an additional oxygen atom compared to cytosporin Q (17). A methyl doublet at δH 1.03 (Me-20) was found in the 1H NMR data of 18 instead of a methyl triplet in 17 (Table 5). Detailed examination of the 2D NMR spectroscopic data revealed 18 to be the 19-OH substituted analog of cytosporin Q (17). Similar NOE interactions were observed in 18 as for 17, indicating that both compounds shared the same relative configuration regarding the ring system. Due to the low amount, cytosporin R (18) was subjected for Mosher’s method directly without protecting the primary alcohol at C-13. The absolute configuration at C-19 of side chain was determined as S (Figure 3). From biogenetic considerations, the remaining stereocenters of 18 are assumed to share the same absolute configuration as cytosporin Q (17).
Table 5.
NMR Data of compounds 18–20.
Compound 19 has the same molecular formula of C19H30O6 as cytosporin R (18) as determined by the HRESIMS data. After detailed analysis of the 1D and 2D NMR spectroscopic data of 19, both compounds were found to differ in the position of the hydroxy group in the side chain. The COSY correlations between the D2O exchangeable proton at δH 4.34 and H-17 (δH 3.46), and between H-14/H-15/H-16ab/H-17 confirmed that C-17 was hydroxylated in 19. Therefore, compound 19 was suggested to be the C-17 epimer of cytosporin K (25) [19]. Similar NOE correlations were observed in 19 and 25, suggesting that both compounds share the same relative configuration regarding the bicyclic ring core. Due to the low amount of cytosporin S (19), Mosher’s reaction was carried out for its 17-epimer cytosporin K (25) [19], whose absolute configuration in the side chain was uncertain before. Comparison of the chemical shifts of (R)- and (S)-MPA esters of 25 (Figure 3) suggested 17S absolute configuration for cytosporin K (25). Thus, 17R absolute configuration was proposed for cytosporin S (19).
Cytosporins T and U (20 and 21) have the same molecular formula of C19H30O6 as deduced by the HRESIMS data. Their NMR data (Table 5 and Table 6) were comparable to those of 19. However, the olefinic proton at H-15 appeared as dd in 20 and 21 rather than as dt in 19, suggesting the hydroxy group in the side chain to be located at C-16 in 20 and 21 rather than at C-17 in 19. This was further supported by the COSY correlations observed between H-14/H-15/H-16/H-17ab, as well as between H-17 and a D2O exchangeable proton in 20 and 21. Detailed analysis of the 2D NMR spectroscopic data of 20 and 21 revealed that the remaining substructure was identical to that of 19. Cytosporins T and U (20 and 21) only differ in the absolute configuration in the side chain, as found for 19 and 25. The absolute configuration at C-16 of both compounds was not determined due to the low amounts.
Table 6.
NMR Data of compounds 21–23.
The NMR data of compound 22 were similar to those of cytosporin F(26) [19], and both compounds have the same molecular formula as evident from the HRESIMS data. However, the shielded methylene group at C-13 (δC 57.6, δH 4.11 and 3.78 in 22 vs. δC 63.2, δH 4.74 and 4.67 in 26), and the deshielded oxygenated methine at C-7 (δH 5.85 in 22 vs. δH 4.63 in 26) in 22 suggested that the acetoxy moiety was attached at C-7 in 22 rather than at C-13 in 26, which was further confirmed by the HMBC correlation from H-7 to the carbonyl carbon (δC 170.0) of the acetoxy moiety. The remaining substructure of 22 was elucidated to be identical to that of 26. Based on the similar NOE correlations and the biogenetic relationship between 22 and 26, compound 22 was suggested to share the same 3R, 5S, 6R, 7R, and 10R absolute configuration as 26.
