Identification, Purification and Molecular Characterization of Chondrosin, a New Protein with Anti-tumoral Activity from the Marine Sponge Chondrosia Reniformis Nardo 1847


 , , , , , ,
, , , , , , 
Abstract
1. Introduction
2. Results and Discussion
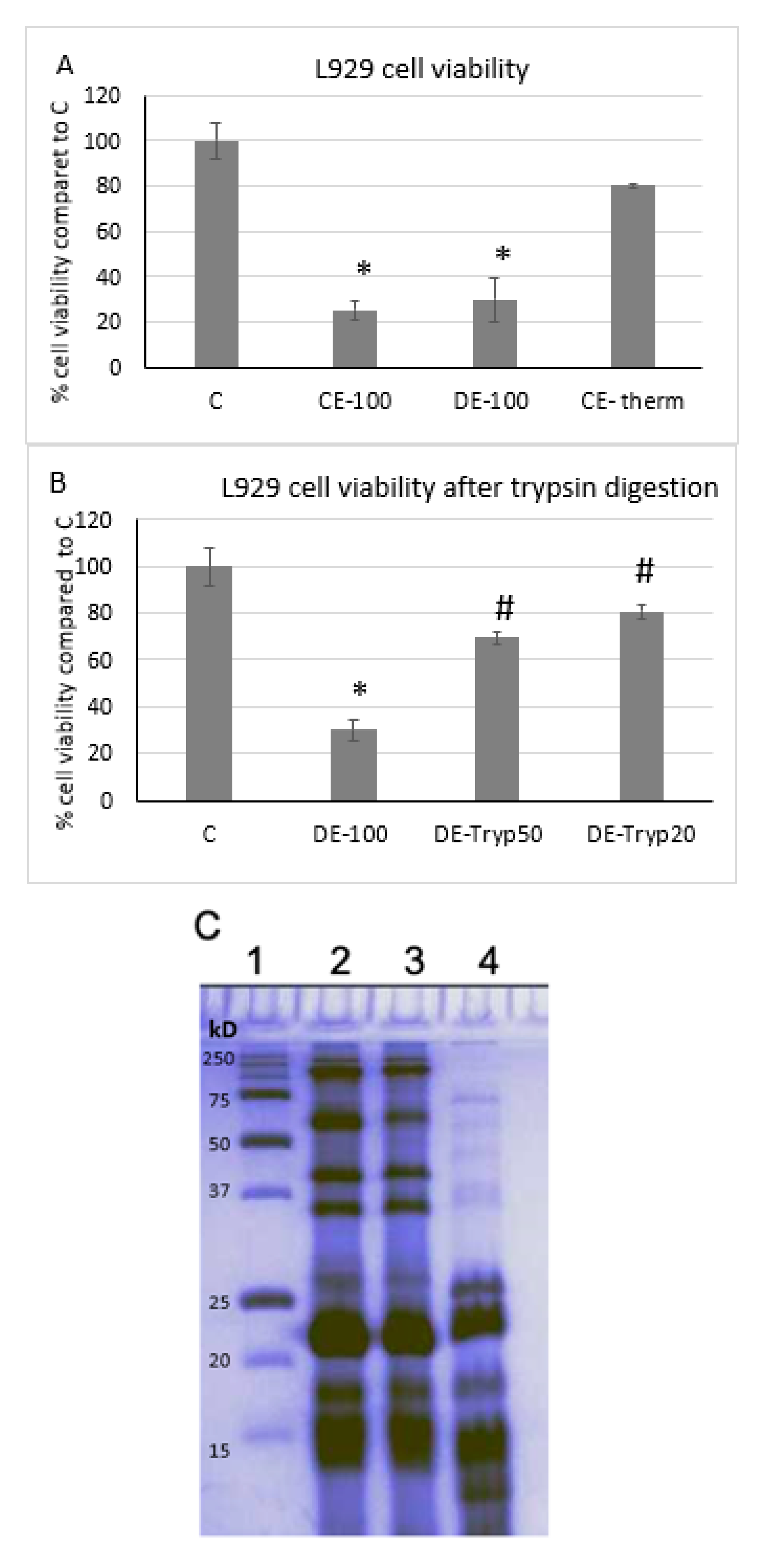
2.1. The Cytotoxicity of a Crude Extract (CE) of C. reniformis is Due to a Protein Fraction
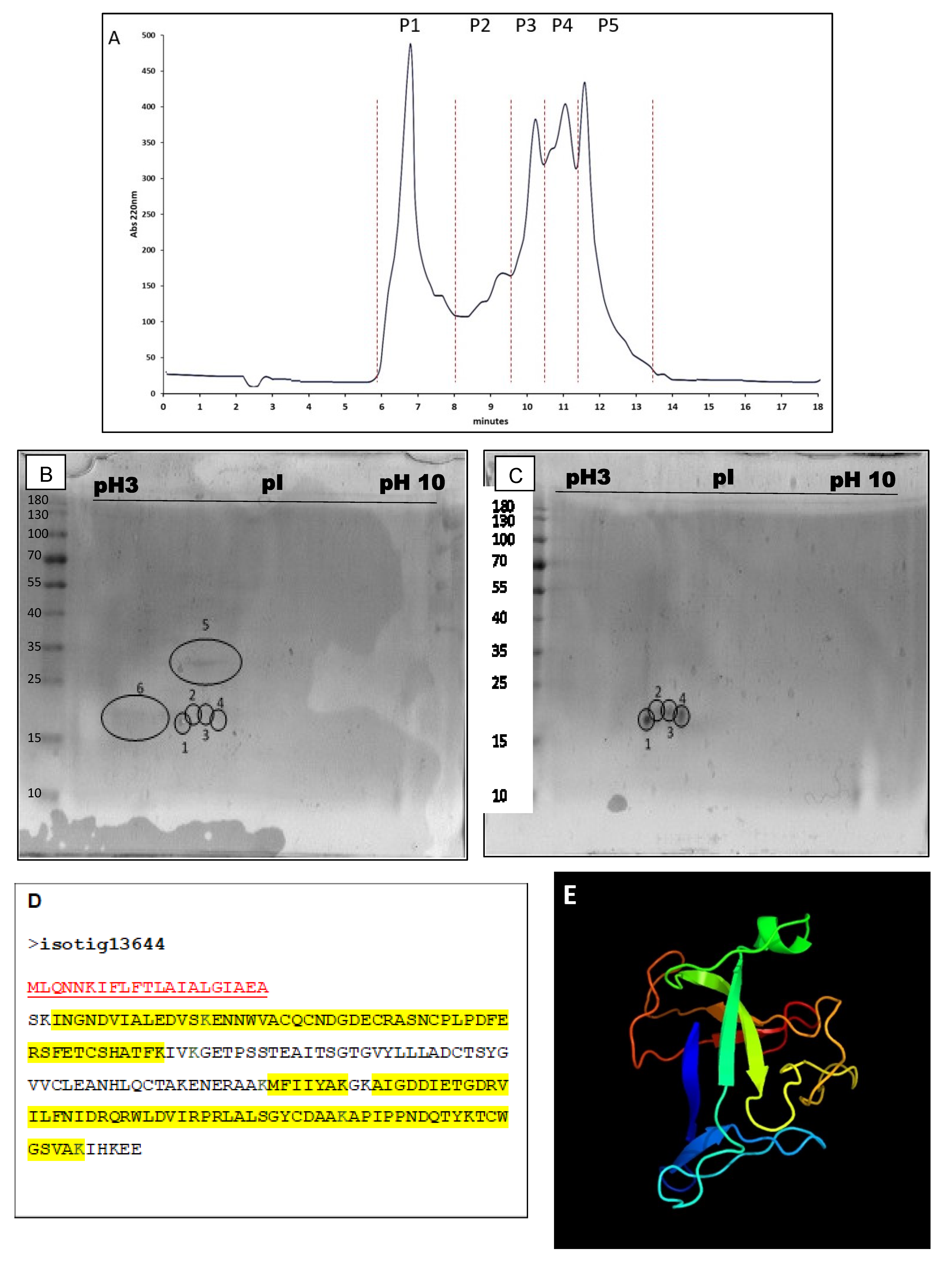
2.2. Identification and Characterization of Chondrosin
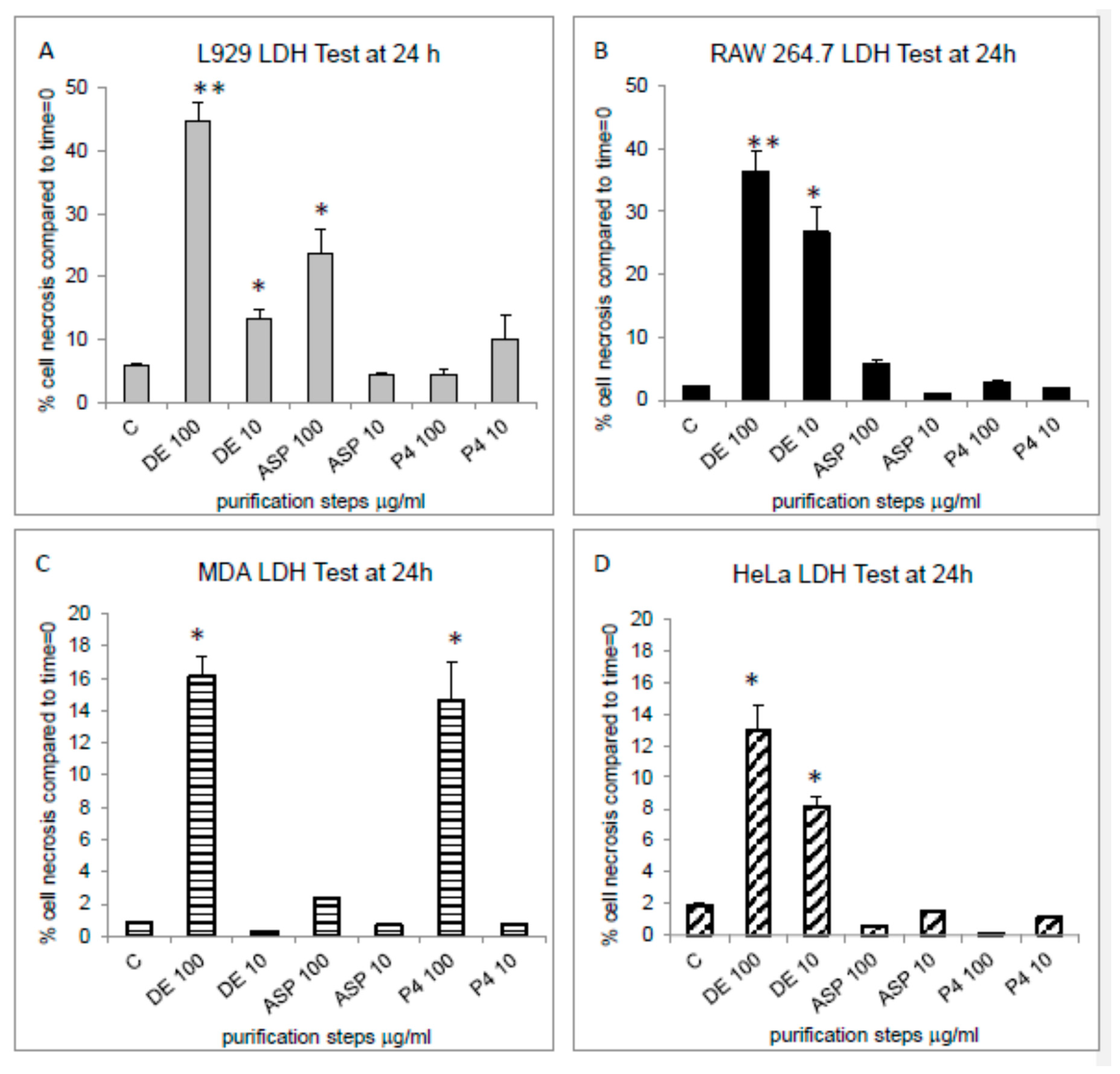
2.3. Anti-Tumour Activity of Protein Purified Fractions Derived from C. reniformis CE
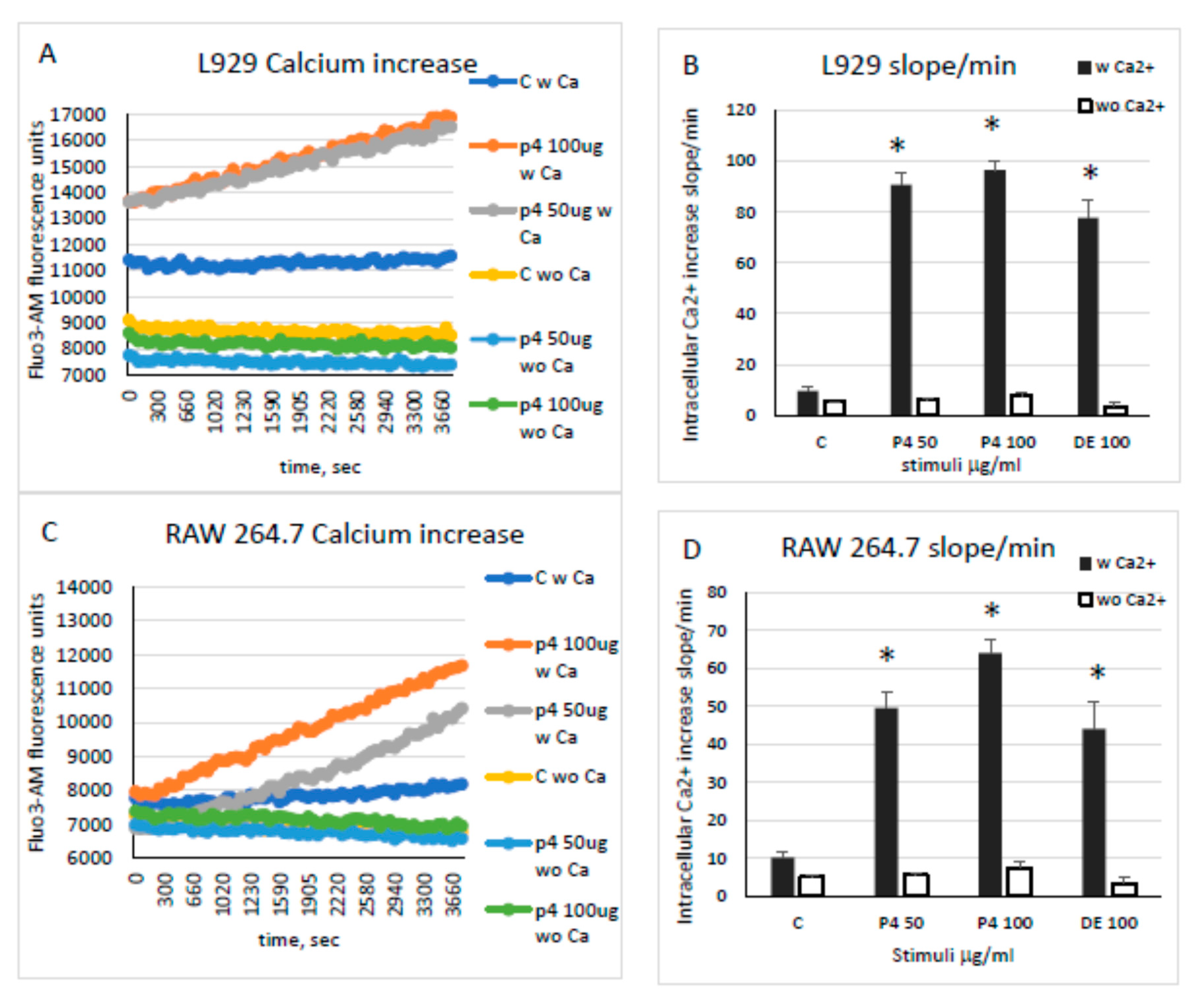
2.4. Mechanisms of Toxicity of Chondrosin on Tumour Cells
2.4.1. Necrosis and Apoptosis Assessment
2.4.2. Cytosolic Calcium Rise and Reactive Oxygen Species (ROS) Production in Challenged Cells
3. Conclusions
4. Materials and Methods
4.1. Preparation of Crude Extracts (CE) of C. reniformis
4.1.1. Chemical Nature Assessment of CE Cytotoxic Activity
4.1.2. Salting out of DE Proteins
4.1.3. HPLC Protein Purification
4.2. Proteomic Analysis
4.2.1. Protein Precipitation
4.2.2. D Gel Electrophoresis
4.2.3. In-Gel Digestion
4.2.4. Nano-LC Mass Spectrometry
4.2.5. Protein Sequence Analysis and 3D Modelling
4.3. Cell Cultures
4.3.1. Cell Toxicity by MTT and LDH Tests
4.3.2. Apoptosis Detection by FACS Analysis and Confocal Microscopy
4.3.3. Intracellular Calcium Detection
4.3.4. Intracellular ROS Detection
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Parma, L.; Fassini, D.; Bavastrello, G.; Wilkie, I.C.; Bonasoro, F.; Carnevali, D.C. Ecology and physiology of mesohyl creep in Chondrosia reniformis. J. Exp. Mar. Biol. Ecol. 2012, 428, 24–31. [Google Scholar] [CrossRef]
