3. Materials and Methods
All reagents were purchased from commercial sources and used without purification unless specified. Acetonitrile, pyridine and CH2Cl2 were refluxed with CaH2 and distilled prior to use. THF was dried with LiAlH4 and distilled prior to use. Thin-layer chromatography was performed using silica gel GF-254 plates (Qingdao Chemical Company, Qingdao) detected by UV (254 nm) or charting with 10% sulfuric acid in ethanol. Column chromatography was performed on silica gel (200–300 mesh, Qingdao Chemical Company, Qingdao). NMR spectra were recorded on a Bruker AV400 spectrometer. Chemical shifts (δ) are reported in ppm and coupling constants (J) are reported in Hz. Then, 1H and 13C NMR spectra were calibrated with TMS as an internal standard. The specific rotation was measured on a Rudolph autopol IV polarimeter. ESI-MS was acquired with a Bruker Dalton microTOFQ II spectrometer.
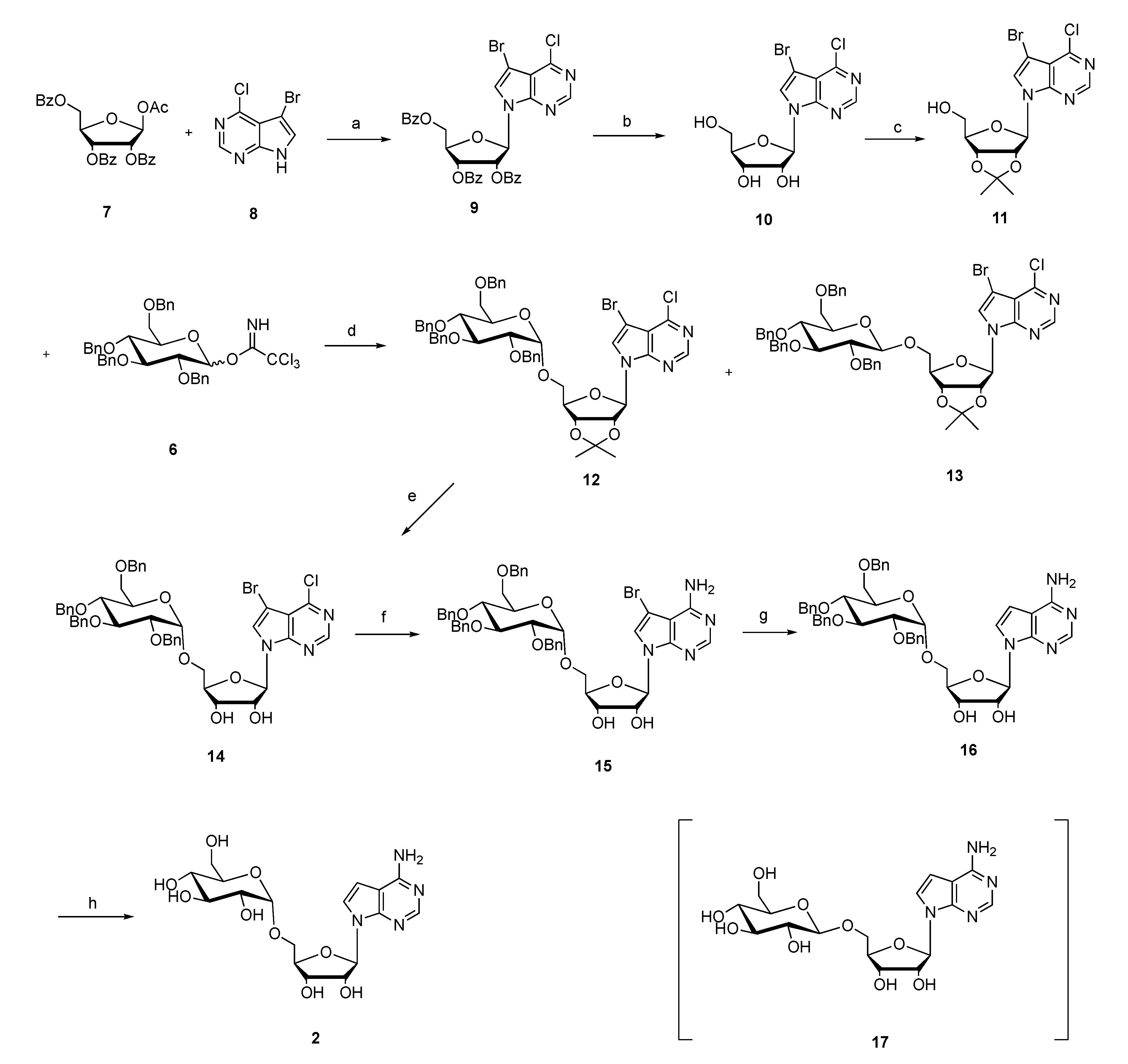
3.1. H NMR, 13C NMR and HRMS of 2,3,4,6-O-Tetrabenzyl-d-glucopyranose
Rf = 0.32 (PE/EA = 3:1); mp 153–155 °C; 1H NMR (400 MHz, CDCl3): δ 7.36–7.28 (m, 18H, Bn), 7.18–7.12 (m, 2H, Bn), 5.31–5.19 (m, 1H, H-1), 4.98–4.48 (m, 8H, 4CH2), 4.09–3.94 (m, 2H, H-4,5), 3.74–3.58 (m, 4H, H-2,3,6), 3.06 (d, J = 2.2 Hz, 1H, 1-OH); 13C NMR (101 MHz, CDCl3): δ 138.7 (C), 138.2 (C), 137.8 (2C), 128.5 (CH), 128.4 (CH), 128.2 (CH), 128.1 (CH), 128.0 (CH), 127.9 (CH), 127.7 (CH), 127.6 (CH), 91.3 (CH-1), 81.7 (CH-5), 80.0 (CH-2), 77.7 (CH-3), 75.7 (CH2), 75.0 (CH2), 73.5 (CH2), 73.3 (CH2), 70.3 (CH-4), 68.6 (CH2-6); HRMS Calcd. For C34H36O6Na+ [M + Na]+: 563.2404, found: 563.2410.