The molecular formula C19H28O5 was suggested for cytosporin W (23) by the HRESIMS data, indicating an additional degree of unsaturation when compared to 17. The absence of one D2O changeable proton in the 1H NMR data (Table 6), along with the HMBC correlations from H-6 (δH 3.54) and H-14 (δH 6.04) to a carbonyl carbon at δC 195.8 indicated a ketone carbonyl to be located at C-7, which accounted for the additional degree of unsaturation. The remaining structure including the relative configuration was determined to be identical to that of 17 after detailed analysis of the 2D NMR spectroscopic data of 23.
The remaining known compounds were identified as truncateols B (10), D (11), and H (12) [16], pestalotheols A (13) and D (14) [17], cytosporins J (24), K (25), and F (26) [19]. Both new compounds pestalotheol O (7) and Q (9) contain acetoxyl group at C-5. In order to exclude the possibility that they might be artifacts formed during the extraction with EtOAc, relative compounds pestalotheol N (6) and pestalotheol A (13) were incubated in EtOAc for 48 h at room temperature. Unchanged retention time and molecular weight from HPLC together with MS results indicated that they were natural products rather than artifacts.
In previous report, cytosporins A–C were known as angiotensin II binding inhibitors [20], while cytosporin L was active against the bacteria Micrococcus lysodeikticus and Enterobacter aerogenes with MIC values of 3.12 µM [21,22]. In the present study, all isolated compounds were tested for their cytotoxicity and antibacterial activity. Only compound 23, with a carbonyl group at C-7 instead of a hydroxy group, showed significant cytotoxicity against the mouse lymphoma cell line L5178Y with an IC50 value of 3.0 μM, even stronger than that of the positive control kahalalide F (IC50 4.3 μM), whereas the other compounds were not active (IC50 > 20 μM), suggesting the importance of α,β-unsaturated ketone moiety in 23 for cytotoxicity. In the antibacterial activity assay, none of the isolated compounds were active against the Gram-positive bacteria Staphylococcus aureus (ATCC 29213) and Mycobacterium tuberculosis (H37Rv) and the Gram-negative bacterium Pseudomonas aeruginosa (ATCC 27853) and drug-resistant Acinetobacter baumannii (ATCC BAA-1605) (MIC > 100 µM). However, in combination with a sublethal colistin concentration of 0.1 µM, compounds 22 and 23 displayed antibacterial activity against drug-resistant Acinetobacter baumannii (ATCC BAA-1605) with MIC values of 50 and 100 µM, respectively.
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations were measured with a Jasco P-2000 polarimeter (JASCO, Tokyo, Japan). Analytical HPLC was performed with a Dionex UltiMate-3400SD system coupled to an LPG-3400SD pump and a DAD300RS photodiode array detector (Dionex Softron, Germering, Germany). The analytical column (125 × 4 mm) was prefilled with Eurosphere-10 C18 (Knauer, Berlin, Germany), following the program: 0 min (10% MeOH); 5 min (10% MeOH); 35 min (100% MeOH); 45 min (100% MeOH). NMR spectra were recorded with Bruker ARX 300 or 600 NMR spectrometers (Bruker, Karlsruhe, Germany). Chemical shifts were referenced to the solvent residual peaks. HRESIMS were recorded on a UHR-QTOF maXis 4G mass spectrometer (Bruker Daltonics, Bremen, Germany). Column chromatography was conducted with Merck MN silica gel 60M (0.04–0.063 mm). TLC plates precoated with silica gel F254 (Merck) were used with detection under 254 and 366 nm. Distilled and spectral grade solvents were used for column chromatography and spectroscopic measurements, respectively. Semipreparative HPLC was performed using a Merck Hitachi HPLC System (UV detector L-5410; pump L-5100; Eurosphere-100 C18, 300 × 8 mm), with a mixture of MeOH-H2O or MeCN-H2O as mobile phase.
3.2. Fungal Material
The fungus was isolated from roots of the mangrove plant Rhizophora racemosa collected around Lagos in October 2017. It was identified as Pseudopestalotiopsis theae (GenBank accession number MN814071) by DNA amplification and sequencing of its ITS region as described before [23].
