Abstract
Further chemical investigation of the EtOAc extract of the soft coral Lobophytum varium resulted in the discovery of eleven new diterpenoids lobovarols F–P (1–11) of lobane– and prenyleudesmane–types, along with two known metabolites (12 and 13). The structures of the new metabolites were established by spectroscopic analyses, including 2D NMR experiments. The absolute configuration of 1 was determined using Mosher’s method. The complete assignment of 1H and 13C NMR spectroscopic data of 12 and 13 and the identification of pyran-derived moieties in the prenyleudesmanes were reported for the first time. Anti-inflammatory activities of the isolated compounds in suppressing elastase release and superoxide anion generation in human neutrophils were disclosed for 1, 2, 4, 12, and 13. A stereospecific biosynthesis for lobanes and prenyleudesmanes from the related prenylgermacranes could explain the coexistence of lobanes and prenylgermacranes in L. varium.
1. Introduction
Soft corals belonging to genus Lobophytum (Alcyoniidae) were shown to be the main biological sources of lobane [1,2,3,4,5,6,7], prenyleudesmane [1,8,9,10,11], and spatane–type diterpenoids [8]. Some lobane and prenyleudesmane diterpenoids have displayed antimicrobial [2], cytotoxic [2,6,8], and anti-inflammatory activities [1]. Cembranoids have also been isolated from this genus, and have been shown to exhibit significant cytotoxic [12,13,14], antiviral [15], and anti-inflammatory activities [14,16,17,18]. Our previous chemical study on the EtOAc extract of a Formosan soft coral, Lobophytum varium has led to the discovery of five new metabolites of lobane- and prenyleudesmane-types, of which some were found to possess anti-inflammatory activity through the suppression of elastase release and/or superoxide anion generation in challenged neutrophils [1]. The present study was aimed to further discover new bioactive diterpenoids from continuing investigation of the organic fractions of L. varium. This study not only led to the isolation of new metabolites but also gave strong evidence for the biosynthetic mechanism of lobane and prenyleudesmane from the corresponding prenylgermacrane biosynthetic intermediate was proposed, which could explain the common coexistence of lobane and prenylgermacranes in L. varium.
2. Results and Discussion
The terpenoids containing fractions of the EtOAc extract of L. varium were repeatedly purified using a series of chromatographic techniques, including HPLC, to afford compounds 1–13 (Figure 1). The structures of the new compounds (1–11) were established on the basis of NMR data (Table 1, Table 2 and Table 3) and spectroscopic analyses (Supplementary Materials, Figures S1 to S79).
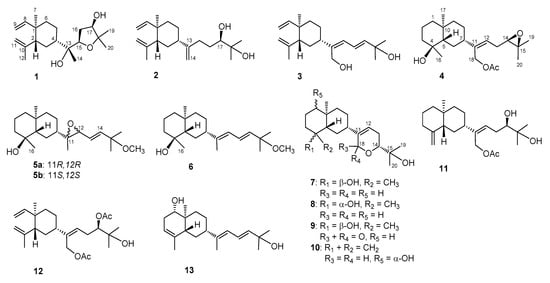
Figure 1.
Structures of compounds 1–13.

Table 1.
13C and 1H NMR spectroscopic data of compounds 1–4.
Table 2.
13C and 1H NMR spectroscopic data of compounds 5–8.
Table 3.
13C and 1H NMR spectroscopic data of compounds 9–11.
Lobovarol F (1) was found to possess a molecular formula C20H34O3 as established by high-resolution electrospray ionization mass spectrometry (HRESIMS, m/z 345.2403, [M + Na]+), indicating four degrees of unsaturation. The IR absorption at 3393 cm−1 designated the presence of the hydroxy group. The NMR signals (Table 1) showed the presence of a vinyl (δC 150.0, CH and 110.0, CH2; δH 5.80, dd, J = 18.0, 10.4 Hz, 4.90 d, J = 10.4 Hz, and 4.89, br d, J = 18.0 Hz), an isopropenyl (δC 147.6, C; 112.1, CH2 and 24.9, CH3; δH 4.83 and 4.59, each 1H, s; and 1.71, 3H, s); a ring-juncture methyl (δC 16.6, CH3; δH 0.99, 3H, s) and methine (δC 52.4, CH; δH 1.98, m) of a β-elemene segment of lobane-type diterpenoids [1,2,3]. Thus, the proton signals at δH 1.30, 1.26, and 1.15 (each 3H, s) should be the methyls attached to the oxygen-bearing carbons of the side chain. The analysis of correlations spectroscopy (COSY) and heteronuclear multiple bond correlation (HMBC) led to the establishment of a 2,2-dimethyl-3-hydroxytetrahydrofuran attached to a methyl-bearing oxygenated carbon (δC 75.0, C-13) to form the side chain of 1 (Figure 2). Accordingly, the HMBC correlations from H3-14 (δH 1.26) and from both H3-19 and H3-20 (δH 1.15 and 1.30) to the downfield shifted oxycarbons (δC 80.4, CH and 84.0, C), respectively, assigned the 15,18-ether linkage of the tetrahydrofuran ring. Moreover, the HMBC correlations observed from H3-14 and from both of H3-19 and H3-20 to oxycarbons (δC 75.0, C and 76.6, CH) positioned the two hydroxy groups at C-13 and C-17, respectively.
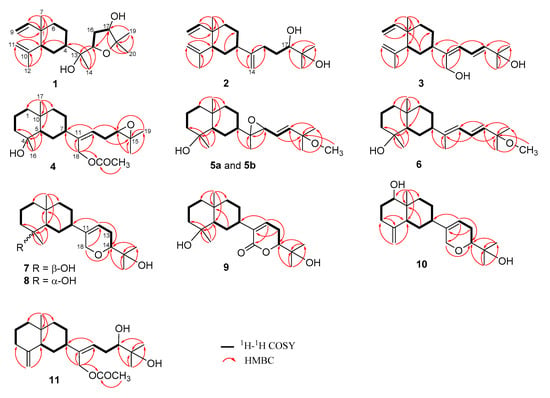
Figure 2.
1H–1H COSY and HMBC correlations of 1–11.
The relative configurations of 1 at C-1, C-2, C-4, C-13, C-15, and C-17 were assigned by the nuclear Overhauser effect spectroscopy (NOESY) correlation analysis (Figure 3a) and supported by MM2 force-field modeling and NMR data. From the NOE correlations displayed for H3-7/H3-12 and H-2/H-4 and by comparison of the chemical shifts of C-1 to C-12 with those of the previously reported lobane–type diterpenoids [1,2,5,6,19], the 1R*,2R*,4S* configurations of 1 was determined. Moreover, H3-14 and H-15 showed the NOE interactions with the β-oriented H-4 and H2-5, leading to the assignment of the corresponding β- and α-orientations of H3-14 and H-15, respectively. The α-orientation for the hydroxy group at C-13 was thus suggested and also supported by the down-field shifted H-3 (δH 1.70, m) relative to those of lobane–type diterpenoids having a β-orientated hydroxy group [1]. A conformation analysis for 1 and its 13-epimer, using MM2 calculations and measuring distances between protons showing key NOEs, also revealed the α-orientation of 13-OH, as the distances between both H-15/H2-5 and H-4/H3-14 were found to be shorter than 2.8 Å in 1. Furthermore, the NOE correlations between H3-19 (δH 1.15, 3H, s) and both H-15 and H-17 disclosed the β-orientations of the hydroxy at C-17. The R*-configurations for C-13, C-15, and C-17 were thus assigned. By using Mosher’s method [20,21], the absolute R configuration for C-17 was proven by the analysis of the calculated ∆δ (δS − δR) values of the neighboring protons to C-17 from the 1H NMR data of 17-(S)- and 17-(R)-α-methoxy-α-(trifluoromethyl)phenylacetyl (MTPA) esters of 1 (1a and 1b), respectively (Figure 3b). Based on the above findings, lobovarol F (1) was determined to be (1R,2R,4S,13R,15R,17R)-15,18-epoxyloba-8,10-dien-13,17-diol. To the best of own knowledge, 1 is the first lobane diterpene with a tetrahydrofuran-containing side chain.