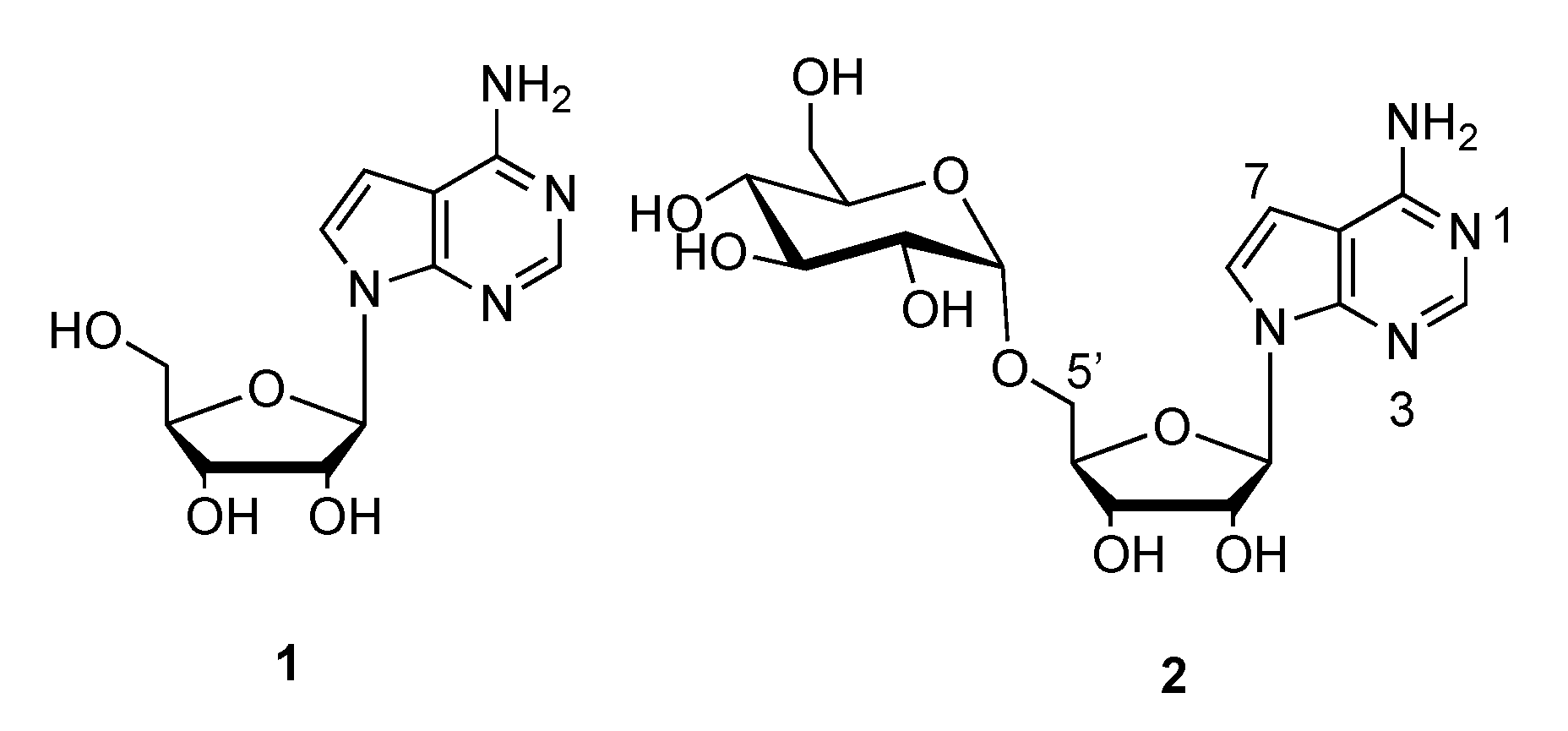
3.2. Synthesis of 2,3,4,6-Tetra-O-benzyl-glucopyranosyl Trichloroacetimidate (6)
To the suspension of anhydrous K2CO3 (10 g, 101 mmol) in dry dichloromethane (100 mL) was added 2,3,4,6-O-tetrabenzyl-d-glucopyranose (10 g, 18.5 mmol) and trichloroacetonitrile (11.3 mL, 112 mmol) sequentially. The mixture was stirred at room temperature for 4 h until the reaction was completed (monitored by TLC, PE/EA = 6:1, Rf = 0.21). After evaporation of the solvent under reduced pressure, the crude product was directly purified by flash column chromatography on silica gel (Et3N/PE/EA = 0.1:3:1) to provide 12.4 g compound 6 as a white solid (98% yield, α:β ≈ 1:4); mp: 77–79 °C; 1H NMR (400 MHz, CDCl3): δ 8.73 (s, 0.8H, β-NH), 8.60 (s, 0.2H, -NH), 7.32 (m, 18H), 7.20 (m, 2H), 6.55 (d, J = 3.2 Hz, 0.2H, α-H-1), 5.84 (d, J = 6.1 Hz, 0.8H, β-H-1), 5.01–4.46 (m, 9H), 3.81–3.76 (m, 4H), 3.67 (d, J = 4.6 Hz, 1H); 13C-NMR (101 MHz, CDCl3): δ 161.2 (C), 138.4 (C), 138.1 (C), 138.0 (C), 138.0 (C), 128.5 (CH), 128.4 (CH), 128.4 (CH), 128.4 (CH), 128.1 (CH), 128.0 (CH), 128.0 (CH), 127.9 (CH), 127.8 (CH), 127.7 (CH), 127.6 (CH), 127.6 (CH), 98.4 (CH), 84.6 (CH), 81.0 (CH), 76.8 (CH), 75.9 (CH2), 75.6 (CH2), 75.0 (CH2), 74.9 (CH2), 73.4 (CH), 68.2 (CH); HRMS Calcd. For C36H36Cl3NO6Na+ [M + Na]+: 706.1500, found: 706.1509.
