Cytotoxic Secondary Metabolites Isolated from the Marine Alga-Associated Fungus Penicillium chrysogenum LD-201810

Abstract
:1. Introduction
2. Results and Discussion
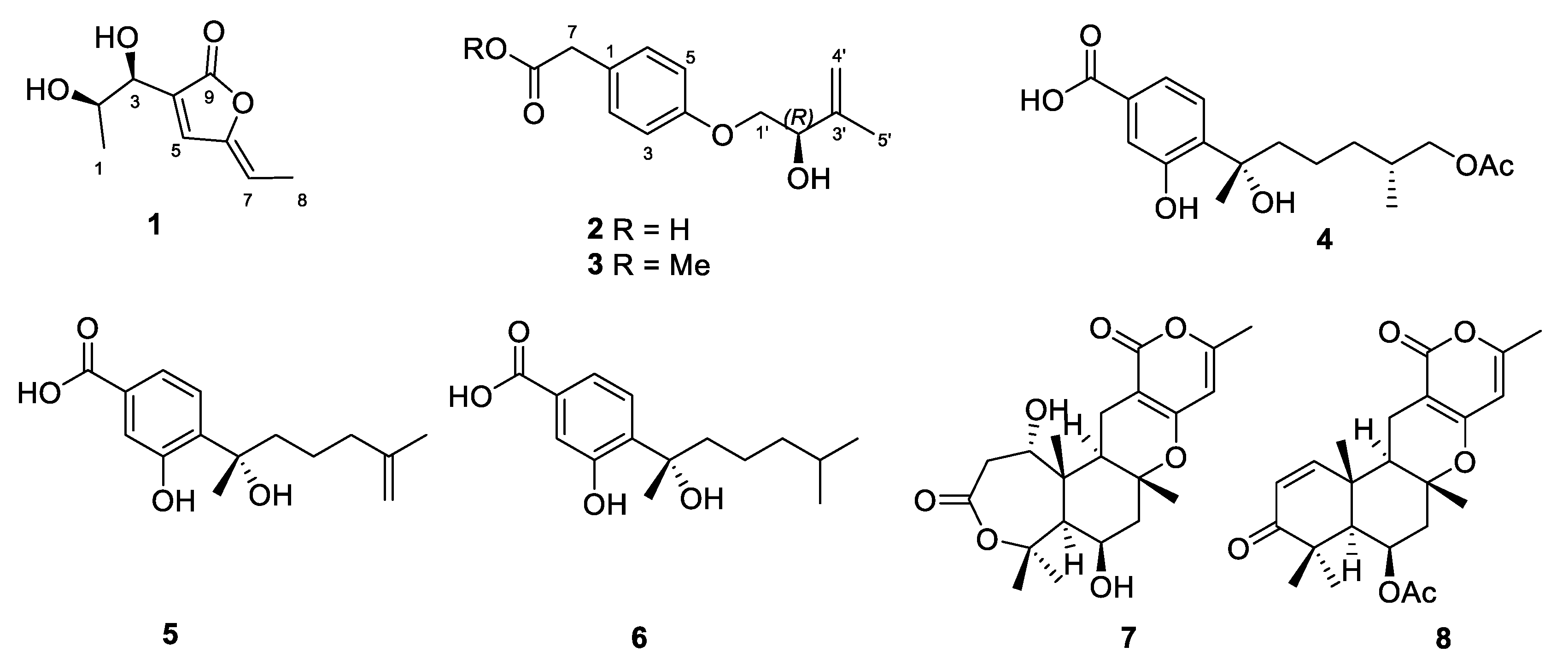
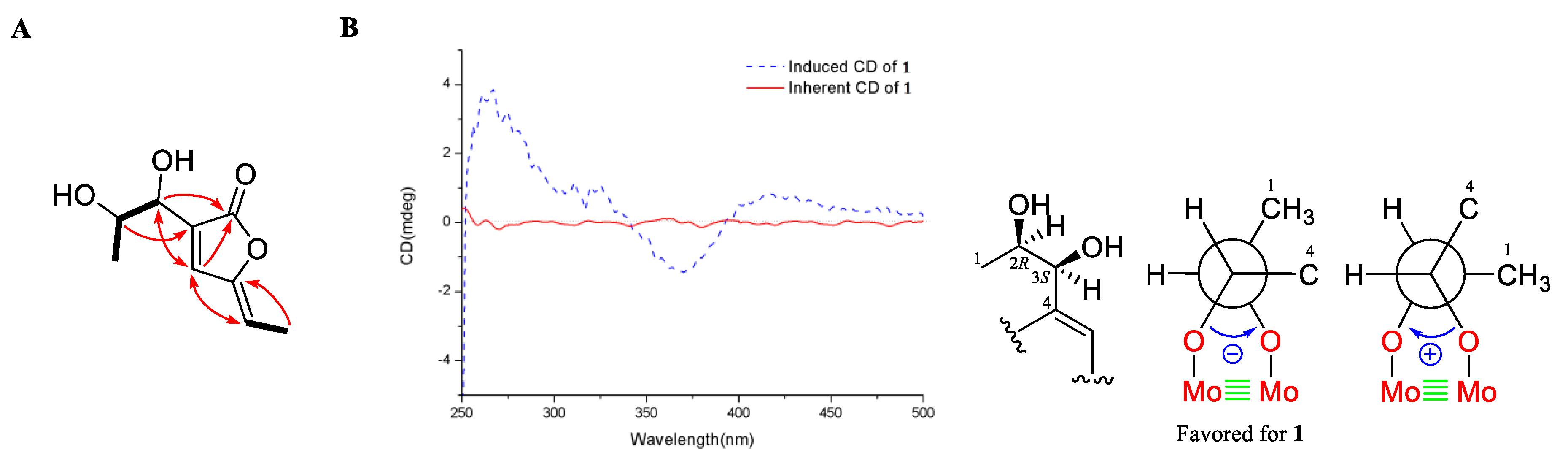
2.1. Structure Elucidation
2.2. Cytotoxicity of Compounds 1–8
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation, Extraction, and Isolation
3.4. Measurement of ICD Spectrum of 1 Using Mo2(OAc)4
3.5. Computational Section
3.6. X-ray Crystallographic Analysis of 2
3.7. Cytotoxic Assays
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hou, X.-M.; Xu, R.-F.; Gu, Y.-C.; Wang, C.; Shao, C.-L. Biological and Chemical Diversity of Coral-Derived Microorganisms. Curr. Med. Chem. 2015, 22, 3707–3762. [Google Scholar] [CrossRef] [PubMed]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2019, 36, 122–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.; Sun, C.; Sun, Z.; Zhang, G.; Che, Q.; Gu, Q.; Zhu, T.; Li, D. Sun Antibacterial Polyketides from Antarctica Sponge-Derived Fungus Penicillium sp. HDN151272. Mar. Drugs 2020, 18, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; Yuan, X.-L.; Li, C.; Li, X.-D. Recent Discovery of Heterocyclic Alkaloids from Marine-Derived Aspergillus Species. Mar. Drugs 2020, 18, 54. [Google Scholar] [CrossRef] [Green Version]
- Rateb, M.E.M.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290. [Google Scholar] [CrossRef]
- Ji, N.-Y.; Wang, B.-G. Mycochemistry of marine algicolous fungi. Fungal Divers. 2016, 80, 301–342. [Google Scholar] [CrossRef]
- Zhang, P.; Li, X.; Wang, B.-G. Secondary Metabolites from the Marine Algal-Derived Endophytic Fungi: Chemical Diversity and Biological Activity. Planta Medica 2016, 82, 832–842. [Google Scholar] [CrossRef] [Green Version]
- Soldatou, S.; Baker, B.J. Cold-water marine natural products, 2006 to 2016. Nat. Prod. Rep. 2017, 34, 585–626. [Google Scholar] [CrossRef]
- Chen, X.-W.; Li, C.-W.; Cui, C.-B.; Hua, W.; Zhu, T.-J.; Gu, Q.-Q. Nine New and Five Known Polyketides Derived from a Deep Sea-Sourced Aspergillus sp. 16-02-1. Mar. Drugs 2014, 12, 3116–3137. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Ding, L.; Li, X.; Wang, N.; Yan, Y.; Yang, M.; Cui, W.; Naman, C.B.; Cheng, K.; Zhang, W.; et al. A new lateral root growth inhibitor from the sponge-derived fungus Aspergillus sp. LS45. Bioorg. Med. Chem. Lett. 2019, 29, 1593–1596. [Google Scholar] [CrossRef]