3.3. Fermentation, Extraction, and Isolation
The fungus was cultivated on solid rice medium in 10 Erlenmeyer flasks (1 L with 100 g rice and 110 mL demineralized water per flask, autoclaved at 121 °C for 20 min before inoculation). EtOAc (700 mL) was added to each flask after fermentation of 14 days at 20 °C. The fungal cultures were subjected to shaking at 140 rpm for 8 h following addition of EtOAc. After removal of the solvent under reduced pressure, the obtained residue (12.4 g) was subjected to liquid–liquid partition between n-hexane and MeOH. After HPLC analysis of both phases, only the methanolic phase was further chromatographically investigated.
The MeOH fraction (8.1 g) was subjected to a vacuum liquid silica gel column chromatography using a gradient solvent system of n-hexane and EtOAc (100:1 to 0:100; v/v) to obtain seven fractions (Fr. 1–7). Following Sephadex LH-20 column (60 × 3 cm) chromatography with MeOH as mobile phase, Fr. 3 (1.8 g) was separated into four subfractions (Fr. 3.1–3.4). Fr. 3.4 (0.6 g) was then subjected to vacuum liquid RP-18 column (60 × 200 mm) chromatography using a solvent gradient (from 100% H2O to 100% MeOH) to give five subfractions (Fr.3.4.1–3.4.5). Compounds 6 (4.0 mg), 7 (2.5 mg), 9 (2.2 mg), 10 (5.0 mg), 11 (15 mg),12 (1.9 mg), 14 (1.7 mg), 22 (3.1 mg), and 23 (2.9 mg) were obtained by semipreparative HPLC (MeOH-H2O: 0–2 min, 40%; 2–27 min, from 40% to 60%; 27–32 min, 100%) from Fr.3.4.1 (110 mg). Fr. 4 (2.7g) was subjected to vacuum liquid RP-18 column chromatography (60 × 200 mm) using a solvent gradient (from 100% H2O to 100% MeCN) to give seven subfractions (Fr. 4.1–4.7). Compounds 1 (2.5 mg), 2 (1.6 mg), 4 (3.0 mg), 5 (1.4 mg), 18 (3.7 mg), and 24 (5.8 mg) were obtained by semipreparative HPLC (MeCN-H2O: 0–2 min, 8%; 2–22 min, from 8% to 20%; 22–30 min, 100%) from Fr.4.3 (210.0 mg), while compounds 15 (3.3 mg), 16 (3.0 mg), 17 (37.2 mg), and 26 (13.0 mg) were obtained by semipreparative HPLC (MeOH-H2O: 0–2 min, 30%; 2–22 min, from 30% to 60%; 22–30 min, 100%) from Fr.4.6 (200.0 mg). Following similar procedures, Fr. 5 (1.8 g) was also separated by vacuum liquid RP-18 column (60 × 200 mm) chromatography to give four subfractions (Fr.5.1–Fr.5.4). Compounds 13 (3.5 mg), 19 (1.5 mg), 20 (1.3 mg), 21(3.8 mg), and 25 (6.2 mg) were obtained from Fr. 5.3 (130 mg) by semipreparative HPLC (MeOH-H2O: 0–2 min, 38%; 2–22 min, from 38% to 58%; 22–30 min, 100%), while compounds 3 (2.5 mg) and 8 (4.1 mg) were obtained by semipreparative HPLC (MeCN-H2O: 0–2 min, 5%; 2–22 min, from 5% to 15%; 22–30 min, 100%) from Fr. 5.4 (68.0 mg).
Pestalotheol I (1): colorless oil; +115 (c 0.25, MeOH); UV (MeOH) λmax 219 nm; 1H and 13C NMR data, Table 1; HRESIMS m/z 297.1694 [M + H]+ (C16H25O5, calcd. 297.1697), 319.1513 [M + Na]+ (C16H24O5Na, calcd. 319.1516).