3.3. Synthesis of 7-Bromo-6-chloro-pyrrole [2,3-d]pyrimidine (8)
To the stirred suspension of 6-chloro-pyrrole[2,3-d]pyrimidine (12.0 g, 78.2 mmol) in dry dichloromethane (500 mL) was added NBS (16.1 g, 91.1 mmol) in batches. After the addition was finished, the mixture was stirred at room temperature for another 10 h until completion (monitored by TLC, DCM/EA = 5:1, Rf = 0.41) and then poured into ice water (500 mL). The resulting brown precipitate was filtered under reduced pressure and the filter cake was washed with water (500 × 3 mL) and dried to afford compound 8 as a brown solid (17.2 g, 95% yield); mp: >300 °C; 1H NMR (400 MHz, DMSO): δ 12.95 (s, 1H, NH), 8.59 (s, 1H, H-2), 7.91 (s, 1H, H-8); 13C NMR (100 MHz, DMSO): δ 151.5 (C), 151.4 (CH), 150.7 (C), 129.1 (CH), 114.1 (C), 86.3 (C); HRMS Calcd. For C6H4BrClN3+ [M + H]+: 231.9272, found: 231.9270.
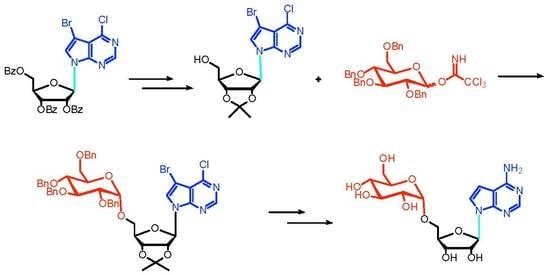
3.4. Synthesis of 7-Bromo-6-chloro-9-(2′,3′,5′-O-tribenzoyl-β-d-ribofuranose-yl)-pyrrole[2,3-d]pyrimidine (9)
To the stirred suspension of 7-bromo-6-chloro-pyrrole[2,3-d]pyrimidine 8 (5.0 g, 21.5 mmol) in dry acetonitrile (40 mL) was added BSA (N,O-bis(trimethylsiyl)acetamide, 5.4 g, 26.0 mmol) at 0 °C under argon. The resulting mixture was stirred at room temperature for 15 min, and then 1-O-ethanoyl-2,3,5-O-tribenzoyl-β-d-ribofuranose (16.0 g, 32.5 mmol) and TMSOTf (8.0 g, 46 mmol) were added sequentially. After the addition was completed, the mixture was stirred at 80 °C for 1 h (monitored by TLC, CH2Cl2: PE, 5:1, Rf 0.27). After the mixture was cooled to room temperature, water (200 mL) was added to quench the reaction and the resulting mixture was extracted with ethyl acetate (200 × 3 mL). The organic layers were separated and washed with saturated NaHCO3 (200 mL) and saturated brine (200 mL) and dried with anhydrous Na2SO4. After the solvent was removed under reduced pressure, the crude product was purified by flash column chromatography on silica gel to provide 10.1 g compound 9 (75.5% yield) as a white solid. mp: 142–143 °C; = −110 (c = 0.30, CHCl3); 1H-NMR (400 MHz, CDCl3): δ (ppm) 8.57 (s, 1H), 8.10 (d, J = 8.0 Hz, 2H), 7.99 (d, J = 8.0 Hz, 2H), 7.91 (d, J = 7.9 Hz, 2H), 7.61–7.32 (m, 10H), 6.67 (d, J = 5.2 Hz, 1H), 6.12 (m, 2H), 4.89 (d, J = 12.1 Hz, 1H), 4.80 (d, J = 3.1 Hz, 1H), 4.67 (dd, J = 12.2, 3.1 Hz, 1H); 13C NMR (101MHz, CDCl3): δ (ppm) 166.1 (C), 165.4 (C), 165.1 (C), 152.7 (C), 151.6 (CH), 150.7 (C), 133.9 (CH), 133.9 (CH), 133.7 (CH), 129.9 (4CH), 129.7 (2CH), 129.2 (C), 128.8 (2CH), 128.6 (2CH; C), 128.6 (2CH), 128.3 (C), 126.5 (CH), 115.9 (C), 90.2 (C), 86.7 (CH), 80.6 (CH), 74.1 (CH), 71.4 (CH), 63.5 (CH); HRMS Calcd. For C32H23BrClN3O7Na+ [M + Na]+: 698.0300, found: 698.0305.