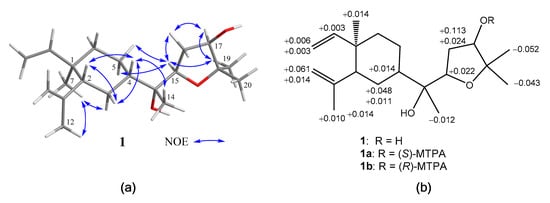
Figure 3.
(a) Key nuclear Overhauser effect (NOE) correlations of 1; (b) 1H NMR chemical shift differences ∆(δS − δR) in ppm for the MTPA esters of 1.
Lobovarol G (2) displayed a sodiated ion peak at m/z 329.2453 in the HRESIMS spectrum, appropriate for a molecular formula C20H34O2. The IR absorption at 3392 cm−1 also indicated the presence of hydroxy group. The NMR data of 2 (Table 1) showed resonances and coupling patterns identical to those of the β-elemene ring system in 1 as well as other lobane-type diterpenoids [1,2,3,4,5,6,7]. The fourth degree of unsaturation has arisen from an 1,1-disubstituted double bond (δC 154.5, C and 107.4, CH2; and δH 4.82 and 4.77, each 1H, s). NMR data showed that 2 differs with (1R,2R,4S,17R)-loba-8,10,13(15)-trien-17,18-diol, isolated from the same organism (L. varium) [1] and L. pauciflorum [2], only in the position of the double bond of the side chain. The analysis of HMBC correlations further confirmed the C-17 and C-18 positions of the hydroxy groups in the side chain (Figure 2). Compound 2 should possess the same 1R,2R,4S-configuration of the β-elemene ring system as that of 1 by the biogenetic relationship with 1 and the previously reported lobane diterpenoids isolated from genus Lobophytum [1,5,19]. Comparison of NMR data of the side chain of 2 (Table 1) with the corresponding data of chokol E (14) [22] and 10-epi-chokol E (15) [23] of known absolute configurations (Figure 4), allowed the elucidation of 17R* configuration in 2. As 1 and 2 should share the same biosynthetic pathway, therefore, the structure of 2 was suggested to be (1R,2R,4S,17R)-loba-8,10,13-trien-17,18-diol.
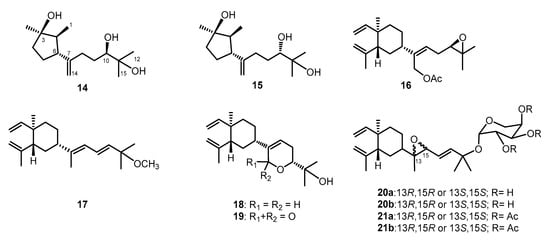
Figure 4.
Known sequiterpenoids chokol E (14) and 10-epi-chokol E (15); lobane diterpenoids (16–19); fuscosides C and D (20a and 20b), and fuscosides C and D acetates (21a and 21b).
Lobovarol H (3) possessed a molecular formula C20H32O2 (HRESIMS: m/z 327.2294 [M + Na]+ and showed the presence of hydroxy and olefinic functionalities from the IR absorptions at 3352 and 1645 cm−1. The NMR data (Table 1) revealed that 3 is a lobane diterpenoid, with the presence of a trisubstituted (δC 145.3, C and 126.1, CH; δH 6.02, d, J = 11.0 Hz) and an 1,2-disubstituted double bond (δC 142.0, CH and 121.9, CH; δH 6.61, dd, J = 15.0, 11.0 Hz and 5.88, d, J = 15.0 Hz); and hydroxymethylene group (δC 60.0 CH2; δH 4.31 and 4.29, each 1H, d, J = 12.5 Hz) in the side chain.
The NMR data of the side chain of 3 was found to be the same as the corresponding data of known compound 13 except the olefinic methyl of 13 was replaced by a hydroxymethylene group (δH 4.31 and 4.29, each 1H, d, J = 12.5 Hz) in 3. The HMBC correlations of 3 further confirmed the C-13, C-13/C-15, C-16/C-17, and C-18 positions of the hydroxymethylene, two conjugated double bonds, and a hydroxy group, respectively (Figure 2). Finally, the strong NOE correlation of H2-14/H-16 and the large J value of H-16 and H-17 (15.0 Hz) determined the Z and E configurations of C-13/C-15 and C-16/C-17 double bonds, respectively. Compound 3 was thus established as (1R,2R,4S,13Z,16E)-loba-8,10,13(15),16-tetraen-14,18-diol.
Lobovarol I (4) displayed a sodiated ion peak [M + Na]+ at m/z 387.2504 in the HRESIMS, appropriate for a molecular formula C22H36O4, and five degrees of unsaturation. The IR absorptions at 3278 and 1740 cm−1 revealed the presence of the hydroxy and ester carbonyl groups. The NMR data (Table 1) showed the presence of a trisubstituted double bond (δC 141.0, C and 124.7, CH; δH 5.58, dd, J = 8.0, 6.8 Hz), a trisubstituted epoxide (δC 58.6, C; 63.7, CH; δH 2.76, 1H, dd, J = 6.4, 6.4 Hz), and an acetoxy group (δC 171.0, C; 21.1 CH3; δH 2.06, 3H, s). The remaining two degrees of unsaturation have arisen from the presence of two rings. A decalin bicarbocyclic structure with an angular methyl (δH 0.90, 3H, s) and a methine (δC 54.9, CH; δH 1.27, m) was elucidated from the COSY and HMBC correlations (Figure 2) which showed 4 to be a prenyleudesmane type diterpene [9,10]. The C-4 position of the hydroxy group was confirmed from the HMBC correlation of H3-16 (δH 1.12, s,) to C-5 (δC 54.9, CH) and C-4 (δC 72.2, C). Moreover, the NMR data of the side chain of 4 were found to be identical in all aspects to those of the side chain of acetoxylobaoxide (16) (Figure 4) [5], the gross structure of 4 was thus established unambiguously.
The relative configuration of 4 was determined based on biogenetic consideration, NOESY correlations (Figure 5a) and NMR data comparison of related metabolites. Accordingly, the steric orientations of H3-17, H-5, and H-7 in the bicarbocyclic ring system of prenyleudesmanes 4 and 5–11 (vide infra) should be α, β, and β, respectively, as the same with the corresponding H3-17, H-2, and H-4 of the ring system of the lobanes (1–3), as the showed by the NOESY analysis of lobovarol I (4) (Figure 5a). Consequently, the NOE interaction displayed between H3-17 and H3-16 indicated the β-orientation of the hydroxy group at C-4. Furthermore, the NMR data of the side chain in 4 were identical to those of the side chain in acetoxylobaoxide (16), of which the R absolute configuration at C-14 has been determined after reduction of the C-14/C-15 epoxide [5]. From the above results, the structure of 4 was determined as (4S,5S,7S,10S,11Z,14R)-14,15-epoxy-18-acetoxyprenyleudesma-11- en-4-ol.