- Fassini, D.; Parma, L.; Lembo, F.; Candia Carnevali, M.D.; Wilkie, I.C.; Bonasoro, F. The reaction of the sponge Chondrosia reniformis to mechanical stimulation is mediated by the outer epithelium and the release of stiffening factor(s). Zoology (Jena) 2014, 117, 282–291. [Google Scholar] [CrossRef]
- Pozzolini, M.; Bruzzone, F.; Berilli, V.; Mussino, F.; Cerrano, C.; Benatti, U.; Giovine, M. Molecular characterization of a nonfibrillar collagen from the marine sponge Chondrosia reniformis Nardo 1847 and positive effects of soluble silicates on its expression. Mar. Biotechnol. 2012, 14, 281–293. [Google Scholar] [CrossRef]
- Pozzolini, M.; Scarfì, S.; Ghignone, S.; Mussino, F.; Vezzulli, L.; Cerrano, C.; Giovine, M. Molecular characterization and expression analysis of the first Porifera tumor necrosis factor superfamily member and of its putative receptor in the marine sponge Chondrosia reniformis. Dev. Comp. Immunol. 2016, 57, 88–98. [Google Scholar] [CrossRef]
- Pozzolini, M.; Scarfì, S.; Gallus, L.; Ferrando, S.; Cerrano, C.; Giovine, M. Silica-induced fibrosis: An ancient response from the early metazoans. J. Exp. Biol. 2017, 220, 4007–4015. [Google Scholar] [CrossRef] [PubMed]
- Nickel, M.; Brümmer, F. In vitro sponge fragment culture of Chondrosia reniformis (Nardo, 1847). J. Biotechnol. 2003, 100, 147–159. [Google Scholar] [CrossRef]