3.5. Synthesis of 7-Bromo-6-chloro-9-(β-d-ribofuranose-yl)-pyrrole[2,3-d]pyrimidine (10)
7-Bromo-6-chloro-9-(2′,3′,5′-O-tribenzoyl-β-d-ribofuranose-yl)-pyrrole[2,3-d]pyrimidine 9 (10 g, 14.77 mmol) was dissolved in methanolic ammonia (methanol was saturated with NH3 at 0 °C, 300 mL) and the mixture was stirred at 0 °C for 12 h until the reaction was completed (monitored by TLC, CH2Cl2:CH3OH, 50:1, Rf 0.45). The mixture was concentrated and the crude product was purified by flash column chromatography on silica gel (CH2Cl2/CH3OH, 70:1) to provide 5.1 g compound 10 (94.0% yield) as a white solid. mp: 179–180 °C; = −64 (c = 0.1, CH3OH); 1H NMR (400 MHz, DMSO): δ 8.72 (s, 1H, H-2), 8.27 (s, 1H, H-8), 6.24 (d, J = 5.8 Hz, 1H, H-1′), 5.49 (d, J = 6.1 Hz, 1H, 2′-OH), 5.25 (d, J = 4.9 Hz, 1H, 3′-OH), 5.15 (t, J = 5.3 Hz, 1H, 5′-OH), 4.40 (dd, J = 11.1, 5.6 Hz, 1H, H-2′), 4.13 (dd, J = 8.3, 4.5 Hz, 1H, H-3′), 3.96 (d, J = 3.4 Hz, 1H, H-4′), 3.71–3.56 (m, 2H, H-5′); 13C NMR (101 MHz, DMSO): δ 151.6 (CH-2), 151.0 (C-6), 151.0 (C-4), 128.75(CH-8), 114.9 (C-5), 87.7 (C-7; CH-1′), 86.0 (CH-4′), 74.9 (CH-2′), 70.8 (CH-3′), 61.7 (CH2-5′); HRMS Calcd. For C11H11BrClN3O4Na+ [M + Na]+: 385.9514, found: 385.9511.
3.6. Synthesis of 7-Bromo-6-chloro-9-(2′,3′-O-isopropylidene-β-d-ribofuranose-yl)-pyrrole[2,3-d] pyrimidine (11)
To the stirred suspension of compound 10 (4.0 g, 11 mmol) in dry acetone (200 mL) was added p-toluenesulfonic acid (200 mg, 1.2 mmol) and 2,2-dimethoxy propane (6.0 g, 55 mmol). The mixture was stirred at room temperature for 4 h until the reaction was completed (monitored by TLC, CH2Cl2:CH3OH, 50:1, Rf 0.57). After that, the mixture was neutralized by triethylamine (70.0 mg, 0.7 mmol) and concentrated under reduced pressure. The crude product was purified by flash column chromatography on silica gel (CH2Cl2/CH3OH, 70:1) to provide 4.4 g compound 11 (98.0% yield) as a white solid. mp: 73–74 °C; = −63 (c = 0.05, CH3OH); 1H NMR (400 MHz, DMSO): δ 8.73 (s, 1H, H-2), 8.24 (s, 1H, H-8), 6.36 (d, J = 2.1 Hz, 1H, H-1′), 5.32–5.04 (m, 2H, H-2′,5′-OH), 5.03–4.83 (m, 1H, H-3′), 4.21 (s, 1H, H-4′), 3.56 (s, 2H, H-5′), 1.54 (s, 3H, Me), 1.31 (s, 3H, Me); 13C NMR (101 MHz, DMSO): δ 151.8 (CH-2), 151.2 (C-4), 150.5 (C-6), 129.2 (CH-8), 115.0 (C-5), 113.6 (C), 90.0 (CH-1′), 87.8 (C-7), 86.9 (CH-4′), 84.4 (CH-2′), 81.4 (CH-3′), 61.9 (CH2-5′), 27.5 (Me), 25.6 (Me); HRMS Calcd. For C14H15BrClN3O4Na+ [M + Na]+: 425.9827, found: 425.9811.
3.7. Synthesis of 7-Bromo-6-chloro-(2′,3′-O-isopropylidene-5′-O-(2″,3″,4″,6″-O-tetrabenzyl-α-d-glucopyranosyl)tubercidin (12) and 7-bromo-6-chloro-(2′,3′-O-isopropylidene-5′-O-(2″,3″,4″,6″-O-tetrabenzyl-β-d-glucopyranosyl)tubercidin (13)
Compound 6 (6.4 g, 9.3 mmol) and compound 11 (2.5 g, 6.2 mmol) were dissolved in dry dichloromethane (300 mL) and the resulting mixture was stirred at room temperature for 5 min under argon. After that, TMSOTf (16.5 mg, 0.07 mmol) was added to the mixture at −20 °C. The mixture was slowly warmed to room temperature and stirred for another 4 h until the reaction was completed (monitored by TLC, PE: EA, 3:1, Rf 0.5). The mixture was concentrated and the crude product was purified by flash column chromatography on silica gel (CH2Cl2/CH3OH, 70:1) to provide 8.0 g of compounds 12 and 13 (79% yield) as a light yellow oil.