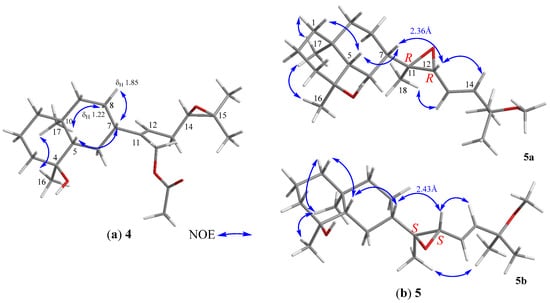
Figure 5.
(a) Selected NOE correlations of 4; (b) Selected NOE correlations of 11,12-bisepimers.
Lobovarol J (5a and 5b) were obtained as an inseparable mixture (≈ 1:1) of two isomers as revealed from the NMR spectra which displayed two sets of signals with an only slight difference in the chemical shifts for certain carbon and proton atoms. The HRESIMS (m/z 359.2555 [M + Na]+) was determining a molecular formula (C21H36O3) for the mixture of two isomers. Comparison of the NMR data of 5a and 5b (Table 2) with those of 4 (Table 1) revealed that lobovarol J possessed the same bicarbocyclic system as that of 4 (Table 2) and the similar side chain of fuscosides C & D triacetates (21a and 21b) [24], except that the sugar moiety in 21a or 21b (Figure 4) was replaced by a methoxy group (δC 50.4, CH3; δH 3.10, 3H, s) in 5. The planar structure of 5a and 5b was then established, as illustrated in Figure 2. The E configuration of the 13,14-double bond and the trans 11,12-epoxide were deduced from the J values of H-13 and H-14 (16.0 Hz) and the NOE correlations (Figure 5b), respectively. However, by using MM2 calculation for either stable conformation of 5a or 5b, it was found that either isomer showed a distance of near 2.4 Å between the NOE-interacting protons H-12 and H-7 (Figure 5b). Therefore, the two new compounds were defined as 11R,12R- (5a) and 11S,12S- (5b) isomers of (4S,5S,7S,10S,13E)-11,12-epoxy-15-methoxyprenyleudesma-13-en-4-ol with the exact NMR spectroscopic data for each isomer remained for further elucidation.
Lobovarol K (6) has a molecular formula C21H36O2, as displayed from the sodiated ion peak in the HRESIMS (m/z 343.2606 [M + Na]+), and showed the presence of a hydroxy group (IR 3431 cm−1). The NMR spectroscopic data of 6 (Table 2) are similar to a methoxylated (δC 50.4, CH3 and δH 3.18, 3H, s) prenyleudesmane, including signals of five methyls. The 1H and 13C NMR data of 6 further revealed that it is a diterpene possessing the same side chain as that of lobane 17 [2]. The chemical shifts of C-18 (δC 15.3, CH3) and J value of H-13 and H-14 (15.6 Hz) indicated the E-geometry for the 11,12- and 13,14-double bonds, respectively. The NOE correlations (Figure 6a) and the above-mentioned biogenetic consideration established 6 as (4S,5S,7S,10S,11E,13E)-15- methoxyprenyleudesma-11,13- dien-4-ol.
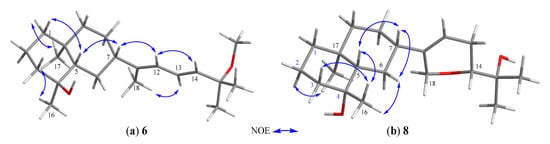
Figure 6.
(a) Selected NOE correlations of 6; (b) selected NOE correlations of 8.
Lobovarol L (7) possessed a molecular formula C20H34O3 (HRESIMS m/z 345.2401 [M + Na]+). Compound 7 displayed the combined NMR data of the eudesmane ring system in 4–6 (Table 2), and the side chain of lobatriene (18) [19]. The planar structure was further established by the analysis of COSY and NMR correlations (Figure 2). The configuration of 7 was established after the structure determination of 8. NOE correlations between the known α-oriented H3-17 and H3-16, and β-oriented H-7 with H-5, but not between H-5 and both H3-16 and H3-17 led to the structure of 7 to be determined as shown in formula 7.
The HRESIMS of lobovarol M (8) showed the same molecular formula C20H34O3 as that of 7, suggesting 8 to be an isomer for 7. The NMR data of 8 were found to be very similar to those of 7 (Table 2); however, 8 exhibited more upfield chemical shifts for carbons around C-4, in particular C-2, C-3, and C-5 (∆δC −2.1, −2.1, and −3.1, respectively) relative to the correspondent carbons in 7, supporting that 8 is the 4-epimer of 7. Unlike the NOE correlations of 7, 8 exhibited correlations (Figure 6b) between H3-17 and H-2α, but not between H3-17 and H3-16. Also, H3-16 showed NOE interactions with both H-3β and H-5, and H-5 displayed correlation with the β-oriented H-7, thus 8 was determined as 4-epimer of 7.
Lobovarol N (9) displayed a sodiated ion peak at m/z 359.2191 [M + Na]+ in the HRESIMS spectrum of a molecular formula C20H32O4 and IR absorptions at 3430 and 1700 cm−1 of the hydroxy and ester carbonyl functionalities. The 13C and 1H NMR data (Table 3) were similar to those of 7 except the replacement of the 18-hydroxymethylene signals (δC 68.2, CH2; δH 4.23 and 4.18, each br d, J = 16.4 Hz) in 7 by a signal at δC 164.7 (C) in 9. Thus, 9 is the 16-oxo derivative of 7. The NMR data of the side chain of 9 are also nearly identical with those of lobatrienolide (19) [5] coexisting in the same organism [1]. A combination of 2D NMR analyses (Figure 2) and biogenetic relationship with the coexisting biogenetically related lobanes 1,19 [5], and 17,18-epoxyloba-8,10,13(15)-trien-16-ol [1] with confirmed absolute configurations established the structure of 9 as (4S,5S,7S,10S,11Z,14R)-14,18-epoxyprenyleudesma-11-en-4,15-diol-18-one.
The sodium adduct ion peak [M + Na]+ of lobovarol O (10) at m/z 343.2242 in the HRESIMS showed a molecular formula C20H32O3 of 10. NMR data (Table 3) of 10 showed that it has the same cyclic ether ring as 7, 8, and lobatriene (18) [19] in the side chain. In comparison of the NMR spectroscopic data of 10 (Table 3) with those of 7 and 8 (Table 2) and analysis of COSY spectrum which showed the proton sequence of H-1 (δH 3.42, dd, J = 11.5, 4.0 Hz) to H-3 and the HMBC correlations of H3-17 (δH 0.70, 3H, s) to C-1 (δC 79.2), C-5 (δC 47.5), C-9 (δC 36.9), and C-10 (δC 40.2), confirmed the presence of a hydroxy group at C-1 and the prenyleudesmane molecular skeleton of 10. Furthermore, NOE correlations of H-1 with the β-oriented H-5 and one proton of H2-9 (δH 1.20, m) showed the β-orientation of H-1 and this H-9. The other proton at C-9 (δH 1.94, m) was thus assigned as H-9α which exhibited NOE correlation with H3-17, but not with both H-1 and H-5, thus H3-17 was assigned as α-oriented.
Lobovarol P (11) has the molecular formula C22H36O4 (HRESIMS: m/z 387.2504 [M + Na]+). The NMR data (Table 3) revealed that it possesses the same decalin bicyclic structures [10] and the same side-chain [3] of known compounds. The gross structure of 11 was thus determined and the relative configuration of 11 was further established by analysis of the NOESY spectrum which exhibited NOE interactions of the β-oriented H-5 (δH 1.83, d, J = 11.5 Hz) with both H-1β (δH 1.28, m) and H-7 (δH 2.10, m), and H-1α (δH 1.28, m) with H3-17 (δH 0.73, 3H, s). Thus, H-7 was determined to be positioned on the β face. By the biogenetic consideration, lobovarol P was found to possess (5R,7S,10S)-configuration, the same as that of lobovarol E [1].