- Di Bari, L.; Pescitelli, G.; Pratelli, C.; Pini, D.; Salvadori, P. Determination of absolute configuration of acyclic 1,2-diols with Mo2(OAc)4. 1. Snatzke’s method revisited. J. Org. Chem. 2001, 66, 4819–4825. [Google Scholar] [CrossRef] [PubMed]
- Frelek, J.; Ruskowska, P.; Suszczynska, A.; Szewczyk, K.; Osuch, A.; Jarosz, S.; Jagodzinski, J. Configurational assignment of sugar erythro-1,2-diols from their electronic circular dichroism spectra with dimolybdenum tetraacetate. Tetrahedron: Asymmetry 2008, 19, 1709–1713. [Google Scholar] [CrossRef]
- Xia, M.-W.; Cheng-Bin, C.; Li, C.-W.; Wu, C.-J. Three New and Eleven Known Unusual C25 Steroids: Activated Production of Silent Metabolites in a Marine-Derived Fungus by Chemical Mutagenesis Strategy using Diethyl Sulphate. Mar. Drugs 2014, 12, 1545–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, C.; Part, N.; Bouhired, S.; Kehraus, S.; König, G.M. Stachylines A−D from the Sponge-Derived Fungus Stachylidium sp. J. Nat. Prod. 2011, 74, 21–25. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Zhu, H.; Fu, P.; Wang, Y.; Zhang, Z.; Lin, H.; Liu, P.; Zhuang, Y.; Hong, K.; Zhu, W. Cytotoxic Polyphenols from the Marine-Derived Fungus Penicillium expansum. J. Nat. Prod. 2010, 73, 911–914. [Google Scholar] [CrossRef]
- Hamasaki, T.; Nagayama, K.; Hatsuda, Y. Two new metabolites, sydonic acid and hydroxysydonic acid from Aspergillus sydowi. Agric. Biol. Chem. 1978, 42, 37–40. [Google Scholar] [CrossRef]
- Yurchenko, A.N.; Smetanina, O.F.; Kalinovsky, A.I.; Pivkin, M.V.; Dmitrenok, P.S.; Kuznetsova, T.A. A new meroterpenoid from the marine fungus Aspergillus versicolor (Vuill.) Tirab. Russ. Chem. Bull. 2010, 59, 852–856. [Google Scholar] [CrossRef]
- Li, H.; Sun, W.; Deng, M.; Qi, C.; Chen, C.; Zhu, H.; Luo, Z.; Wang, J.; Xue, Y.; Zhang, Y. Asperversins A and B, two novel meroterpenoids with an unusual 5/6/6/6 ring from the marine-derived fungus Aspergillus versicolor. Mar. Drugs 2018, 16, 177. [Google Scholar] [CrossRef] [Green Version]