12: = −7 (c = 0.1, CH3OH); 1H NMR (400 MHz, DMSO): δ 8.71 (s, 1H), 8.23 (s, 1H), 7.34–7.22 (m, 18H), 7.13–7.04 (m, 2H), 6.41 (d, J = 2.6 Hz, 1H), 5.20 (dd, J = 6.1, 2.7 Hz, 1H), 4.97 (dd, J = 6.1, 3.2 Hz, 1H), 4.90 (m, 2H), 4.72–4.61 (m, 4H), 4.40 (m, 4H), 3.78–3.67 (m, 2H), 3.61 (dd, J = 10.9, 4.1 Hz, 1H), 3.56–3.37 (m, 5H), 1.54 (s, 3H), 1.29 (s, 3H); 13C NMR (101 MHz, DMSO): δ 151.8 (CH), 151.3 (C), 150.4 (C), 139.3 (C), 138.9 (C), 138.7 (C), 138.6 (CH), 129.2 (CH), 128.7 (CH), 128.6 (CH), 128.2 (CH), 128.0 (CH), 128.0 (CH), 127.9 (CH), 127.8 (CH), 115.1 (C), 114.0 (C), 96.4 (CH), 89.6 (CH), 88.3 (C), 84.5 (CH), 84.3 (CH), 81.5 (CH), 81.4 (CH), 79.8 (CH), 77.7 (CH), 75.0 (CH2), 74.4 (CH2), 72.8 (CH2), 71.8 (CH2), 70.4 (CH), 68.8 (CH2), 68.0 (CH2), 27.5 (Me), 25.8 (Me); HRMS Calcd. For C48H49BrClN3O9Na+ [M + Na]+: 948.2233, found: 948.2236.
13: = −19 (c = 0.1, CH3OH); 1H NMR (400 MHz, DMSO): δ 8.67 (s, 1H), 8.17 (s, 1H), 7.24 (m, 14H), 7.14 (m, 6H), 6.36 (d, J = 2.5 Hz, 1H), 5.19 (dd, J = 6.1, 2.6 Hz, 1H), 4.92 (dd, J = 6.1, 2.8 Hz, 1H), 4.78–4.64 (m, 4H), 4.56–4.30 (m, 6H), 4.04–3.98 (m, 1H), 3.76–3.38 (m, 6H), 3.24 (m, 1H), 1.50 (s, 3H), 1.23 (s, 3H); 13C NMR (101 MHz, DMSO): δ 151.7 (CH), 151.2 (C), 150.3 (C), 139.0 (C), 138.9 (C), 138.7 (C), 138.6 (C), 129.3 (CH), 128.7 (CH), 128.6 (CH), 128.4 (CH), 128.2 (CH), 128.0 (CH), 128.0 (CH), 127.9 (CH), 127.9 (CH), 127.8 (CH), 115.1 (C), 113.8 (C), 102.9 (CH), 90.4 (CH), 87.9 (C), 85.0 (CH), 84.4 (CH), 84.1 (CH), 82.1 (CH), 81.4 (CH), 78.1 (CH), 74.9 (CH2), 74.4 (CH2), 74.4 (CH2), 74.0 (CH2), 72.7 (CH), 69.7 (CH2), 69.2 (CH2), 27.4 (Me), 25.6 (Me); MS (ESI): HRMS Calcd. For C48H49BrClN3O9Na+ [M + Na]+: 948.2233, found: 948.2235.
3.8. Synthesis of 7-Bromo-6-chloro-5′-O-(2″,3″,4″,6″-O-tetrabenzyl-α-d-glucopyranosyl) Tubercidin (14)
Compound 12 (1.0 g, 1.08 mmol) was added to 80% acetic acid aqueous solution (50 mL) and the solution was stirred at room temperature for 30 min. After that, the mixture was transferred to preheated oil and stirred at 50 °C for another 12 h until the reaction was completed (monitored by TLC, CH2Cl2:CH3OH,20:1, Rf 0.35). The mixture was concentrated and the crude product was purified by flash column chromatography on silica gel (CH2Cl2/CH3OH, 50:1) to provide 0.9 g compound 14 (85.0% yield) as a white solid. mp: 49–50 °C; = −26° (c = 0.3, CH3OH); 1H NMR (400 MHz, DMSO): δ 8.71 (s, 1H, H-2), 8.37 (s, 1H, H-8), 7.42–7.21 (m, 18H, H-Ph), 7.16–7.09 (m, 2H, H-Ph), 6.35 (d, J = 6.4 Hz, 1H, H-1′), 5.60 (d, 1H, 2′-OH), 5.37 (s, 1H, 3′-OH), 5.01 (d, J = 11.1 Hz, 1H, H-1″), 4.84 (m, 2H, H-CH2), 4.77–4.65 (m, 3H, H-CH2; H-2′), 4.45 (m, 4H, H-CH2), 4.19 (d, J = 2.3 Hz, 1H, H-3′), 4.12 (s, 1H, H-5′), 3.82–3.62 (m, 3H, H-5′; H-4′), 3.58–3.56 (m, 3H, H-3″; H-6″), 3.52–3.40 (m, 2H, H-2″; H-4″); 13C NMR (101 MHz, DMSO): δ 151.7 (CH-2), 151.3 (C-6), 151.1 (C-4), 139.3 (C), 138.8 (C), 138.6 (2C), 129.6 (CH), 128.7 (CH), 128.6 (CH), 128.5 (CH), 128.4 (CH), 128.2 (CH), 128.1 (CH), 127.9 (CH), 127.89 (CH), 114.9 (C-5), 96.4 (CH-1″), 88.4 (C-7), 87.3 (CH-1′), 83.8 (CH-5″), 82.0 (CH-4′), 79.5 (CH-4″), 77.8 (CH-3″), 75.3 (CH2), 75.1 (CH-2′), 74.6 (CH2), 72.8 (CH2), 71.9 (CH2), 71.6 (CH-3′), 70.6 (CH-2″), 69.0 (CH2-6″), 67.9 (CH2-5′); HRMS Calcd. For C45H46BrClN3O9+ [M + H]+: 886.2100, found: 886.2098.