- Pozzolini, M.; Gallus, L.; Ghignone, S.; Ferrando, S.; Candiani, S.; Bozzo, M.; Bertolino, M.; Costa, G.; Bavestrello, G.; Scarfì, S. Insights into the evolution of metazoan regenerative mechanisms: Roles of TGF superfamily members in tissue regeneration of the marine sponge Chondrosia reniformis. J. Exp. Biol. 2019, 222, jeb207894. [Google Scholar] [CrossRef] [PubMed]
- Swatschek, D.; Schatton, W.; Kellermann, J.; Müller, W.E.G.; Kreuter, J. Marine sponge collagen: Isolation, characterization and effects on the skin parameters surface-pH, moisture and sebum. Eur. J. Pharm. Biopharm. 2002, 53, 107–113. [Google Scholar] [CrossRef]
- Nicklas, M.; Schatton, W.; Heinemann, S.; Hanke, T.; Kreuter, J. Enteric coating derived from marine sponge collagen. Drug Dev. Ind. Pharm. 2009, 35, 1384–1388. [Google Scholar] [CrossRef]
- Nicklas, M.; Schatton, W.; Heinemann, S.; Hanke, T.; Kreuter, J. Preparation and characterization of marine sponge collagen nanoparticles and employment for the transdermal delivery of 17β-estradiol-hemihydrate. Drug Dev. Ind. Pharm 2009, 35, 1035–1042. [Google Scholar] [CrossRef]
- Pozzolini, M.; Scarfì, S.; Gallus, L.; Castellano, M.; Vicini, S.; Cortese, K.; Gagliani, M.; Bertolino, M.; Costa, G.; Giovine, M. Production, characterization and biocompatibility evaluation of collagen membranes derived from marine sponge Chondrosia reniformis Nardo, 1847. Mar. Drugs 2018, 16, 111. [Google Scholar] [CrossRef]
- Pozzolini, M.; Millo, E.; Oliveri, C.; Mirata, S.; Salis, A.; Damonte, G.; Arkel, M.; Scarfì, S. Elicited ROS scavenging activity, photoprotective, and wound-healing properties of collagen-derived peptides from the marine sponge Chondrosia reniformis. Mar. Drugs 2018, 16, 465. [Google Scholar] [CrossRef] [PubMed]