Pestalotheol J (2): colorless oil; −12 (c 0.20, MeOH); UV (MeOH) λmax 268 nm; 1H and 13C NMR data, Table 1; HRESIMS m/z 297.1700 [M + H]+ (C16H25O5, calcd. 297.1702).
Pestalotheol K (3): colorless oil; +111 (c 0.25, MeOH); UV (MeOH) λmax 246 nm; 1H and 13C NMR data, Table 1; HRESIMS m/z 297.1700 [M + H]+ (C16H25O5, calcd. 297.1702).
Pestalotheol L (4): colorless oil; −60 (c 0.30, MeOH); UV (MeOH) λmax 260 nm; 1H and 13C NMR data, Table 2; HRESIMS m/z 317.1362 [M + Na]+ (C16H22O5Na, calcd. 317.1359).
Pestalotheol M (5): colorless oil; +17 (c 0.25, MeOH); UV (MeOH) λmax 262 nm; 1H and 13C NMR data, Table 2; HRESIMS m/z 317.1360 [M + Na]+ (C16H22O5Na, calcd. 317.1359).
Pestalotheol N (6): colorless oil; +47 (c 0.15, MeOH); UV (MeOH) λmax 219 nm; 1H and 13C NMR data, Table 2; HRESIMS m/z 319.1518 [M + Na]+ (C16H24O5Na, calcd.319.1516).
Pestalotheol O (7): colorless oil; +35 (c 0.22, MeOH); UV (MeOH) λmax 218 nm; 1H and 13C NMR data, Table 3; HRESIMS m/z 361.1619 [M + Na]+ (C18H26O6Na, calcd. 361.1622).
Pestalotheol P (8): colorless oil; +89 (c 0.22, MeOH); UV (MeOH) λmax 282 nm; 1H and 13C NMR data, Table 3; HRESIMS m/z 329.1595 [M + H]+ (C16H25O7, calcd. 329.1595).
Pestalotheol Q (9): colorless oil; +121 (c 0.15, MeOH); UV (MeOH) λmax 279 nm; 1H and 13C NMR data, Table 3; HRESIMS m/z 355.1754 [M + H]+ (C18H27O7, calcd. 355.1757).
Cytosporin O (15): colorless oil; −8 (c 0.30, MeOH); UV (MeOH) λmax 243 nm; 1H and 13C NMR data, Table 4; HRESIMS m/z 363.2144 [M + Na]+ (C19H32O5Na, calcd. 363.2142).
Cytosporin P (16): colorless oil; −4 (c 0.35, MeOH); UV (MeOH) λmax 240 nm; 1H and 13C NMR data, Table 4; HRESIMS m/z 379.2094 [M + Na]+ (C19H32O6Na, calcd. 379.2091).
Cytosporin Q (17): colorless oil; +16 (c 0.22, MeOH); UV (MeOH) λmax 237 nm; 1H and 13C NMR data, Table 4; HRESIMS m/z 356.2432 [M + NH4]+ (C19H34O5N, calcd. 356.2431).
Cytosporin R (18): colorless oil; +6 (c 0.20, MeOH); UV (MeOH) λmax 239 nm; 1H and 13C NMR data, Table 5; HRESIMS m/z 355.2117 [M + H]+ (C19H31O6, calcd. 355.2121).
Cytosporin S (19): colorless oil; +14 (c 0.10, MeOH); UV (MeOH) λmax 239 nm; 1H and 13C NMR data, Table 5; HRESIMS m/z 377.1934 [M + Na]+ (C19H30NaO6, calcd. 377.1935).
Cytosporin T (20): colorless oil; −1 (c 0.20, MeOH); UV (MeOH) λmax 238 nm; 1H and 13C NMR data, Table 5; HRESIMS m/z 377.1931[M + Na]+ (C19H30O6Na, calcd. 377.1935).