Although 5,6-dihydro-2H-pyran and 5,6-dihydro-2H-pyran-2-one moieties have been known to be present in the side chain of lobane diterpenoid isolated from genus Lobophytum [1,2,3,5], this is the first report about discovering both pyran and pyranone systems in the structures of prenyleudesmanes. The structures of two known compounds loba-8,10,13(15)-trien-14,17,18-triol-14,17-diacetate (12) [3] and the eudesmane derivative (13) [9] were established mainly based on the MS and partial 1H NMR data. The detailed 1D and 2D NMR spectroscopic analysis enabled us to fully assign the 1H and 13C NMR data of these two diterpenoids for the first time.
Cytotoxicity for compounds 1, 2, 4, 7, 8, 12, and 13 against the growth of human colon adenocarcinoma (DLD-1), human colon carcinoma (HT-29), and human liver bile duct carcinoma (HuCCT-1); and for 3, 5, 6, and 9–11 against DLD-1 and mouse lymphocytic leukemia (P388) cancer cell lines using the Alamar Blue assay has been screened [25,26]. The results showed that all of the tested metabolites did not exhibit cytotoxicity towards the above cell lines (IC50 > 40 μg/mL) compared to doxorubicin HCl (IC50 0.9~7.4 μg/mL).
The anti-inflammatory activities of diterpenoids 2–13 against the fMLF/CB-induced pro-inflammatory responses on human neutrophils were evaluated [27,28], too. The results (Table 4) showed that 12 and 13 have potent inhibitory effect against the elastase release (IC50 6.9 ±2.7 and 4.4 ± 0.7 μM, respectively). Compounds 2 and 4 could suppress elastase release (IC50 18.8 ± 1.8 and 20.00 ± 3.0 μM), too, while 13 is the only compound also could significantly suppress the superoxide anion generation (IC50 13.7 ± 4.4 μM). Thus 2, 4, 12, and 13, in particular 12 and 13, have the potential to become anti-inflammatory agents. Compound 1 was found to strongly suppress elastase release in the absence of fMLF/CB, and is worthy for further biological study.
Table 4.
Inhibitory effects of compounds 1–13 on the generation of superoxide anion and release of elastase in fMLF/CB-stimulated human neutrophils.
Although the structures of lobanes 1–3, and 12 and eudesmanes 4–11, and 13, along with the previously reported related metabolites from the same organism [1] possess different ring structures, they are coexisted in L. varium and are biogenetically related to each other
A prenylgermacrene has been regarded as the same biosynthetic precursor [8,9] and we further propose a stereospecific biosynthesis for lobane and prenyleudesmane diterpenoids through Cope rearrangement and acid-catalyzed cyclization, respectively [9,29], as illustrated in Scheme 1. Thus, the co-occurrence of lobanes and prenyleudesmanes could be found in the same soft coral as in the cases of L. varium [1], Lobophytum sp. [9], Eunicea fusca [24], Sinularia gyrosa [30], and Sinularia polydactyla [31], is scientifically reasonable.
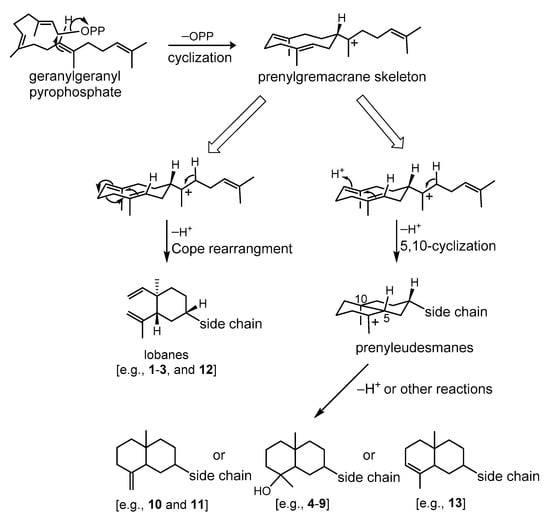
Scheme 1.
Plausible biosynthetic pathway of lobanes and prenyleudesmane-type diterpenoids.
3. Experimental Section
3.1. General Experimental Procedures
Optical rotations and IR spectra were measured on a JASCO P-1020 polarimeter and FT/IR-4100 infrared spectrophotometer (Jasco Corporation, Tokyo, Japan), respectively. ESIMS and HRESIMS experiments were performed on VG Quattro GC/MS and Bruker APEX II mass spectrometers, respectively. The NMR spectra were recorded on a Varian Unity Inova 500 FT-NMR (Varian Inc., Palo Alto, CA, USA) at 500 and 125 MHz for 1H and 13C, respectively; or on a Varian 400 FT-NMR at 400 and 100 MHz for 1H and 13C, respectively. Silica gel 60 or reversed-phase silica gel (RP-18; 230–400 mesh) and precoated silica gel plates (Kieselgel 60 F254, 0.2 mm) (Merck, Darmstadt, Germany) were used for open column chromatography (CC) and analytical TLC analysis, respectively. Isolation and purification of isolates by HPLC were performed by Hitachi L-2455 instrument equipped with an RP-18 column (ODS-3, 5 µm, 250 × 20 mm; Sciences Inc., Tokyo, Japan).
3.2. Aminal Material
The soft coral L. varium (Tixier-Durivault, 1970) was collected in March 2013 at a depth of 10 to 15 m at Jihui Fish Port, Taitung, Taiwan (23°7′2″ N, 121°23′49.2″ E) and identified by Professor Chang-Feng Dai, Institute of Oceanography, National Taiwan University, Taipei, Taiwan [1].
3.3. Extraction and Isolation
The sliced frozen soft coral L. varium (1.3 kg, wet weight) was exhaustively extracted with EtOAc, and the resulting solvent-free extract (55.40 g) was fractionated by silica gel column chromatography into 24 fractions (F1 to F24) as described before1. F19 (1.39 g), eluting with 50% EtOAc in n-hexane, was permeated through a column of Sephadex LH-20 using acetone to afford two diterpenoid-rich fractions F19-1 (105.0 mg) and F19-2 (94.0 mg). F19-1 was chromatographed over silica gel and eluted with EtOAc–n-hexane (1:3) to give two subfractions F19-1a (70.5 mg) and F19-1b (15.0 mg). F19-1a was further divided by reverse phase (RP-18) column chromatography, using MeOH–H2O, into two subfractions F19-1a1 and F19-1a2 which were purified separately by RP-18 HPLC, using CH3CN–H2O (2:1), to yield 1 (2.4 mg) and 2 (3.1 mg) from F19-1a1, and 12 (10.0 mg) from F19-1a2, respectively. F19-1b (15.0 mg) was purified by RP-18 HPLC using CH3CN–H2O (1.5:1) to afford 4 (1.1 mg). F19-2 was also primarily chromatographed over silica gel column using EtOAc–n-hexane (1:3) where the eluted diterpenoid-rich fraction (50.1 mg) was further isolated by RP-18 column chromatography, using MeOH–H2O (9:1 then 2:1), to yield 13 (1.4 mg), 7 (1.5 mg), and 8 (4.8 mg), respectively. F20 (1.20 g), eluted with 66.7% EtOAc in n-hexane, was fractionated over silica gel column, using EtOAc–n-hexane (5:95 to 0:100, gradient), to yield four fractions (F20-1 to F20-4). Isolation of F20-1 over RP-18 silica gel column, using MeOH–H2O (9:1), gave 5 (2.2 mg) and 6 (3.8 mg), respectively. F20-2 was further separated by RP-HPLC, using MeOH–H2O (6:1), to afford 10 (2.0 mg) and 11 (3.2 mg), respectively. F21 (0.50 g), eluted with 100% EtOAc, was similarly further fractionated, as for fractionation of F20, to give F21-1 and F21-2. RP-18 silica gel column chromatography was then applied to purify F21-1, using MeOH–H2O (5:1), to afford 3 (5.0 mg) and to further purify F21-2, using MeOH–H2O (6:1), to yield 9 (4.0 mg).