- Pracht, P.; Bohle, F.; Grimme, S. Automated exploration of the low-energy chemical space with fast quantum chemical methods. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. [Google Scholar] [CrossRef]
- Grimme, S. Exploration of Chemical Compound, Conformer, and Reaction Space with Meta-Dynamics Simulations Based on Tight-Binding Quantum Chemical Calculations. J. Chem. Theory Comput. 2019, 15, 2847–2862. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Pescitelli, G.; Bruhn, T. Good Computational Practice in the Assignment of Absolute Configurations by TDDFT Calculations of ECD Spectra. Chirality 2016, 28, 466–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2011, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXTL, Structure Determination Software Programs; Bruker Analytical X-ray System Inc.: Madison, WI, USA, 1997. [Google Scholar]
- Sheldrick, G.M. SHELXL-97 and SHELXS-97, Program for X-ray Crystal Structure Solution and Refinement; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Yuan, X.-L.; Zhang, P.; Liu, X.-M.; Du, Y.-M.; Hou, X.-D.; Cheng, S.; Zhang, Z.-F. Cytological Assessments and Transcriptome Profiling Demonstrate that Evodiamine Inhibits Growth and Induces Apoptosis in a Renal Carcinoma Cell Line. Sci. Rep. 2017, 7, 12572. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Compound 1 | No. | Compound 2 | Compound 3 | |||
|---|---|---|---|---|---|---|---|
| δH (Mult, J in Hz) | δC, Type | δH (Mult, J in Hz) | δC, Type | δH (Mult, J in Hz) | δC, Type | ||
| 1 | 1.39 (d, 6.6) | 19.7, CH3 | 1 | 127.4, C | 126.3, C | ||
| 2 | 4.36 (m) | 59.3, CH | 2/6 | 7.14 (d, 8.3) | 130.8, CH | 7.15 (d, 8.6) | 130.4, CH |
| 3 | 4.47 (d, 5.4) | 69.1, CH | 3/5 | 6.86 (d, 8.3) | 114.8, CH | 6.88 (d, 8.6) | 114.5, CH |
| 4 | 132.1, C | 4 | 157.8, C | 157.6, C | |||
| 5 | 7.67 (s) | 140.4, CH | 7 | 3.46 (br s) | 40.2, CH2 | 3.58 (d, 5.3) | 39.3, CH2 |
| 6 | 148.6, C | 8 | 173.4, C | 171.9, C | |||
| 7 | 5.60 (q, 7.4) | 112.2, CH | 1’ | 3.92 (dd, 9.9, 4.4) 3.84 (m) | 71.3, CH2 | 3.93 (dd, 9.9, 4.5) 3.86 (dd, 9.9, 6.9) | 70.9, CH2 |
| 8 | 1.88 (d, 7.4) | 11.7, CH3 | 2’ | 4.24 (t, 5.3) | 72.7, CH | 4.25 (m) | 72.3, CH |