- Pozzolini, M.; Scarfì, S.; Mussino, F.; Salis, A.; Damonte, G.; Benatti, U.; Giovine, M. Pichia pastoris production of a prolyl 4-hydroxylase derived from Chondrosia reniformis sponge: A new biotechnological tool for the recombinant production of marine collagen. J. Biotechnol. 2015, 208, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Pozzolini, M.; Scarfì, S.; Mussino, F.; Ferrando, S.; Gallus, L.; Giovine, M. Molecular cloning, characterization, and expression analysis of a prolyl 4-hydroxylase from the marine sponge Chondrosia reniformis. Mar. Biotechnol. (Ny) 2015, 17, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Gökalp, M.; Wijgerde, T.; Sarà, A.; De Goeij, J.M.; Osinga, R. Development of an integrated mariculture for the collagen-rich sponge Chondrosia reniformis. Mar. Drugs 2019, 17, 29. [Google Scholar] [CrossRef]
- Pronzato, R.; Manconi, R. Mediterranean commercial sponges: Over 5000 years of natural history and cultural heritage. Mar. Ecol. 2008, 29, 146–166. [Google Scholar] [CrossRef]
- Mebs, D.; Weiler, I.; Heinke, H.F. Bioactive proteins from marine sponges: Screening of sponge extracts for hemagglutinating, hemolytic, ichthyotoxic and lethal properties and isolation and characterization of hemagglutinins. Toxicon 1985, 23, 955–962. [Google Scholar] [CrossRef]
- Cheung, R.C.F.; Wong, J.H.; Pan, W.; Chan, Y.S.; Yin, C.; Dan, X.; Ng, T.B. Marine lectins and their medicinal applications. Appl. Microbiol. Biotechnol 2015, 99, 3755–3773. [Google Scholar] [CrossRef]
- Lazcano-Perez, F.; A. Roman-Gonzalez, S.; Sanchez-Puig, N.; Arreguin-Espinosa, R. Bioactive peptides from marine organisms: A short overview. Protein Pept. Lett. 2012, 19, 700–707. [Google Scholar] [CrossRef]