Lobovarol F (1): Colorless oil; + 2.8 (c 2.4, CHCl3); IR (neat) vmax 3393, 2926, 1643, 1532, 1462, 1147, 1024, and 897 cm−1; 13C and 1H NMR data (see Table 1); ESIMS m/z 345 [M + Na]+; HRESIMS m/z 345.2403 [M + Na]+ (calcd for C20H34O3Na, 345.2400).
Lobovarol G (2): Colorless oil; + 44.5 (c 3.1, CHCl3); IR (neat) vmax 3392, 2926, 2860, 1642, 1523, and 891 cm−1; 13C and 1H NMR data (see Table 1); ESIMS m/z 329 [M + Na]+; HRESIMS m/z 329.2453 [M + Na]+ (calcd for C20H34O2Na, 329.2451).
Lobovarol H (3): Colorless oil; −46.7 (c 0.57, CHCl3); IR (neat) vmax 3352, 2926, 1645, 1456, 1374, and 898 cm−1; 13C and 1H NMR data (see Table 1); ESIMS m/z 327 [M + Na]+; HRESIMS m/z 327.2294 [M + Na]+ (calcd for C20H32O2Na, 327.2295).
Lobovarol I (4): Colorless oil; + 66.0 (c 1.1, CHCl3); IR (neat) vmax 3278, 2919, 2852, 1740, and 1531 cm−1; 13C and 1H NMR data (see Table 1); ESIMS m/z 387 [M + Na]+; HRESIMS m/z 387.2504 [M + Na]+ (calcd for C22H36O4Na, 387.2506).
Lobovarol J (mixture of 5a and 5b): Colorless oil; + 14.2 (c 0.63, CHCl3); IR (neat) vmax 3443, 2929, 1531, 1456, 1378, and 1068 cm−1; 13C and 1H NMR data (see Table 2); ESIMS m/z 359 [M + Na]+; HRESIMS m/z 359.2555 [M + Na]+ (calcd for C21H36O3Na, 359.2557).
Lobovarol K (6): Colorless oil; −10.9 (c 0.46, CHCl3); IR (neat) vmax 3431, 2922, 1670, 1535, 1458, 1377, and 1071 cm−1; 13C and 1H NMR data (see Table 2); ESIMS m/z 343 [M + Na]+; HRESIMS m/z 343.2606 [M + Na]+ (calcd for C21H36O2Na, 343.2608).
Lobovarol L (7): Colorless oil; + 99.2 (c 1.5, CHCl3); IR (neat) vmax 3378, 2922, 2852, 1540, 1458, 1089, 906, and 670 cm−1; 13C and 1H NMR data (see Table 2); ESIMS m/z 345 [M + Na]+; HRESIMS m/z 345.2401 [M + Na]+ (calcd for C20H34O3Na, 345.2400).
Lobovarol M (8): Colorless oil; + 87.7 (c 4.8, CHCl3); IR (neat) vmax 3459, 2927, 2852, 1451, 1378, 1161, 1088, and 756 cm−1; 13C and 1H NMR data (see Table 2); ESIMS m/z 345 [M + Na]+; HRESIMS m/z 345.2401 [M + Na]+ (calcd for C20H34O3Na, 345.2400).
Lobovarol N (9): Colorless oil; + 59.7 (c 0.54, CHCl3); IR (neat) vmax 3430, 2924, 1700, 1429, 1376, and 1096 cm−1; 13C and 1H NMR data (see Table 3); ESIMS m/z 359 [M + Na]+; HRESIMS m/z 359.2191 [M + Na]+ (calcd for C20H32O4Na, 359.2193).
Lobovarol O (10): Colorless oil; + 48.1 (c 0.57, CHCl3); IR (neat) vmax 3433, 2926, 1696, 1532, 1024, and 670 cm−1; 13C and 1H NMR data (see Table 3); ESIMS m/z 343 [M + Na]+; HRESIMS m/z 343.2242 [M + Na]+ (calcd for C20H32O3Na, 343.2244).
Lobovarol P (11): Colorless oil; −54.0 (c 0.71, CHCl3); IR (neat) vmax 3503, 2925, 1731, 1373, and 1237 cm−1; 13C and 1H NMR data (see Table 3); ESIMS m/z 387 [M + Na]+; HRESIMS m/z 387.2504 [M + Na]+ (calcd for C22H36O4Na, 387.2506).
Compound 12: Colorless oil; + 57.0 (c 14.3, CHCl3); IR (neat) vmax 3496, 2927, 2937, 1736, 1640, 1442, 1373, and 1027 cm−1; 1H NMR (CDCl3, 400 MHz): δH 5.80 (1H, dd, J = 17.6, 10.4 Hz, H-8), 5.48 (1H, dd, J = 7.2, 7.2 Hz, H-15), 4.89 (1H, d, J = 17.6 Hz, H-9a), 4.89 (1H, d, J = 10.4 Hz, H-9b), 4.85 (1H, dd, J = 9.2, 4.0 Hz, H-17), 4.81 (1H, s, H-11a), 4.66 (1H, d, J = 12.0 Hz, H-14a), 4.58 (1H, d, J = 12.0 Hz, H-14b), 4.56 (1H, s, H-11b), 2.46, (2H, m, H-6, H2-16), 2.06 (3H, s, 17-OCOCH3), 2.05 (3H, s, 14-OCOCH3) 2.03, (1H, m, H-4), 1.99 (1H, dd, J = 11.6, 3.6 Hz, H-2), 1.69 (3H, s, H3-12), 1.53, (2H, m, H2-5), 1.49 (2H, m, H2-3), 1.41 (2H, m, H2-6), 1.22 (6H, s, H3-19 and H3-20), 0.98 (3H, s, H3-7); 13C NMR (CDCl3, 100 MHz): δC 171.0 (C, 14-OCOCH3), 170.6 (C, 17-OCOCH3), 150.0 (CH, C-8), 147.4 (C, C-10), 140.7 (C, C-13), 125.6 (CH, C-15), 112.2 (CH2, C-11), 110.0 (CH2, C-9), 78.9 (CH, C-17), 72.1 (C, C-18), 61.1 (CH2, C-14), 52.6 (CH, C-2), 43.7 (CH, C-4), 39.8 (CH2, C-6), 39.7 (C, C-1), 33.1 (CH2, C-3), 28.2 (CH2, C-16), 27.1 (CH2, C-5), 26.5 (CH3, C-19), 25.3 (CH3, C-20), 24.8 (CH3, C-12), 21.1 (CH3, 14-OCOCH3), 20.9 (CH3, 17-OCOCH3), 16.5 (CH3, C-7); ESIMS m/z 429 [M + Na]+; HRESIMS m/z 429.2609 [M + Na]+ (calcd for C24H38O5Na, 429.2612).