| 9 | 168.4, C | 3’ | 145.8, C | 145.4, C | |||
| 4’ | 4.86 (br s) 5.02 (br s) | 112.1, CH2 | 4.87 (br s) 5.03 (br s) | 111.7, CH2 | |||
| 5’ | 1.72 (s) | 18.9, CH3 | 1.73 (s) | 18.5, CH3 | |||
| 8-OMe | 3.58 (s) | 51.6, CH3 | |||||
| 2’-OH | 5.23 (br s) | ||||||
| Compound | A549 | BT-549 | HeLa | HepG2 | MCF-7 | THP-1 |
|---|---|---|---|---|---|---|
| 1 | >100 | >100 | >100 | >100 | >100 | >100 |
| 2 | >100 | 87.3 ± 3.5 | 96.6 ± 1.5 | >100 | >100 | >100 |
| 3 | 70.0 ± 1.2 | >100 | >100 | 22.0 ± 1.2 | >100 | >100 |
| 4 | 63.6 ± 2.6 | >100 | 78.7 ± 2.9 | >100 | >100 | 78.7 ± 1.9 |
| 5 | 21.2 ± 2.3 | >100 | 61.7 ± 2.2 | >100 | >100 | 18.2 ± 1.2 |
| 6 | >100 | >100 | >100 | >100 | >100 | >100 |
| 7 | >100 | >100 | >100 | >100 | >100 | >100 |
| 8 | >100 | >100 | >100 | >100 | >100 | >100 |
| Epirubicin a | 6.6 ± 0.5 | 4.4 ± 0.2 | 2.2 ± 0.3 | 3.7 ± 0.2 | 3.9 ± 0.1 | 4.8 ± 0.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, L.-L.; Tang, J.-X.; Bo, Y.-H.; Li, Y.-Z.; Feng, T.; Zhu, H.-W.; Yu, X.; Zhang, X.-X.; Zhang, J.-L.; Wang, W. Cytotoxic Secondary Metabolites Isolated from the Marine Alga-Associated Fungus Penicillium chrysogenum LD-201810. Mar. Drugs 2020, 18, 276. https://doi.org/10.3390/md18050276
Jiang L-L, Tang J-X, Bo Y-H, Li Y-Z, Feng T, Zhu H-W, Yu X, Zhang X-X, Zhang J-L, Wang W. Cytotoxic Secondary Metabolites Isolated from the Marine Alga-Associated Fungus Penicillium chrysogenum LD-201810. Marine Drugs. 2020; 18(5):276. https://doi.org/10.3390/md18050276
Chicago/Turabian StyleJiang, Lin-Lin, Jin-Xiu Tang, Yong-Heng Bo, You-Zhi Li, Tao Feng, Hong-Wei Zhu, Xin Yu, Xing-Xiao Zhang, Jian-Long Zhang, and Weiyi Wang. 2020. "Cytotoxic Secondary Metabolites Isolated from the Marine Alga-Associated Fungus Penicillium chrysogenum LD-201810" Marine Drugs 18, no. 5: 276. https://doi.org/10.3390/md18050276
APA StyleJiang, L. -L., Tang, J. -X., Bo, Y. -H., Li, Y. -Z., Feng, T., Zhu, H. -W., Yu, X., Zhang, X. -X., Zhang, J. -L., & Wang, W. (2020). Cytotoxic Secondary Metabolites Isolated from the Marine Alga-Associated Fungus Penicillium chrysogenum LD-201810. Marine Drugs, 18(5), 276. https://doi.org/10.3390/md18050276