3.9. Synthesis of 7-Bromo-6-chloro-5′-O-(2″,3″,4″,6″-O-tetrabenzyl-β-d-glucopyranosyl) Tubercidin (14′)
Compound 13 was converted into 14′ (79.0% yield) as described for the synthesis of 14: Rf 0.34 (CH2Cl2/CH3OH, 20:1); mp: 49–50 °C; = −12 (c = 0.3, CH3OH); 1H NMR (400 MHz, DMSO): δ 8.67 (s, 1H), 8.22 (s, 1H), 7.24 (s, 14H), 7.17–7.05 (m, 6H), 6.23 (d, J = 5.4 Hz, 1H), 5.54 (d, J = 6.1 Hz, 1H), 5.34 (s, 1H), 4.78 (d, J = 11.1 Hz, 2H), 4.69 (d, J = 8.8 Hz, 2H), 4.60–4.37 (m, 7H), 4.12 (m, 3H), 3.86–3.72 (m, 1H), 3.72–3.50 (m, 4H), 3.45 (m, 1H), 3.29–3.23 (m, 1H); 13C NMR (101 MHz, DMSO) δ 151.6 (CH-2), 151.0 (C-6), 150.9 (C-4), 139.1 (C), 138.8 (C), 138.7 (C), 138.6 (C), 128.8 (CH), 128.7 (CH), 128.6 (CH), 128.6 (CH), 128.5 (CH), 128.4 (CH), 128.2 (CH), 128.1 (CH), 128.0 (CH), 127.9 (CH), 127.9 (CH), 127.8 (CH), 127.8 (CH), 115.0 (C-5), 103.0 (CH-1″), 88.2 (C-7), 87.9 (CH-1′), 84.1 (CH-4′), 83.7 (CH-5″), 82.4 (CH-4″), 78.1 (CH-3″), 74.9 (CH-2′), 74.7 (CH-3′), 74.5 (CH2), 74.4 (CH2), 74.1 (CH2), 72.7 (CH2), 71.0 (CH-2″), 70.0 (CH2-5′), 69.2 (CH2-6″); HRMS Calcd. For C45H46BrClN3O9+ [M + H]+: 886.2100, found: 886.2099.
3.10. Synthesis of 7-Bromo-5′-O-(2″,3″,4″,6″-O-tetrabenzyl-α-d-glucopyranosyl)tubercidin (15)
Compound 14 (100 mg, 0.11 mmol) was dissolved in methanolic ammonia (methanol was saturated with NH3 at 0 °C, 50 mL) and the mixture was stirred at 130 °C for 12 h (monitored by TLC, CH2Cl2:CH3OH, 25:1, Rf 0.36). After cooling to room temperature, the mixture was concentrated and the crude product was purified by flash column chromatography on silica gel (CH2Cl2/CH3OH, 30:1) to provide 92.9 mg compound 15 (94.9% yield) as a white solid. mp: 68–69 °C, = −33 (c = 0.1, CH3OH); 1H NMR (400 MHz, DMSO): δ 8.10 (s, 1H, H-2), 7.87 (s, 1H, H-8), 7.40–7.18 (m, 18H, H-Bn), 7.18–7.07 (m, 2H, H-Bn), 6.76 (br s, 2H, H-NH2), 6.19 (d, J = 6.6 Hz, 1H, H-1′), 5.46 (d, J = 6.3 Hz, 1H, 2′-OH), 5.26 (d, J = 4.1 Hz, 1H, 3′-OH), 4.98 (d, J = 11.1 Hz, 1H, H-1″), 4.81–4.66 (m, 5H, H-2′; 2CH2), 4.49–4.32 (m, 4H, 2CH2), 4.10–4.09 (m, 1H, H-3′), 4.06–4.04 (m, 1H, H-5″), 3.78–3.50 (m, 6H, H-4′,5′,3″,6″), 3.48–3.39 (m, 2H, H-2″,4″); 13C NMR (101 MHz, DMSO): δ 157.4 (C-4), 153.0 (C-6), 150.6 (CH-2), 139.3 (C), 138.8 (C), 138.7 (2C), 128.7 (CH), 128.7 (CH), 128.4 (CH), 128.2 (CH), 128.1 (CH), 128.0 (CH), 127.9 (CH), 121.7 (C-5), 96.3 (CH-1″), 87.9 (C-7), 86.5 (CH-1′), 83.3 (CH-5″), 82.0 (CH-4′), 79.5 (CH-4″), 77.8 (CH-3″), 75.3 (CH2), 74.9 (CH-2′), 74.6 (CH2), 72.8 (CH2), 71.8 (CH2), 71.7 (CH-3′), 70.6 (CH-2″), 69.0 (CH2-6′), 68.1 (CH2-5′); HRMS Calcd. For C45H48BrN4O9+ [M + H]+: 867.2599, found: 867.2598.