Compound 13: Colorless oil; − 81.0 (c 1.4, CHCl3); IR (neat) vmax 3254, 2925, 2857, 1640, 1450, and 1023 cm−1; 1H NMR (CDCl3, 500 MHz): δH 6.50 (1H, dd, J = 15.5, 11.0 Hz, H-13), 5.90 (1H, d, J = 11.0 Hz, H-12), 5.78 (1H, d, J = 15.5 Hz, H-14), 5.29 (1H, s, H-3), 4.58 (1H, dd, J = 7.0, 7.0 Hz, H-1), 3.55 (1H, dd, J = 7.0, 7.0 Hz, H-1), 2.31 (1H, m, H-2a), 1.98 (2H, m, H-2b and H-7), 1.95 (1H, m, H-5), 1.92 (1H, m, H-9a), 1.71 (1H, m, H-6a), 1.80 (3H, s, H3-18), 1.60 (2H, m, H2-8), 1.58 (3H, s, H3-16), 1.36 (6H, s, H3-19 and H3-20), 1.29 (1H, m, H-6b), 1.14 (1H, m, H-9b), 0.79 (3H, s, H3-17); 13C NMR (CDCl3, 125 MHz): δC 143.6 (C, C-11), 139.3 (CH, C-14), 135.2 (C, C-4), 123.0 (CH2, C-13), 122.7 (CH, C-12), 119.5 (CH, C-3), 76.4 (CH, C-1), 71.0 (C, C-15), 52.8 (CH2, C-2), 48.0 (CH, C-7), 46.6 (CH, C-5), 37.4 (C, C-10), 35.0 (CH2, C-9), 29.9 (2 x CH3, C-19 and C-20), 28.2 (CH2, C-6), 26.0 (CH2, C-8), 20.8 (CH3, C-16), 15.0 (CH3, C-18), 9.5 (CH3, C-17); ESIMS m/z 327 [M + Na]+; HRESIMS m/z 327.2296 [M + Na]+ (calcd for C20H32O2Na, 327.2295).
Preparation of (S)- and (R)-MTPA Esters of 1
To a solution of 1 (1.0 mg, 3.1 μmol) in anhydrous pyridine (200 μL) was added S-(+)- -α-methoxy-α-(trifluoromethyl)phenylacetyl (MTPA) chloride (20 μL), and the mixture was further reacted for 20 h at room temperature. The reaction mixture was then processed as previously described [20,21] to yield the (R)-MTPA ester 1a (0.5 mg, 0.93 μmol, 30%). The correspondent (S)-MTPA ester 1b was similarly yielded from the reaction of R-(–)-MTPA chloride with 1. 1H NMR (CDCl3, 400 MHz) of 1a: δH 5.8005 (1H, dd, J = 17.6, 10.4 Hz, H-8), 4.892 (1H, d, J = 12 Hz, H-9b), 5.114 (1H, dd, J = 5.6, 5.6 Hz, H-17), 4.8895 (1H, d, J = 16.4 Hz, H-9a), 4.822 (1H, s, H-11b), 4.591 (1H, s, H-11a), 3.949 (1H, dd, J = 8.4, 6.8 Hz, H-15), 2.395 (1H, ddd, J = 10.0, 6.8, 6.8 Hz, H-16b), 1.994 (1H, m, H-16a), 1.9475 (1H, m, H-2), 1.708 (3H,s, H3-12), 1.689 (1H, d, J = 15.2, H-3b), 1.522 (1H, m, H-3a), 1.367 (1H, m, H-4), 1.394-1.229 (4H, m, H2-5 and H2-6), 1.279 (3H, s, H3-20), 1.210 (3H, s, H3-14), 1.158 (3H, s, H3-19), 0.981 (3H, s, H3-7); 1H NMR (CDCl3, 400 MHz) of 1b: δH 5.803 (1H, dd, J = 17.6, 10.4 Hz, H-8), 4.895 (1H, d, J = 12 Hz, H-9b), 5.136 (1H, dd, J = 6.0, 6.0 Hz, H-17), 4.8945 (1H, d, J = 16.4 Hz, H-9a), 4.883 (1H, s, H-11b), 4.605 (1H, s, H-11a), 3.971 (1H, dd, J = 8.0, 7.6 Hz, H-15), 2.419 (1H, ddd, J = 9.6, 6.8, 6.8 Hz, H-16b), 2.107 (1H, m, H-16a), 1.989 (1H, m, H-2), 1.718 (3H,s, H3-12), 1.7665 (1H, br d, J = 14.0, H-3b), 1.5325 (1H, m, H-3a), 1.415 (1H, m, H-4), 1.394-1.256 (4H, m, H2-5 and H2-6), 1.236 (3H, s, H3-20), 1.198 (3H, s, H3-14), 1.106 (3H, s, H3-19), 0.995 (3H, s, H3-7).
3.4. Cyotoxic Testing
Cytotoxicities of 1–11 were assayed using Almar Blue assay [25,26]. Doxorubicin HCl, employed as positive control, displayed cytotoxic activity toward DLD-1, HT-29, HuCCT-1, and P388 cell lines with IC50 0.9, 4.4, 2.6, and 7.4 μg/mL, respectively.
3.5. In Vitro Anti-Inflammatory Testing
The experiment for measuring superoxide anion generation and elastase release were manipulated according to previously reported method [28,32,33].
4. Conclusions
In conclusion, our further chemical investigation and biological evaluation on the EtOAc extract of the soft coral L. varium disclosed three new lobane and eight prenyleudesmane diterpenoids along with two known derivatives. Compound 1 is the first labane possessing a tetrahydrofuran ring at the end of the side chain, and displayed ability to inhibit elastase release in the absence of fMLF/CB. Compounds 2, 4, 12, and 13, in particular 12 and 13, displayed the potential to be the leads for anti-inflammatory medicines. Our proposed stereospecific biosynthetic pathway can explain the common coexistence of both lobanes and prenyleudesmanes in the soft coral L. varium.