Cytosporin U (21): colorless oil; + 4 (c 0.20, MeOH); UV (MeOH) λmax 237 nm; 1H and 13C NMR data, Table 6; HRESIMS m/z 372.2381[M + NH4]+ (C19H34NO6, calcd. 372.2381), 377.1935 [M + Na]+ (C19H30O6Na, calcd. 377.1935).
Cytosporin V (22): colorless oil; −21 (c 0.30, MeOH); UV (MeOH) λmax 237 nm; 1H and 13C NMR data, Table 6; HRESIMS m/z 398.2537[M + NH4]+ (C21H36NO6, calcd. 398.2537), 403.2089 [M + Na]+ (C21H32O6Na, calcd. 403.2091).
Cytosporin W (23): colorless oil; −4 (c 0.10, MeOH); UV (MeOH) λmax 215, 281 nm; 1H and 13C NMR data, Table 6; HRESIMS m/z 359.1829 [M + Na]+ (C19H28O5Na, calcd. 359.1829).
3.4. Preparation of (R)- and (S)-MTPA Esters
Compound 1 (0.8 mg) was dissolved in prydine-d6 (500 μL) and then transferred to an NMR tube. (S)-MTPACl (10.0 µL, 0.050 mmol) was added quickly. By shaking the NMR tube, the reagent and the dissolved compound were mixed. After 10 h at room temperature, 1H NMR data were obtained for the (R)-MTPA ester (1a). Following the same protocol, the (S)-MTPA ester (1b) was produced. A similar procedure was applied for compound 8.
3.5. Preparation of Compound 17a
Compound 17 (10 mg, 0.030 mmol) was dissolved in anhydrous CH2Cl2 (3 mL) together with catalytic amount of DMAP and 10.0 mg of 4,4′-dimethoxytrytil chloride (0.030 mmol, DMTCl) at room temperature for 2 h. After removal of the solvent, preparative thin-layer chromatography was used for purification, using CH2Cl2-MeOH (95:5), to give compound 17a.
3.6. Preparation of (R)- and (S)-MPA Esters
Compound 7 (0.8 mg, 0.002 mmol), along with DMAP (0.5 mg, 0.004 mmol), N,N′-dicyclohexylcarbodiimide (0.8 mg, 0.004 mmol, DCC) and (R)-MPA (0.7 mg, 0.004 mmol), was mixed in anhydrous CH2Cl2 (0.3 mL). The solvent was removed after 2 h at room temperature. The (R)-MPA ester (7a) was obtained from the crude product by semipreparative HPLC (MeOH-H2O: 0-2 min, 40%; 2–15 min; 15–25 min, 100%). Following a similar procedure, the (S)-MPA ester (7b) was obtained by using (S)-MPA. The above steps were also applied to compounds 17a, 18, 25, and 26.
3.7. Cytotoxicity and Antibacterial Assay
The MTT method was applied for the cytotoxicity assay against the mouse lymphoma cell line L5178Y as described before [24]. Antibacterial activity against Gram-positive bacteria Staphylococcus aureus (ATCC 29213) and Mycobacterium tuberculosis (H37Rv), and the Gram-negative bacteria Pseudomonas aeruginosa (ATCC 27853) and drug-resistant Acinetobacter baumannii (ATCC BAA-1605), was evaluated using the microdilution method in alignment with the CLSI guidelines using Muller Hinton broth for bacteria and 7H9 broth for Mycobacteria [25]. Additionally, the antibacterial activity was performed in combination with sublethal concentrations of the antibiotic colistin (0.1 µM) for drug-resistant A. baumannii (ATCC BAA-1605) to elucidate synergistic effects of the combinatory therapy.
Supplementary Materials
The following are available online at https://www.mdpi.com/1660-3397/18/2/129/s1, UV, HRMS, 1D and 2D NMR spectra of all the new compounds 1–9 and 15–23 as well as spectra for Mosher’s esters of 1, 7, 8, 17, 18, 25 and 26.