- Görg, A.; Weiss, W.; Dunn, M.J. Current two-dimensional electrophoresis technology for proteomics. Proteomics 2004, 4, 3665–3685. [Google Scholar] [CrossRef]
- Deng, Z.; Bu, S.; Wang, Z.-Y. Quantitative analysis of protein phosphorylation using two-dimensional difference gel electrophoresis. Methods Mol. Biol. 2012, 876, 47–66. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gao, B.; Zhu, S. Exon shuffling and origin of scorpion venom biodiversity. Toxins 2016, 9, 10. [Google Scholar] [CrossRef]
- Fry, B.G.; Roelants, K.; Winter, K.; Hodgson, W.C.; Griesman, L.; Kwok, H.F.; Scanlon, D.; Karas, J.; Shaw, C.; Wong, L.; et al. Novel venom proteins produced by differential domain-expression strategies in beaded lizards and Gila Monsters (genus Heloderma). Mol. Biol. Evol. 2010, 27, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.R.; Eid, R.; Miller, K.A.; Boucher, E.; Mandato, C.A.; Greenwood, M.T. Intracellular second messengers mediate stress inducible hormesis and programmed cell death: A review. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 773–792. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Hu, N.; Feng, X.; Sun, Y.; Pu, D.; Lv, X.; Hao, Z.; Li, Y.; Xue, W.; He, L. Cav1.3 is upregulated in osteoporosis rat model and promotes osteoclast differentiation from preosteoclast cell line RAW264.7. J. Cell. Physiol. 2019, 234, 12821–12827. [Google Scholar] [CrossRef]
- Kim, S.; Yun, H.-M.; Baik, J.-H.; Chung, K.C.; Nah, S.-Y.; Rhim, H. Functional interaction of neuronal Cav 1.3 L-type Calcium channel with ryanodine receptor type 2 in the rat hippocampus. J. Biol. Chem. 2007, 282, 32877–32889. [Google Scholar] [CrossRef]