Supplementary Materials
HRESIMS, 1H, 13C, COSY, heteronuclear single quantum coherence spectroscopy (HSQC), HMBC, and NOESY spectra of new compounds 1–11 and 1H NMR spectrum of MTPA ester of 1 are available online at https://www.mdpi.com/1660-3397/18/4/223/s1, Figure S1: HRESIMS spectrum of 1, Figure S2: 1H NMR spectrum of 1 in CDCl3 at 400 MHz, Figure S3: 13C NMR spectrum of 1 in CDCl3 at 100 MHz, Figure S4: 1H-1H COSY spectrum of 1 in CDCl3, Figure S5: HSQC spectrum of 1 in CDCl3, Figure S6: HMBC spectrum of 1 in CDCl3, Figure S7: NOESY spectrum of 1 in CDCl3, Figure S8: 1H NMR spectrum of (S)-MTPA ester of 1 (1a) in CDCl3 at 400 MHz, Figure S9: 1H NMR spectrum of (R)-MTPA ester of 1 (1b) in CDCl3 at 400 MHz, Figure S10: HRESIMS spectrum of 2, Figure S11: 1H NMR spectrum of 2 in CDCl3 at 400 MHz, Figure S12: 13C NMR spectrum of 2 in CDCl3 at 100 MHz, Figure S13: 1H-1H COSY spectrum of 2 in CDCl3, Figure S14: HSQC spectrum of 2 in CDCl3, Figure S15: HMBC spectrum of 2 in CDCl3, Figure S16: NOESY spectrum of 2 in CDCl3, Figure S17: HRESIMS spectrum of 3, Figure S18: 1H NMR spectrum of 3 in CDCl3 at 500 MHz, Figure S19: 13C NMR spectrum of 3 in CDCl3 at 125 MHz, Figure S20: 1H-1H COSY spectrum of 3 in CDCl3, Figure S21: HSQC spectrum of 3 in CDCl3, Figure S22: HMBC spectrum of 3 in CDCl3, Figure S23: NOESY spectrum of 3 in CDCl3, Figure S24: HRESIMS spectrum of 4, Figure S25: 1H NMR spectrum of 4 in CDCl3 at 400 MHz, Figure S26: 13C NMR spectrum of 4 in CDCl3 at 100 MHz, Figure S27: 1H-1H COSY spectrum of 4 in CDCl3, Figure S28: HSQC spectrum of 4 in CDCl3, Figure S29: HMBC spectrum of 4 in CDCl3, Figure S30: NOESY spectrum of 4 in CDCl3, Figure S31: HRESIMS spectrum of 5a and 5b, Figure S32: 1H NMR spectrum of 5a and 5b acetone-d6 at 500 MHz, Figure S33: 13C NMR spectrum of 5a and 5b in acetone-d6 at 125 MHz, Figure S34: 1H-1H COSY spectrum of 5a and 5b in acetone-d6, Figure S35: HSQC spectrum of 5a and 5b in acetone-d6, Figure S36: HMBC spectrum of 5a and 5b in acetone-d6, Figure S37: NOESY spectrum of 5a and 5b in acetone-d6, Figure S38: HRESIMS spectrum of 6, Figure S39: 1H NMR spectrum of 6 in CDCl3 at 400 MHz, Figure S40: 13C NMR spectrum of 6 in CDCl3 at 100 MHz, Figure S41: 1H-1H COSY spectrum of 6 in CDCl3, Figure S42: HSQC spectrum of 6 in CDCl3, Figure S43: HMBC spectrum of 6 in CDCl3, Figure S44: NOESY spectrum of 6 in CDCl3, Figure S45: HRESIMS spectrum of 7, Figure S46: 1H NMR spectrum of 7 in CDCl3 at 400 MHz, Figure S47: 13C NMR spectrum of 7 in CDCl3 at 100 MHz, Figure S48: 1H-1H COSY spectrum of 7 in CDCl3, Figure S49: HSQC spectrum of 7 in CDCl3, Figure S50: HMBC spectrum of 7 in CDCl3, Figure S51: NOESY spectrum of 7 in CDCl3, Figure S52: HRESIMS spectrum of 8, Figure S53: 1H NMR spectrum of 8 in CDCl3 at 400 MHz, Figure S54: 13C NMR spectrum of 8 in CDCl3 at 100 MHz, Figure S55: 1H-1H COSY spectrum of 8 in CDCl3, Figure S56: HSQC spectrum of 8 in CDCl3, Figure S57: HMBC spectrum of 8 in CDCl3, Figure S58: NOESY spectrum of 8 in CDCl3, Figure S59: HRESIMS spectrum of 9, Figure S60: 1H NMR spectrum of 9 in CDCl3 at 400 MHz, Figure S61: 13C NMR spectrum of 9 in CDCl3 at 100 MHz, Figure S62: 1H-1H COSY spectrum of 9 in CDCl3, Figure S63: HSQC spectrum of 9 in CDCl3, Figure S64: HMBC spectrum of 9 in CDCl3, Figure S65: NOESY spectrum of 9 in CDCl3, Figure S66: HRESIMS spectrum of 10, Figure S67: 1H NMR spectrum of 10 in CDCl3 at 500 MHz, Figure S68: 13C NMR spectrum of 10 in CDCl3 at 125 MHz, Figure S69: 1H-1H COSY spectrum of 10 in CDCl3, Figure S70: HSQC spectrum of 10 in CDCl3, Figure S71: HMBC spectrum of 10 in CDCl3, Figure S72: NOESY spectrum of 10 in CDCl3, Figure S73: HRESIMS spectrum of 11, Figure S74: 1H NMR spectrum of 11 in CDCl3 at 500 MHz, Figure S75: 13C NMR spectrum of 11 in CDCl3 at 125 MHz, Figure S76: 1H-1H COSY spectrum of 11 in CDCl3, Figure S77: HSQC spectrum of 11 in CDCl3, Figure S78: HMBC spectrum of 11 in CDCl3, Figure S79: NOESY spectrum of 11 in CDCl3.
Author Contributions
J.-H.S. designed and guided the whole experiment; C.-H.C. and T.-S.Y. isolated the compounds and performed structure elucidation; A.F.A. and Y.-C.L. performed structure elucidation and manuscript preparation; C.-Y.H. performed cytotoxicity assay; T.-L.H. performed the anti-inflammatory activity assay. All authors have read and agreed to the published version of the manuscript.
Funding
The research was funded by the Ministry of Science and Technology of Taiwan (MOST 102-2628-B-110-002-MY2, 104-2320-B-110-001-MY2, 105-2113-M-110-002- and 105-2811-M-110-013-) and the Deanship of Scientific Research at King Saud University, Saudi Arabia (RG-1440-127).
Acknowledgments
This work was mainly supported by grants from Ministry of Science and Technology (MOST 102-2628-B-110-002-MY2, 104-2320-B-110-001-MY2, 105-2113-M-110-002- and 105-2811-M-110-013-) awarded to J.-H.S., A.F.A. would like to extend appreciation to the Deanship of Scientific Research at King Saud University for further funding this work through research group RG-1440-127.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ahmed, A.F.; Teng, W.T.; Hung, C.Y.; Dai, C.F.; Hwang, T.L.; Sheu, J.H. Anti-inflammatory lobane and prenyleudesmane diterpenoids from the soft coral Lobophytum varium. Mar. Drugs 2017, 15, 300. [Google Scholar] [CrossRef] [PubMed]
- Edrada, R.A.; Proksch, P.; Wray, V.; Witte, L.; van Ofwegen, L. Four new bioactive lobane diterpenes of the soft coral Lobophytum pauciflorum from Mindoro, Philippines. J. Nat. Prod. 1998, 61, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, R.W.; Wells, R.J. Isolation of some novel diterpenes from a soft coral of the genus Lobophytum. Aust. J. Chem. 1979, 32, 1345–1351. [Google Scholar] [CrossRef]
- Lakshmana Raju, B.; Subbaraju, G.V.; Bheemasankara Rao, C.; Trimurtulu, G. Two new oxygenated lobanes from a soft coral of Lobophytum species of the Andaman and Nicobar coasts. J. Nat. Prod. 1993, 56, 961–966. [Google Scholar] [CrossRef]
- Hamada, T.; Kusumi, T.; Ishitsuka, M.O.; Kakisawa, H. Structures and absolute configurations of new lobane diterpenoids from the Okinawan soft coral Sinularia flexibilis. Chem. Lett. 1992, 21, 33–36. [Google Scholar] [CrossRef]
- Govindam, S.V.; Yoshioka, Y.; Kanamoto, A.; Fujiwara, T.; Okamoto, T.; Ojika, M. Cyclolobatriene, a novel prenylated germacrene diterpene, from the soft coral Lobophytum pauciflorum. Bioorg. Med. Chem. 2012, 20, 687–692. [Google Scholar] [CrossRef]