Author Contributions
Investigation, X.Y., W.E.G.M., D.M., R.K., Z.G., and K.Z.; resources, B.O.U.; writing—original draft preparation, X.Y.; writing—review and editing and supervision, Z.L. and P.P. All authors have read and agreed to the published version of the manuscript.
Funding
This project was supported by grants of the DFG (GRK 2158, project number 270650915) to P.P. and R.K. and by the Manchot Foundation to P.P.
Acknowledgments
Xiaoqin Yu is grateful to China Scholarship Council, the Ministry of Education of China for a doctoral scholarship.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Surup, F.; Stadler, M. Natural products in the chemical industry. Von Bernd Schaefer. Angew. Chem. 2015, 127, 8999–9000. [Google Scholar] [CrossRef]
- Petersen, A.B.; Rønnest, M.H.; Larsen, T.O.; Clausen, M.H. The Chemistry of Griseofulvin. Chem. Rev. 2014, 114, 12088–12107. [Google Scholar] [CrossRef] [PubMed]
- Tobert, J.A. Lovastatin and beyond: The history of the HMG-CoA reductase inhibitors. Nat. Rev. Drug Discov. 2003, 2, 517–526. [Google Scholar] [CrossRef]
- Watts, R.; Clunie, G.; Hall, F.; Marshall, T. Rheumatology; Oxford University Press: New York, NY, USA, 2009; p. 558. [Google Scholar]
- Kusari, S.; Pandey, S.P.; Spiteller, M. Untapped mutualistic paradigms linking host plant and endophytic fungal production of similar bioactive secondary metabolites. Phytochem. 2013, 91, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, S.; Prakash, V.; Ranjan, N. Recent advances in the discovery of bioactive metabolites from Pestalotiopsis. Phytochem. Rev. 2017, 71, 882. [Google Scholar] [CrossRef]
- Maharachchikumbura, S.; Hyde, K.; Groenewald, J.; Xu, J.; Crous, P. Pestalotiopsis revisited. Stud. Mycol. 2014, 79, 121–186. [Google Scholar] [CrossRef]
- Xu, J.; Ebada, S.S.; Proksch, P. Pestalotiopsis a highly creative genus: Chemistry and bioactivity of secondary metabolites. Fungal Divers. 2010, 44, 15–31. [Google Scholar] [CrossRef]
- Yang, X.-L.; Zhang, J.-Z.; Luo, D.-Q. The taxonomy, biology and chemistry of the fungal Pestalotiopsis genus. Nat. Prod. Rep. 2012, 29, 622. [Google Scholar] [CrossRef]
- Li, E.; Jiang, L.; Guo, L.; Zhang, H.; Che, Y. Pestalachlorides A–C, antifungal metabolites from the plant endophytic fungus Pestalotiopsis adusta. Bioorganic Med. Chem. 2008, 16, 7894–7899. [Google Scholar] [CrossRef]
- Liu, L.; Liu, S.; Jiang, L.; Chen, X.; Guo, L.; Che, Y. ChemInform Abstract: Chloropupukeananin, the First Chlorinated Pupukeanane Derivative, and Its Precursors from Pestalotiopsis fici. Chemin 2008, 39, 1397–1400. [Google Scholar] [CrossRef]
- Ding, G.; Zhang, F.; Chen, H.; Guo, L.; Zou, Z.; Che, Y. Pestaloquinols A and B, Isoprenylated Epoxyquinols fromPestalotiopsissp. J. Nat. Prod. 2011, 74, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Ge, H.M.; Li, F.; Song, Y.C.; Tan, R.X. New Phytotoxic Metabolites fromPestalotiopsissp. HC02, a Fungus Residing inChondracris roseeGut. Chem. Biodivers. 2008, 5, 2402–2407. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Dai, H.; Makhloufi, G.; Heering, C.; Janiak, C.; Hartmann, R.; Mandi, A.; Kurtán, T.; Müller, W.E.G.; Kassack, M.U.; et al. Cytotoxic 14-Membered Macrolides from a Mangrove-Derived Endophytic Fungus, Pestalotiopsis microspora. J. Nat. Prod. 2016, 79, 2332–2340. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, N.; Proksch, P.; Zhou, D.; Lin, W. Truncateols O-V, further isoprenylated cyclohexanols from the sponge-associated fungus Truncatella angustata with antiviral activities. Phytochemistry 2018, 155, 61–68. [Google Scholar] [CrossRef]
- Zhao, Y.; Si, L.; Liu, N.; Proksch, P.; Zhou, D.; Lin, W. Truncateols A–N, new isoprenylated cyclohexanols from the sponge-associated fungus Truncatella angustata with anti-H1N1 virus activities. Tetrahedron 2015, 71, 2708–2718. [Google Scholar] [CrossRef]
- Li, E.; Tian, R.; Liu, S.; Chen, X.; Guo, L.; Che, Y. Pestalotheols A–D, Bioactive Metabolites from the Plant Endophytic FungusPestalotiopsis theae. J. Nat. Prod. 2008, 71, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Ciavatta, M.L.; López-Gresa, M.P.; Gavagnin, M.; Nicoletti, R.; Manzo, E.; Mollo, E.; Guo, Y.-W.; Cimino, G. Cytosporin-related compounds from the marine-derived fungus Eutypella scoparia. Tetrahedron 2008, 64, 5365–5369. [Google Scholar] [CrossRef]
- Akone, S.H.; El Amrani, M.; Lin, W.; Lai, D.; Proksch, P. Cytosporins F–K, new epoxyquinols from the endophytic fungus Pestalotiopsis theae. Tetrahedron Lett. 2013, 54, 6751–6754. [Google Scholar] [CrossRef]
- Stevens-Miles, S.; Goetz, M.A.; Bills, G.; Giacobbe, R.A.; Tkacz, J.S.; Chang, R.S.L.; Mojena, M.; Martín, I.; Díez, M.T.; Pelae, F.; et al. Discovery of an Angiotensin II Binding Inhibitor from a Cytospora sp. Using Semi-automated Screening Procedures. J. Antibiot. 1996, 49, 119–123. [Google Scholar] [CrossRef]
- Liao, H.-X.; Sun, D.-W.; Zheng, C.-J.; Wang, C. A new hexahydrobenzopyran derivative from the gorgonian-derived Fungus Eutypella sp. Nat. Prod. Res. 2017, 31, 1640–1646. [Google Scholar] [CrossRef]
- Rivera-Chávez, J.; Zacatenco-Abarca, J.; Morales-Jiménez, J.; Martínez-Aviña, B.; Hernández-Ortega, S.; Aguilar-Ramírez, E. Cuautepestalorin, a 7,8-Dihydrochromene–Oxoisochromane Adduct Bearing a Hexacyclic Scaffold from Pestalotiopsis sp. IQ-011. Org. Lett. 2019, 21, 3558–3562. [Google Scholar] [CrossRef] [PubMed]
- Kjer, J.; Debbab, A.; Aly, A.H.; Proksch, P. Methods for isolation of marine-derived endophytic fungi and their bioactive secondary products. Nat. Protoc. 2010, 5, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Ashour, M.; Edrada, R.; Ebel, R.; Wray, V.; Wätjen, W.; Padmakumar, K.; Müller, W.E.G.; Lin, W.H.; Proksch, P. Kahalalide Derivatives from the Indian Sacoglossan MolluskElysiagrandifolia. J. Nat. Prod. 2006, 69, 1547–1553. [Google Scholar] [CrossRef] [PubMed]
- Hecht, D.W.; Citron, D.M.; Cox, M.; Jacobus, N.; Jenkins, S.G.; Onderdonk, A.; Wexler, H.M. Methods for Antimicrobial Susceptibiluty Testing of Anaerobic Bacteria: Approved Standard; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2007; pp. 1–47. [Google Scholar]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).