- Starkov, A.A.; Chinopoulos, C.; Fiskum, G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium 2004, 36, 257–264. [Google Scholar] [CrossRef]
- Pozzolini, M.; Mussino, F.; Cerrano, C.; Scarfì, S.; Giovine, M. Sponge cell cultivation: Optimization of the model Petrosia ficiformis (Poiret 1789). J. Exp. Mar. Biol. Ecol. 2014, 454, 70–77. [Google Scholar] [CrossRef]
- Neuhoff, V.; Arold, N.; Taube, D.; Ehrhardt, W. Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis 1988, 9, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Pozzolini, M.; Vergani, L.; Ragazzoni, M.; Delpiano, L.; Grasselli, E.; Voci, A.; Giovine, M.; Scarfì, S. Different reactivity of primary fibroblasts and endothelial cells towards crystalline silica: A surface radical matter. Toxicology 2016, 361–362, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Pozzolini, M.; Scarfi, S.; Benatti, U.; Giovine, M. Interference in MTT cell viability assay in activated macrophage cell line. Anal. Biochem. 2003, 313, 338–341. [Google Scholar] [CrossRef]
- Magnone, M.; Bauer, I.; Poggi, A.; Mannino, E.; Sturla, L.; Brini, M.; Zocchi, E.; De Flora, A.; Nencioni, A.; Bruzzone, S. NAD+ levels control Ca2+ store replenishment and mitogen-induced increase of cytosolic Ca2+ by Cyclic ADP-ribose-dependent TRPM2 channel gating in human T lymphocytes. J. Biol. Chem. 2012, 287, 21067–21081. [Google Scholar] [CrossRef] [PubMed]