- Minh, C.V.; Kiem, P.V.; Nhiem, N.X.; Cuong, N.X.; Thao, N.P.; Nam, N.H.; Anh, H.L.T.; Thung, D.C.; Thuy, D.T.T.; Kang, H.K.; et al. Cytotoxic and antioxidant activities of diterpenes and sterols from the Vietnamese soft coral Lobophytum compactum. Bioorg. Med. Chem. Lett. 2011, 21, 2155–2159. [Google Scholar] [CrossRef]
- Li, L.; Sheng, L.; Wang, C.Y.; Zhou, Y.B.; Huang, H.; Li, X.B.; Li, J.; Mollo, E.; Gavagnin, M.; Guo, Y.W. Diterpenes from the Hainan soft coral Lobophytum cristatum Tixier-Durivault. J. Nat. Prod. 2011, 74, 2089–2094. [Google Scholar] [CrossRef]
- Coll, J.C.; Bowden, B.F.; König, G.M.; Braslau, R.; Price, I.R. Studies of Australian soft corals. XXXX.1 The natural products chemistry of Alcyonacean soft corals with special reference to the genus Lobophytum. Bull. Soc. Chim. Belg. 1986, 95, 815–834. [Google Scholar] [CrossRef]
- Matthee, G.F.; König, G.M.; Wright, A.D. Three new diterpenes from the marine soft coral Lobophytum crassum. J. Nat. Prod. 1998, 61, 237–240. [Google Scholar] [CrossRef]
- Bowden, B.F.; Coll, J.C.; Liyanage, N.; Mitchell, S.J.; Stokie, G.J.; van Altena, I. A novel bicyclic diterpene was obtained from a soft coral Lobophytum hedleyi. Aust. J. Chem. 1978, 31, 163–170. [Google Scholar] [CrossRef]
- Chao, C.H.; Wen, Z.H.; Wu, Y.C.; Yeh, Y.C.; Sheu, J.H. Cytotoxic and anti-inflammatory cembranoids from the soft coral Lobophytum crassum. J. Nat. Prod. 2008, 71, 1819–1824. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.L.; Su, J.H. Tetrahydrofuran cembranoids from the cultured soft coral Lobophytum crassum. Mar. Drugs 2011, 9, 2526–2536. [Google Scholar] [CrossRef]
- Roy, P.K.; Roy, S.; Ueda, K. New cytotoxic cembranolides from an Okinawan soft coral, Lobophytum sp. Fitoterapia 2019, 136, 104162. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.A.; Gustafson, K.R.; Boyd, M.R. HIV-inhibitory cembrane derivatives from a Philippines collection of the soft coral Lobophytum species. J. Nat. Prod. 2000, 63, 531–533. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.Y.; Su, J.H.; Lu, M.C.; Hwang, T.L.; Wang, W.H.; Chen, J.J.; Sheu, J.H.; Kuo, Y.H.; Weng, C.F.; Fang, L.S.; et al. New cembrane-type diterpenoids from the soft coral Lobophytum crassum. Mar. Drugs 2011, 9, 1319–1331. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.H.; You, Y.J.; Lin, C.C.; El-Shazly, M.; Liao, Z.J.; Su, J.H. Anti-Inflammatory cembranoids from the soft coral Lobophytum crassum. Mar. Drugs 2017, 15, 327. [Google Scholar] [CrossRef]
- Thao, N.P.; Luyen, B.T.T.; Ngan, N.T.T.; Song, S.B.; Cuong, N.X.; Nam, N.H.; Kiem, P.V.; Kim, Y.H.; Minh, C.V. New anti-inflammatory cembranoid diterpenoids from the Vietnamese soft coral Lobophytum crassum. Bioorg. Med. Chem. Lett. 2014, 24, 228–232. [Google Scholar] [CrossRef]
- Kusumi, T.; Hamada, T.; Ishitsuka, M.O.; Ohtani, I.; Kakisawa, H. Elucidation of the relative and absolute stereochemistry of lobatriene, a marine diterpene, by a modified mosher method. J. Org. Chem. 1992, 57, 1033–1035. [Google Scholar] [CrossRef]
- Peng, C.-C.; Huang, C.-Y.; Ahmed, A.F.; Hwang, T.-L.; Dai, C.-F.; Sheu, J.-H. New cembranoids and a biscembranoid peroxide from the soft coral Sarcophyton cherbonnieri. Mar. Drugs 2018, 16, 276. [Google Scholar] [CrossRef]
- Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. High-Field FT NMR application of Mosher’s Method. The absolute configurations of marine terpenoids. J. Am. Chem. Soc. 1991, 113, 4092–4096. [Google Scholar] [CrossRef]
- Hiroyuki, K.; Satoshi, T.; Shun-ichi, T.; Yoshihara, T.; Sakamura, S.; Shimanuki, T.; Sato, T.; Tajimi, A. New fungitoxic sesquiterpenoids, chokols A-G, from stromata of Epichloe typhina and the absolute configuration of chokol E. Agric. BioI. Chem. 1989, 53, 789–796. [Google Scholar] [CrossRef]
- Pérez Morales, C.; Catalán, J.; Domingo, V.; González Delgado, J.A.; Dobado, J.A.; Herrador, M.M.; Quílez del Moral, J.F.; Barrero, A.F. Protecting-group-free synthesis of chokols. J. Org. Chem. 2011, 76, 2494–2501. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Fenical, W. Fuscosides A-D: Anti-inflammatory diterpenoid glycosides of new structural classes from the caribbean gorgonian Eunicea fusca. J. Org. Chem. 1991, 56, 3153–3158. [Google Scholar] [CrossRef]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef]
- Nakayama, G.R.; Caton, M.C.; Nova, M.P.; Parandoosh, Z. Assessment of the Alamar Blue assay for cellular growth and viability in vitro. J. Immunol. Methods 1997, 204, 205–208. [Google Scholar] [CrossRef]
- Hwang, T.L.; Yeh, S.H.; Leu, Y.L.; Chern, C.Y.; Hsu, H.C. Inhibition of superoxide anion and elastase release in human neutrophils by 3′-isopropoxychalcone via a cAMP-dependent pathway Br. J. Pharmacol. 2006, 148, 78–87. [Google Scholar]
- Hwang, T.L.; Su, Y.C.; Chang, H.L.; Leu, Y.L.; Chung, P.J.; Kuo, L.M.; Chang, Y.J. Suppression of superoxide anion and elastase release by C18 unsaturated fatty acids in human neutrophils. J. Lipid Res. 2009, 50, 1395–1408. [Google Scholar] [CrossRef]
- De Kraker, J.W.; Franssen, M.C.R.; de Groot, A.; König, W.A.; Bouwmeester, H.J. (+)-Germacrene a biosynthesis: The committed step in the biosynthesis of bitter sesquiterpene lactones in chicory. Plant Physiol. 1998, 117, 1381–1392. [Google Scholar] [CrossRef]
- Cheng, S.Y.; Chuang, C.T.; Wang, S.K.; Wen, Z.H.; Chiou, S.F.; Hsu, C.H.; Dai, C.F.; Duh, C.Y. Antiviral and anti-inflammatory diterpenoids from the soft coral Sinularia gyrosa. J. Nat. Prod. 2010, 73, 1184–1187. [Google Scholar] [CrossRef]
- Ye, F.; Zhu, Z.D.; Gu, Y.C.; Li, J.; Zhu, W.L.; Guo, Y.W. Further New Diterpenoids as PTP1B Inhibitors from the Xisha Soft Coral Sinularia polydactyla. Mar. Drugs 2018, 16, 103. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.C.; Chung, P.J.; Ho, C.M.; Kuo, C.Y.; Hung, M.F.; Huang, Y.T.; Chang, W.Y.; Chang, Y.W.; Chan, K.H.; Hwang, T.L. Propofol inhibits superoxide production, elastase release, and chemotaxis in formyl peptide-activated human neutrophils by blocking formyl peptide receptor 1. J. Immunol. 2013, 190, 6511–6519. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.P.; Hsieh, P.W.; Chang, Y.J.; Chung, P.J.; Kuo, L.M.; Hwang, T.L. 2-(2-Fluorobenzamido)benzoate ethyl ester (EFB-1) inhibits superoxide production by human neutrophils and attenuates hemorrhagic shock-induced organ dysfunction in rats. Free Radic. Biol. Med. 2011, 50, 1737–1748. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).