3.11. Synthesis of 7-Bromo-5′-O-(2″,3″,4″,6″-O-tetrabenzyl-β-d-glucopyranosyl) Tubercidin (15′)
Compound 14′ was converted into 15′ (79.0% yield) as described for the synthesis of 15; Rf 0.40 (CH2Cl2/CH3OH, 20:1); mp: 145–147 °C; = −15 (c = 0.1, CH3OH); 1H NMR (400 MHz, DMSO) δ 8.11 (s, 1H, H-2), 7.68 (s, 1H, H-8), 7.40–7.19 (m, 14H, H-Bn), 7.19–7.10 (m, 6H, H-Bn), 6.77 (br s, 2H, NH2), 6.13 (d, J = 5.4 Hz, 1H, H-1′), 5.44 (d, J = 6.1 Hz, 1H, 2′-OH), 5.26 (d, J = 5.0 Hz, 1H, 3′-OH), 4.81 (m, 2H, H-2′,1″), 4.71 (d, J = 11.0 Hz, 2H, CH2), 4.60–4.08 (m, 6H, 3CH2), 4.15–4.04 (m, 3H, H-3′,5′,5″), 3.78–3.42 (m, 6H, H-4′,5′,2″,3″,6″), 3.29 (s, 1H, H-4″); 13C NMR (101 MHz, DMSO) δ 157.4 (C-6), 153.0 (CH-2), 150.2 (C-4), 139.1 (CH), 138.8 (CH), 138.7 (CH), 138.6 (CH), 128.7 (CH), 128.7 (CH), 128.6 (CH), 128.5 (CH), 128.5 (CH), 128.2 (CH), 128.1 (CH), 128.0 (CH), 127.8 (CH), 122.0 (C-5), 103.0 (C-7), 101.5 (CH-1″), 87.5 (CH-1′), 87.4 (CH-4′), 84.1 (CH-5″), 83.0 (CH-4″), 82.3 (CH-3″), 78.1 (CH-2′), 74.9 (CH-3′), 74.4 (CH2), 74.4 (CH2), 74.2 (CH2), 72.7 (CH2), 71.0 (CH-2″), 70.3 (CH2-5′), 69.2 (CH2-6″); HRMS Calcd. For C45H48BrN4O9+ [M + H]+: 867.2599, found: 867.2599.
3.12. Synthesis of 5′-O-(2″,3″,4″,6″-O-Tetrabenzyl-α-d-glucopyranosyl) Tubercidin (16)
To the stirred suspension of compound 15 (200 mg, 0.23 mmol) in THF (15 mL) and methanol (15 mL) was added triethylamine (0.3 mL) and 20% Pd(OH)2/C (50 mg, 0.04 mmol), and the mixture was stirred at room temperature for 5 h under a continuous hydrogen environment until the reaction was completed (monitored by TLC, CH2Cl2:CH3OH,20:1, Rf 0.24). The mixture was filtered, and the filtrate was concentrated to provide the crude product, which was purified by flash column chromatography on silica gel (CH2Cl2/CH3OH, 20:1) to provide 151.8 mg compound 16 (83.5% yield) as a white solid. mp: 74–75 °C; = 1.8 (c = 0.1, CH3OH); 1H NMR (400 MHz, DMSO) δ 8.06 (s, 1H, H-2), 7.63 (d, J = 3.6 Hz, 1H, H-8), 7.41–7.21 (m, 18H, H-Bn), 7.17–7.10 (m, 2H, H-Bn), 7.00 (s, 2H, NH2), 6.53 (d, J = 3.5 Hz, 1H, H-7), 6.16 (d, J = 5.9 Hz, 1H, H-1′), 5.41 (d, J = 6.2 Hz, 1H, 2′-OH), 5.22 (s, 1H, 3′-OH), 4.95–4.41 (m, 9H, H-1″,Bn), 4.36–4.32 (m, 1H, H-2′), 4.12–4.08 (m, 2H, H-3′,5″), 3.83–3.77 (m, 2H, H-4′,5′), 3.70–3.41 (m, 6H, H-2″,3″,4″,5′,6″); 13C NMR (101 MHz, DMSO): δ 157.5 (C-6), 151.7 (CH-2), 150.8 (C-4), 139.2 (CH), 138.9 (CH), 138.7 (CH), 128.7 (CH), 128.7 (CH), 128.2 (CH), 128.1 (CH), 128.1 (CH), 128.0 (CH), 127.9 (CH), 122.1 (CH-8), 103.1 (C-5), 100.4 (CH-7), 96.3 (CH-1″), 87.0 (CH-1′), 82.7 (CH-5″), 82.0 (CH-4′), 79.9 (CH-4″), 77.8 (CH-3″), 75.2 (CH2), 74.9 (CH-2′), 74.6 (CH2), 72.8 (CH2), 72.0 (CH2), 71.4 (CH-3′), 70.5 (CH-2″), 69.0 (CH2-6″), 68.0 (CH2-5′); HRMS Calcd. For C45H49N4O9+ [M + H]+: 789.3494, found: 789.3499.
3.13. Synthesis of 5′-O-(2″,3″,4″,6″-O-Tetrabenzyl-β-d-glucopyranosyl) Tubercidin (16′)
Compound 15′ was converted into 16′ (79.0% yield) as described for the synthesis of 16; Rf 0.23 (CH2Cl2/CH3OH, 20:1); mp: 166–167 °C; = −21 (c = 0.1, CH3OH); 1H NMR (400 MHz, DMSO): δ 8.03 (s, 1H, H-2), 7.38–7.18 (m, 15H, H-8, Bn), 7.19–7.05 (m, 6H, H-Bn), 6.98 (s, 2H, NH2), 6.59 (d, J = 3.6 Hz, 1H, H-7), 6.09 (d, J = 5.2 Hz, 1H, H-1′), 5.36 (d, J = 6.3 Hz, 1H, 2′-OH), 5.22 (d, J = 5.3 Hz, 1H, OH-3′), 4.94–4.26 (m, 10H, H-1″,2′,Bn), 4.11–4.05 (m, 3H, H-3′,5′,5″), 3.70–3.40 (m, 6H, H-2″,3″,4′,5′,6″), 3.26 (d, J = 8.0 Hz, 1H, H-4″); 13C NMR (101 MHz, DMSO): δ 157.9 (C-6), 152.3 (CH-2), 150.8 (C-4), 139.1 (CH), 138.8 (CH), 138.7 (CH), 138.6 (CH), 128.7 (CH), 128.7 (CH), 128.6 (C-Bn), 128.6 (CH), 128.5 (CH), 128.2 (CH), 128.1 (CH), 128.1 (CH), 128.1 (CH), 128.0 (CH), 127.9 (CH), 127.8 (CH), 127.8 (CH), 122.0 (CH-8), 103.3 (CH-1″), 103.2 (C-5), 100.5 (CH-7), 87.6 (CH-1′), 84.1 (CH-4′), 82.6 (CH-5″), 82.1 (CH-4″), 78.1 (CH-3″), 74.9 (CH-2′), 74.4 (CH2), 74.3 (CH2), 74.2 (CH2), 74.1 (CH-3′), 72.8 (CH2), 71.1 (CH-2″), 70.6 (CH2-5′), 69.1 (CH2-6″); HRMS Calcd. For C45H49N4O9+ [M + H]+: 789.3494, found: 789.3499.