- Scarfì, S.; Magnone, M.; Ferraris, C.; Pozzolini, M.; Benvenuto, F.; Benatti, U.; Giovine, M. Ascorbic acid pre-treated quartz stimulates TNF-alpha release in RAW 264.7 murine macrophages through ROS production and membrane lipid peroxidation. Respir. Res. 2009, 10, 25. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| P4 | ||||||
|---|---|---|---|---|---|---|
| Spot | C. R. Database Id | Score | Coverage | Proteins | Unique Peptides | Peptides |
| 1 | isotig13644 | 207.90 | 58.29 | 1 | 10 | 11 |
| 2 | isotig13644 | 133.15 | 58.29 | 1 | 8 | 11 |
| 3 | isotig13644 | 137.50 | 58.29 | 1 | 8 | 11 |
| 4 | isotig13644 | 237.20 | 58.29 | 1 | 11 | 11 |
| 5 | isotig11703 | 66.86 | 37.37 | 1 | 4 | 10 |
| 6 | isotig13644 | 19.68 | 45.23 | 1 | 7 | 8 |
| 6 | isotig02770 | 74.19 | 45.81 | 2 | 2 | 6 |
| 6 | isotig02771 | 47.81 | 45.81 | 2 | 2 | 6 |
| P5 | ||||||
| Spot | C. R. Database Id | Score | Coverage | Proteins | Unique peptides | Peptides |
| 1 | isotig13644 | 379.61 | 58.29 | 1 | 12 | 12 |
| 2 | isotig13644 | 340.54 | 58.29 | 1 | 10 | 12 |
| 3 | isotig13644 | 181.69 | 58.29 | 1 | 11 | 11 |
| 4 | isotig13644 | 309.34 | 58.29 | 1 | 12 | 12 |
| C. R. Database Id | aa | MW (kDa) | IP | Accession Number | Description | Organism | E Value |
|---|---|---|---|---|---|---|---|
| isotig13644 | 199 | 21.9 | 5.27 | WP_142038611.1 | DNA-binding response regulator | Arthrobacter sp. | 5.9 |
| isotig11703 | 281 | 31.9 | 5.76 | ABR53885.1 | Astrosclerin-1 | Astrosclera willeyana | 7.30 × 10−61 |
| isotig02770 | 203 | 23.3 | 4.59 | XP_016993171.1 | PREDICTED: ubiquitin-conjugating enzyme E2 H | Drosophila takahashii | 4.4 |
| isotig02771 | 203 | 23.3 | 4.55 | XP_016993171.1 | PREDICTED: ubiquitin-conjugating enzyme E2 H | Drosophila takahashii | 4.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scarfì, S.; Pozzolini, M.; Oliveri, C.; Mirata, S.; Salis, A.; Damonte, G.; Fenoglio, D.; Altosole, T.; Ilan, M.; Bertolino, M.; et al. Identification, Purification and Molecular Characterization of Chondrosin, a New Protein with Anti-tumoral Activity from the Marine Sponge Chondrosia Reniformis Nardo 1847. Mar. Drugs 2020, 18, 409. https://doi.org/10.3390/md18080409
Scarfì S, Pozzolini M, Oliveri C, Mirata S, Salis A, Damonte G, Fenoglio D, Altosole T, Ilan M, Bertolino M, et al. Identification, Purification and Molecular Characterization of Chondrosin, a New Protein with Anti-tumoral Activity from the Marine Sponge Chondrosia Reniformis Nardo 1847. Marine Drugs. 2020; 18(8):409. https://doi.org/10.3390/md18080409
Chicago/Turabian StyleScarfì, Sonia, Marina Pozzolini, Caterina Oliveri, Serena Mirata, Annalisa Salis, Gianluca Damonte, Daniela Fenoglio, Tiziana Altosole, Micha Ilan, Marco Bertolino, and et al. 2020. "Identification, Purification and Molecular Characterization of Chondrosin, a New Protein with Anti-tumoral Activity from the Marine Sponge Chondrosia Reniformis Nardo 1847" Marine Drugs 18, no. 8: 409. https://doi.org/10.3390/md18080409
APA StyleScarfì, S., Pozzolini, M., Oliveri, C., Mirata, S., Salis, A., Damonte, G., Fenoglio, D., Altosole, T., Ilan, M., Bertolino, M., & Giovine, M. (2020). Identification, Purification and Molecular Characterization of Chondrosin, a New Protein with Anti-tumoral Activity from the Marine Sponge Chondrosia Reniformis Nardo 1847. Marine Drugs, 18(8), 409. https://doi.org/10.3390/md18080409