3.14. Synthesis of 5′-O-α-d-Glucopyranosyl tubercidin (2)
To the stirred suspension of compound 16 (100 mg, 0.13 mmol) in THF (10 ml) and methanol (10 mL) was added 20% Pd(OH)2/C (50 mg, 0.04 mmol), and the mixture was stirred at 40 °C for 12 h under a continuous hydrogen environment. The mixture was filtered and the filtrate was concentrated to provide the crude product, which was purified by isocratic HPLC (H2O/CH3OH, 4:1) to provide 43.5 mg compound 2 (80.1% yield) as a white solid; mp: 120–122 °C; = 10.5 (c = 0.08, H2O); 1H NMR (400 MHz, DMSO): δ 8.09 (s, 1H, H-2), 7.76 (d, J = 3.2 Hz, 1H, H-8), 7.20 (s, 2H, NH2), 6.61 (d, J = 3.4 Hz, 1H, H-7), 6.14 (d, J = 7.0 Hz, 1H, H-1′), 5.21 (d, J = 4.6 Hz, 2H, 2″, 3′-OH), 5.10 (d, J = 4.3 Hz, 1H, 2′-OH), 4.92 (d, J = 4.9 Hz, 1H, 3″-OH), 4.84 (d, J = 3.6 Hz, 1H, 4″-OH), 4.72 (d, J = 3.1 Hz, 1H, H-1″), 4.49 (s, 1H, 6″-OH), 4.43 (m, 1H, H-2′), 4.10 (s, 1H, H-4′), 4.08 (d, J = 1.7 Hz, 1H, H-3′), 3.78 (dd, J = 10.9, 2.8 Hz, 1H, H-5′), 3.66–3.63 (m, 1H, H-6′), 3.47–3.42 (m, 3H, H-5′,4″,6″), 3.40–3.34 (m, 2H, H-2″,5″), 3.13–3.07 (m, 1H, H-3″); 13C NMR (101 MHz, DMSO): δ 157.3 (C-6), 151.5 (CH-2), 150.6 (C-4), 122.4 (CH-8), 102.8 (C-5), 100.0 (CH-7), 98.5 (CH-1″), 85.9 (CH-1′), 83.2 (CH-4′), 74.2 (CH-2′), 73.3 (CH-4″), 72.7 (CH-5″), 71.5 (CH-2″), 70.9 (CH-3′), 70.0 (CH-3″), 67.0 (CH2-5′), 60.9 (CH2-6″); HRMS Calcd. For C17H25N4O9+ [M + H]+: 429.1616, found: 429.1620.
3.15. Synthesis of 5′-O-β-d-Glucopyranosyl Tubercidin (17)
Compound 16′ was converted into 17 (79.0% yield) as described for the synthesis of 2; mp: 156–157 °C; = −37 (c = 0.08, H2O); 1H NMR (400 MHz, DMSO): δ 8.07 (s, 1H, H-2), 7.38 (d, J = 3.7 Hz, 1H, H-8), 7.09 (s, 2H, 6-NH2), 6.61 (d, J = 3.6 Hz, 1H, H-7), 6.08 (d, J = 5.6 Hz, 1H, H-1′), 5.26 (d, J = 5.5 Hz, 1H, 2′-OH), 5.17 (d, J = 4.6 Hz, 1H, 3′-OH), 5.03 (d, J = 3.7 Hz, 1H, 2″-OH), 4.95 (s, 1H, 3″-OH), 4.95 (s, 1H, 4″-OH), 4.51 (s, 1H, 6″-OH), 4.36 (d, J = 5.0 Hz, 1H, H-2′), 4.21 (d, J = 7.8 Hz, 1H, H-1″), 4.14 (d, J = 4.4 Hz, 1H, H-3′), 4.02–3.95 (m, 2H, H-4′,5′), 3.68 (d, J = 9.2 Hz, 1H, H-6″), 3.59 (dd, J = 11.9, 5.7 Hz, 1H, H-5′), 3.44 (d, J = 6.7 Hz, 1H, H-6″), 3.15 (d, J = 9.9 Hz, 1H, H-3″), 3.12–3.04 (m, 2H, H-4″,5″), 3.00 (d, J = 7.8 Hz, 1H, H-2″); 13C NMR (101 MHz, DMSO): δ 157.5 (C-6), 151.7 (CH-2), 150.7 (C-4), 122.2 (CH-8), 103.5 (CH-1″), 103.1 (C-5), 100.5 (CH-7), 87.1 (CH-1′), 83.1 (CH-4′), 77.4 (CH-2″), 77.2 (CH-2′), 74.2 (CH-4″), 74.1 (C-5″), 71.1 (CH-3′), 70.5 (CH-3″), 69.5 (CH2-5′), 61.5 (CH2-6″); HRMS Calcd. For C17H25N4O9+ [M + H]+: 429.1616, found: 429.1619.


{kind=link}
{kind=link}
{kind=link}
{kind=link}


